- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RI-SEC-seq: Comprehensive Profiling of Nonvesicular Extracellular RNAs with Different Stabilities
Published: Vol 11, Iss 4, Feb 20, 2021 DOI: 10.21769/BioProtoc.3918 Views: 6094
Reviewed by: RAMESH KUDIRAJuliang QinShaoxin XU
Original research article
The authors used this protocol in:

Dec 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Exosomes and other extracellular vesicles (EVs) are considered the main vehicles transporting RNAs in extracellular samples, including human bodily fluids. However, a major proportion of extracellular RNAs (exRNAs) do not copurify with EVs and remain in ultracentrifugation supernatants of cell-conditioned medium or blood serum. We have observed that nonvesicular exRNA profiles are highly biased toward those RNAs with intrinsic resistance to extracellular ribonucleases. These highly resistant exRNAs are interesting from a biomarker point of view, but are not representative of the actual bulk of RNAs released to the extracellular space. In order to understand exRNA dynamics and capture both stable and unstable RNAs, we developed a method based on size-exclusion chromatography (SEC) fractionation of RNase inhibitor (RI)-treated cell-conditioned medium (RI-SEC-seq). This method has allowed us to identify and study extracellular ribosomes and tRNAs, and offers a dynamical view of the extracellular RNAome which can impact biomarker discovery in the near future.
Graphical abstract:
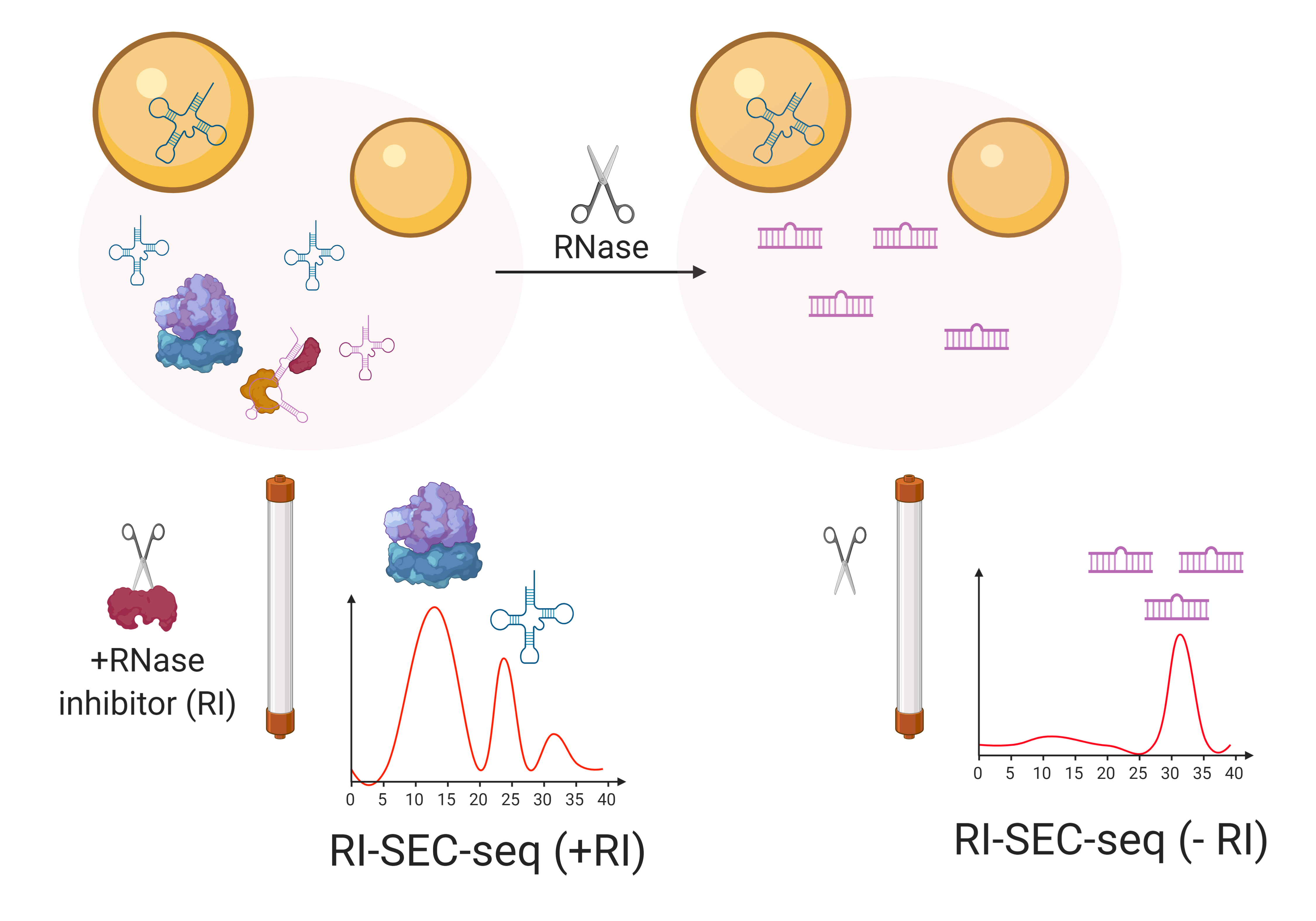
Overview of the RI-SEC-seq protocol: sequencing of size-exclusion chromatography fractions from nonvesicular extracellular samples treated or not with RNase inhibitors (+/- RI)
Background
Extracellular RNAs (exRNAs) are involved in intercellular communication and are promising disease biomarkers in minimally invasive liquid biopsies (O’Brien et al., 2020). To be transported in human bodily fluids, exRNAs need to achieve some degree of protection against extracellular ribonucleases, which are ubiquitous in the extracellular milieu. By far, the most studied and best understood mechanism is RNA encapsulation inside extracellular vesicles (EVs) and release of these endogenous lipid nanoparticles into the extracellular space (Kalluri and LeBleu, 2020). Beyond simply protecting exRNAs from degradation, EVs contain a set of surface proteins which are involved in cellular uptake (Hoshino et al., 2015), behaving as vehicles for RNA transfer between cells. This can occur both physiologically (Thomou et al., 2017) or under therapeutic biotechnological setups (Kamerkar et al., 2017; Kojima et al., 2018).
Much less is known about those exRNAs which are not associated with EVs (Li et al., 2018). However, early work has shown that the majority of microRNAs in human plasma are forming soluble ribonucleoprotein complexes with Argonaute 2 and are not transported inside EVs (Arroyo et al., 2011; Turchinovich et al., 2011). Later work has shown that the RNA content of EVs is not as high as previously suggested (Chevillet et al., 2014) and we have observed that most exRNAs in cell culture media remain in ultracentrifugation supernatants (Tosar et al., 2015).
We were surprised to find that the majority of the small RNAs that we could sequence in ultracentrifugation supernatants of cell-conditioned medium were 5’ tRNA halves derived from glycine and glutamic acid issoaceptors (Tosar et al., 2015). These sequences were also the most abundant in blood plasma, serum and other biofluids (Srinivasan et al., 2019), where they also seemed to be transported outside EVs (Dhahbi et al., 2013). We later observed these sequences could form dimers and that these dimers were highly resistant to single-stranded ribonucleases (Tosar et al., 2018). These observations suggested that extracellular nonvesicular RNA profiles are prone to survivorship bias (Tosar et al., 2021), where we tend to see only those RNAs that are more resistant to degradation. Although this could be desirable for biomarker studies, it can be problematic for understanding RNA release mechanisms. Thus, we developed a method to study nonvesicular exRNAs irrespectively of their extracellular stability. This method, called RI-SEC-seq (for: size-exclusion chromatography [SEC] fractionation of RNase inhibitor [RI]-treated cell-conditioned medium), enabled us to study RNAs in the form they were effectively released from cells. Following this procedure, we discovered the presence of extracellular ribosomes and tRNAs, and the extracellular biogenesis of extracellular tRNA-derived fragments (Tosar et al., 2020). The latter was independently validated by others, using cell lines devoid of RNase I (Nechooshtan et al., 2020).
Here, we provide the experimental procedure so that readers can also obtain snapshots of the extracellular nonvesicular RNAome under low, intermediate and high RNase activities. This will help to characterize exRNA profiles in a comprehensive and dynamic manner, and to identify other stable RNAs with biomarker potential.
Materials and Reagents
T-75 flask (Corning, catalog number: 430641U )
0.22 µm or 0.45 µm membrane (Millipore, catalog number: GSWP04700 )
MCF-7 cell line (ATCC, ATCC® HTB-22TM), storage: liquid nitrogen when not in use. Cryopreservation: follow ATCC recommendations
1 ml syringe
14 ml, Open-Top Thinwall Ultra-Clear Tube, 14 × 95 mm, 50Pk (Beckman Coulter Life Sciences, catalog number: 344060 ), storage: room temperature
Vivaspin 20 centrifugal concentrator, MWCO 10 kDa, PES membrane (Sartorius, catalog number: VS2001 ), storage: room temperature
96 Well Standard Assay Block, 2 ml, Certified RNase/DNase free, for collection of chromatographic fraction (Costar, Corning Incorporated, catalog number: 3961 ), storage: room temperature
Superdex® 200 10/300 GL (GE Life Sciences, Sigma/Merck, catalog number: GE17-5175-01, discontinued product; alternative: Superdex® 200 Increase 10/300 GL, catalog number: GE28-9909-44 ), storage: room temperature
DMEM, high glucose, GlutaMAX TM Supplement (Gibco, Thermo Scientific, catalog number: 10569010 ), storage: 4 °C
Fetal bovine serum (FBS), qualified, origin: Brazil (Gibco, Thermo Scientific, catalog number: 12657029 ), storage: aliquoted (sterile) at -20 °C
MEGMTM Mammary Epithelial Cell Growth Medium BulletKitTM (Lonza, catalog number: CC-3150 ), storage: the basal MEBM medium is stored at 4 °C
Note: The MEGM SingleQuotsTM Supplements are stored at -20 °C. Once mixed, the complete MEGM medium is stored at 4 °C until expiry. We do not add the bovine pituitary extract or the antibiotics included in the kit.
DPBS, no calcium, no magnesium (Gibco, Thermo Scientific, catalog number: 14190144 ), storage: 4 °C. Use this sterile solution for cell culture
DPBS (10×), no calcium, no magnesium (Gibco, Thermo Scientific, catalog number: 14200075 ). Make 1× solution with ultrapure or DEPC (Diethyl pyrocarbonate)-treated water, filter, and use as mobile phase in size-exclusion chromatography
Trypsin-EDTA (0.05%), phenol red (Gibco, Thermo Scientific, catalog number: 25300062 ), storage: -20 °C
RNase inhibitor, murine (New England Biolabs, catalog number: M0314S ), storage: -20 °C
TRIzolTM reagent (Invitrogen, Thermo Scientific, catalog number: 15596018 ), storage: 4 °C
TRIzolTM LS reagent (Invitrogen, ThermoScientific, catalog number: 10296028 ), storage: 4 °C
2-propanol, for molecular biology (Sigma-Aldrich, catalog number: I9516-500ML ), storage: room temperature. Reagent needed for RNA purification based on the TRIzol method
Ethyl alcohol, pure, for molecular biology, storage: room temperature. Reagent needed for RNA purification based on the TRIzol method
Glycogen, RNA grade (Thermo Scientific, catalog number: R0551 ), storage: -20 °C. Reagent needed for RNA purification based on the TRIzol method
UltraPureTM DNase/RNase-Free Distilled Water (Invitrogen, ThermoScientific, catalog number: 10977035 ), storage: room temperature, sterile
Note: We do not use this water for large volumes of buffers such as those used in size-exclusion chromatography. For those purposes, we use MilliQ water that we validate as RNase-free in house. Autoclaved DEPC-treated water can be used instead. In this case, add DEPC (diethylpyrocarbonate, Sigma, catalog number: D5758 ) to a final concentration of 0.1% v/v, incubate overnight at room temperature and then autoclave to inactivate DEPC.
10× Tris-Borate-EDTA Electrophoresis Buffer (ThermoScientific, catalog number: B52 ), storage: room temperature
Acrylamide/bis-Acrylamide 30% solution, bioreagent, suitable for electrophoresis, 29:1 (Sigma-Aldrich, catalog number: A3574 ), storage: 4 °C. Hazardous (read MSDS)
SYBRTM Gold nucleic acid gel stain (10,000× concentrate in DMSO) (Invitrogen, ThermoScientific, catalog number: S11494 ), storage: -20 °C
RNA loading dye, 2× (New England Biolabs, catalog number: B0363S ), storage: -20 °C
QubitTM RNA HS Assay Kit (Invitrogen, ThermoScientific, catalog number: Q32852 ), storage: 4 °C
NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (New England Biolabs, catalog number: E7300S ), storage: -20 °C
MiSeq Reagent Kit v2 (300-cycles) (Ilumina, catalog number: MS-102-2002 ), storage: -20 °C (box 1) and 4 °C (box 2)
Equipment
NovexTM TBE-Urea Gels (ThermoFisher, catalog number: EC6875BOX )
XCell SureLock Mini-Cell (ThermoFisher, catalog number: EI0001 )
ÄKTA pure FPLC system (Cytiva, former GE Life Sciences) with multiple wavelength detectors (at least capable of simultaneous detection at 280 and 260 nm) and an automatic fraction collector with capacity for 96 well assay blocks (Costar, 2 mL, RNase-free, catalog number: 3961 )
QubitTM Fluorometer 2.0 (Thermo Scientific, catalog number: Q32866 , discontinued product. Replaced by Catalog number Q33216 )
MiSeq Next Generation Sequencer (Illumina)
Software
Note: All software used for sequencing data analysis was run using the Galaxy Europe web interface (https://usegalaxy.eu).
fastx_clipper (fastx_toolkit)
Fastqc
RNA STAR
BAM filter
bamCoverage
featureCounts
Procedure
Note: The chances of RNase contamination should be minimized throughout this protocol even if not specifically indicated. Use disposable RNase-free plastics and certified RNase-free solutions whenever possible. Use gloves in all steps even when handling non-hazardous reagents and work in RNase-decontaminated surfaces with RNase-decontaminated materials.
Cell culture
Note: For this procedure to be successful, cells must be grown in serum-free medium (SFM), to avoid the presence of serum RNases. In this protocol, we will use Mammary Epithelial Growth Medium (MEGM, Lonza) and MCF-7 cells, but this can be adapted to other cell lines. However, finding a suitable SFM for each cell line is not trivial. Serum deprivation can induce cell stress and this can affect both intracellular and extracellular RNA profiles. As described in Tosar et al. (2020), we monitor this by Western blot, looking at eIF2 subunit α (Ser 51) phosphorylation levels, and by nonradioactive Northern blot, monitoring the levels of stress-induced tRNA-derived fragments (Yamasaki et al., 2009). For certain cell types, commercial SFM formulations are available. We have observed that MCF-7 cells can be grown for short periods of time in MEGM, which is the recommended growth medium by ATCC for other mammary epithelial cell lines such as MCF-10A. However, we do not add the Bovine Pitutary Extract contained in the MEGM kit to keep this as a chemically-defined medium. We also do not add antibiotics to our cell cultures unless needed. In theory, this protocol could be applied to suspension cells, 3D cultures and organoids with slight modifications, although we have not tried these applications yet.
Seed aprox. One million cells in each of two T-75 flask. Grow cells up to 60-70% confluency in complete medium (e.g., DMEM + 10% FBS) at 37 °C and 5% CO2. Confluency should be adapted depending on the growth rate of each cell line. The aim is 80-90% confluency 48 h later.
Wash cells briefly with 3-5 ml pre-warmed DMEM or PBS.
Add 10 ml of pre-warmed MEGM. Grow cells for 24 h.
Wash cells again with pre-warmed MEGM. Add 10 ml of pre-warmed MEGM to one of the flasks (hereafter: control). Add 10 ml of pre-warmed MEGM + 80U (2 µl) of murine RNase inhibitor to the second flask (hereafter: RI-treated) and incubate for further 24 h.
Aspirate cell-conditioned medium from both flasks and discharge in 15 ml nuclease-free Falcon conical tubes. Add 1 µl of murine RNase inhibitor to the RI-treated flask (only). Spin down cells at 800 × g for 5 min at room temperature.
While centrifuging cell-conditioned medium, move the empty flasks containing the cell monolayers to a fume hood and lyse cells in 2 ml of Trizol. Gently tilt the flasks to lyse all cells, incubate 5 min at room temperature, close the lid and store the flask at -20 °C. This will be later used to obtain intracellular RNA profiles.
Place the supernatant from Step A5 to a new 15 ml nuclease-free Falcon conical tube and centrifuge at 2,000 × g for 20 min at 4 °C. We routinely store the supernatant from this spin (saved in a new tube) at -20 °C for later use.
Preparation of extracellular nonvesicular samples by ultracentrifugation
Note: Ultracentrifugation requires specific training in situ. This protocol is not intended to be substitute for this training. Improper use of ultracentrifuges can provoke sample lost, permanent damage to expensive equipment and put the safety of the laboratory personnel at risk.
Thaw the tubes from Step A7 by placing the tubes in a beaker containing water at room temperature. Exchange the water several times, as it will cool rapidly.
Add 1 µl of murine RNase inhibitor to the tube containing the RI-treated sample. Because recombinant RNase inhibitors contain many cysteines and are prone to oxidation, we usually add this boosters to ensure lack of RNase activity.
Spin the samples again at 2,000 × g for 20 min at 4 °C to remove precipitates that could have formed during freeze/thaw.
Place the samples in two identical ultraclear ultracentrifugation tubes. If necessary, adjust the volume of the samples with ice-cold nuclease-free PBS so that the tubes are up to their full capacity minus 2-3 mm from the top of the tube. All opposing tubes for a run must be filled to the same level with liquid of the same density.
Place the tubes in the buckets. We also check that buckets containing filled tubes show a difference in weight equal or less than 2 mg if they were placed in opposite positions of the rotor. Place the buckets in an already cooled rotor (stored in a cold room overnight). Place the rotor in the ultracentrifuge, set all relevant parameters and start vaccum.
Note: Forgetting to cool the rotors overnight played an important role in the development of this protocol. A science journalist wrote a story about this episode that gives some historical context (Khamsi, 2020).
Run for 2.5 h at 100,000 × g at 4 °C with maximum acceleration and break.
Transfer the supernatants from Step B6 to new tubes. Keep the tubes on ice.
Concentrate to 500 µl by ultrafiltration at 4 °C, using Vivaspin 20 units with a molecular weight cut-off of 10 kDa. Run ultrafiltration units with nuclease free PBS or water before loading the samples in order to clean the membranes. Transfer the concentrate to a new, nuclease-free 1.5 ml tube.
Size-exclusion chromatography
Note: This procedure needs to be done twice, with the RI-treated and control samples. Working with an FPLC system also requires specific training in situ as high pressures will be used. Avoid introduction of air to the column at all stages and the injection of samples or solvents containing dust or other particulates.
Filter 500 ml of ultrapure water and 500 ml of 1× PBS (prepared in ultrapure water) through a 0.22 µm or a 0.45 µm membrane using a vacuum filtration device. Degass the solutions with a magnetic stirrer under reduced pressure.
Set an alarm in the FPLC system lower or equal to the maximum pressure admitted by the column (in this case, 1.5 MPa). Connect a Superdex 200 10/300 column to the FPLC system. Remove ethanol from the column by passing at least 1.5 volumes of filtered and degassed water. Equilibrate the column in 1× PBS.
Connect a 500 µl loop to the system. Clean the loop with water, and then with filtered PBS. Avoid the introduction of air bubbles.
Spin the sample (from Step B8) at 10,000 × g for 10 min at 4 °C. Transfer the supernatant to a new tube.
Using a 1 ml syringe, inject the sample from Step C4 (500 µl) in the column.
Run at a flow rate of 1 ml/min by monitoring absorbance at 280 nm and 260 nm, simultaneously.
Collect fractions of 0.25 ml using a new, RNase-free 96-well plate. Start collection at least 2 ml before elution of the fractions corresponding to the void volume.
Identify fractions were ABS260 > ABS280 in both samples. In contrast to proteins, nucleic acids absorb more UV light at 260 nm than at 280 nm. Thus, these fractions correspond to the elution of nucleic acids or ribonucleoprotein complexes. At least in MCF-7 cells, the control sample should show a main 260 nm peak at Ve = 15 ml (that we call “P1”: adjusted Ve’ = 7.5 ml) and a lower peak at Ve = 16.5 ml (that we call “P2”: adjusted Ve’ = 9.0 ml). In contrast, the P2 peak should not be seen in the RI-treated sample, and a new peak should appear in the void volume (that we call “P0”: Ve = 7.5 ml; adjusted Ve’ = 0 ml) (Figure 1).
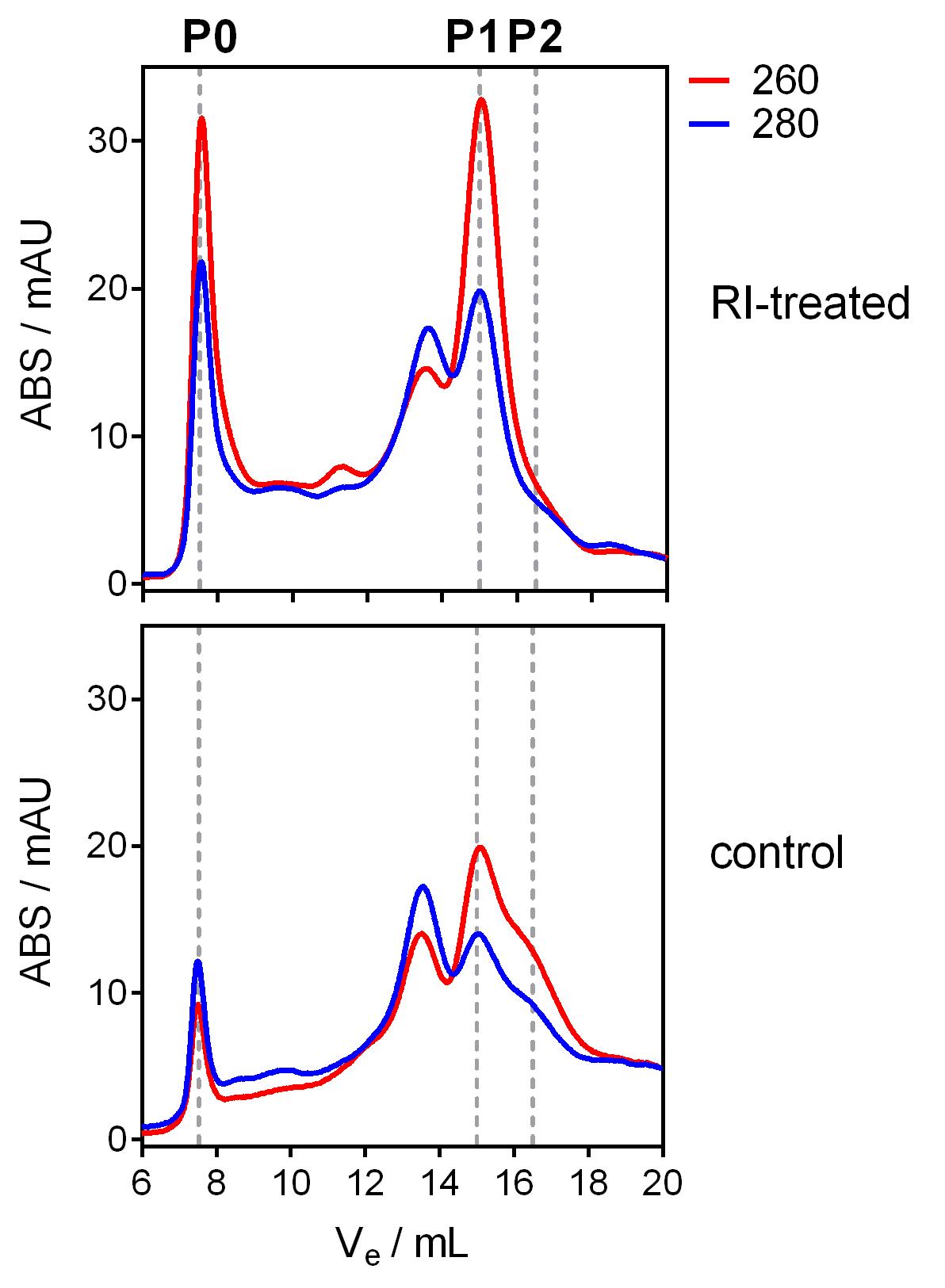
Figure 1. Representative chromatograms of RI-treated (top) and control (bottom) samples. The absorbance at 260 nm and 280 nm is shown in red and blue, respectively. Peaks with 280/260 > 1 correspond to the elution of proteins, while peaks with 280/260 < 1 correspond to the elution of nucleic acids and/or ribonucleoprotein complexes. Ve: elution volume. Modified from Tosar et al. (2020).Transfer fractions corresponding to peaks P0, P1 and P2 from both samples to new 1.5 ml RNase-free tubes. Make sure that paired fractions correspond to the same elution volumes, even if no evident peaks are present (e.g., P0 in control or P2 in RI-treated).
In a fume hood, add 750 µl of Trizol LS to each collected fraction (250 µl). Proceed with RNA extraction following the instructions of the manufacturers.
RNA analysis and next-generation sequencing
Resuspend the RNA pellet in 9 µl of RNase-free water. Check RNA concentration using 1 µl of each sample and a Qubit fluorimeter (following the instructions of the vendor). If using a Bioanalyzer to check RNA integrity, rembember these are not intracellular samples and therefore the concept of RNA Integrity Number (RIN) does not apply here. Nevertheless, the P0 peak in the RI-treated samples should contain intact ribosomal RNAs. Instead of using the Bioanalyzer, we characterize the RNAs in each fraction by denaturing gel electrophoresis using custom-made 6% polyacrylamide-8 M urea gels (P0) or 10% polyacrylamide-8M urea gels (P1 and P2) in 0.5× Tris-Borate-EDTA buffer. Mix 1 µl of each sample with 1 µl of a 2× loading dye suitable for denaturing polyacrylamide gel electrophoresis (Figure 2). Pre-cast gels can be used as a time-effective alternative to custom-made gels (e.g., Novex TBE-Urea, ThermoFisher, catalog number: EC6875BOX ; run on the XCell SureLock Mini-Cell, ThermoFisher, catalog number: EI0001 ).
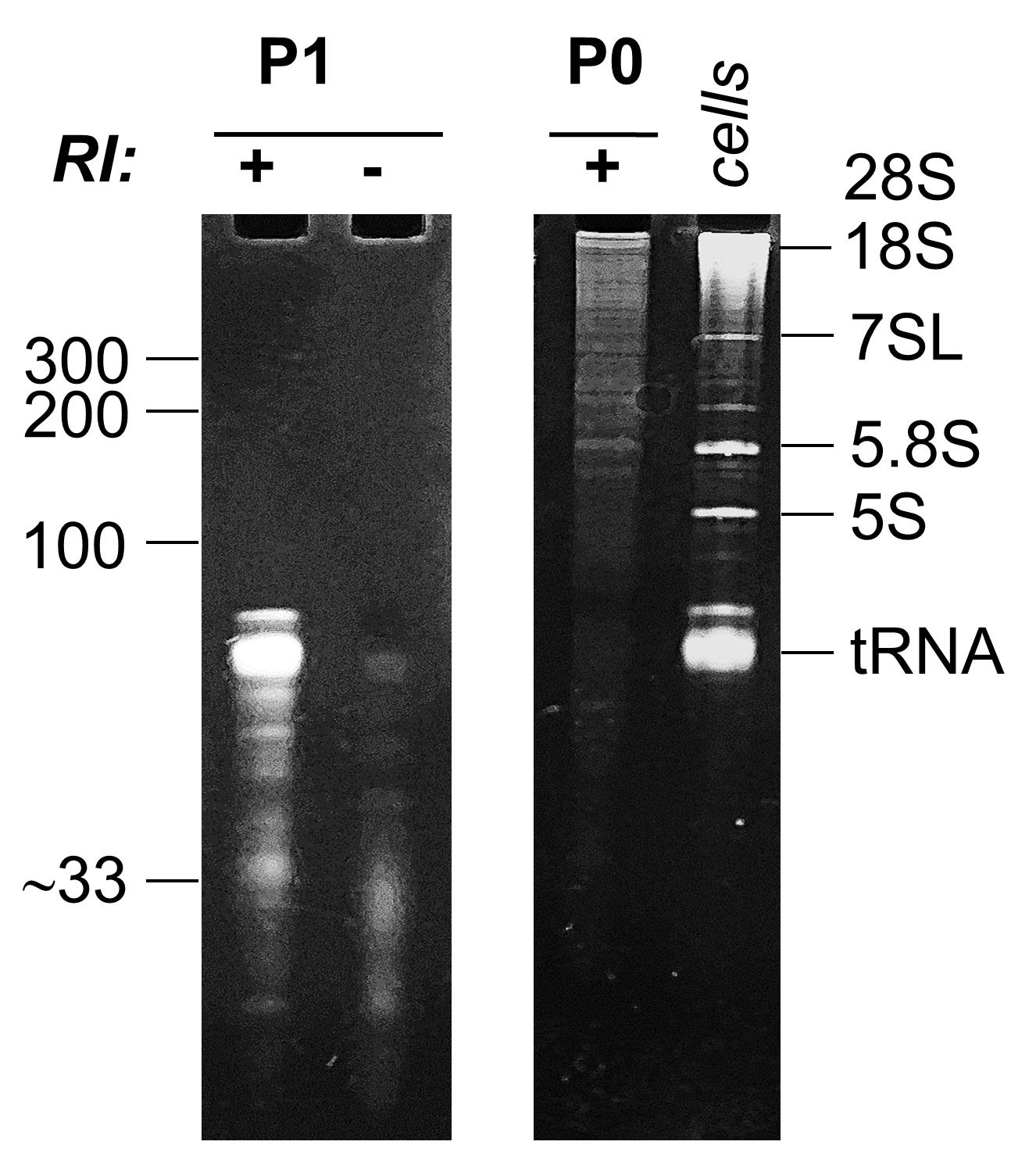
Figure 2. Analysis of the P1 peak either with (+) or without (−) RI treatment (left gel) or the P0 peak (right gel) in a denaturing polyacrylamide gel. Cells: intracellular RNA extracted from MCF-7 cells. Bands corresponding to tRNAs, 5S rRNA, 5.8S rRNA, 7SL RNA, 18S rRNA and 28S rRNA are indicated. Numbers at the left (100, 200, 300) correspond to the migration of a RNA ladder (not shown). The gel shows that RNAs in the P1 peak are shifted from full-length tRNAs (+RI) to small RNAs of around 30 nt (-RI). In contrast, RNAs in P0 are considerably larger, and bands of the size of 28S, 18S and 5.8S rRNAs are evident. Modified from Tosar et al. (2020).Using the remaining 7 µl of each sample, proceed to sequencing library preparation using a small RNA kit such as the NebNext Small RNA sequencing kit for Illumina. Perform single-end sequencing in an Illumina platform. In our study (Tosar et al., 2020) we sequenced for 200 cycles in order to be able to detect some full-length RNAs (e.g., 5.8S rRNA) and not only their fragments, based on gel electrophoresis results (Figure 2). However, doing so has a major drawback. Since most of the clusters in the chip contain inserts of small size, sequencing for longer than 150 cycles provokes sequencing the entire adapter, polymerase run off, and abortive termination of the sequencing reaction before proceeding to demultiplexation, at least in a MiSeq benchtop sequencer as the one we used. Data analysis can still be performed because the index will be contained as part of the sequence of each read. However, extracting data from an aborted run is not straightforward, and we recommend not to proceed for longer than 100-150 cycles unless understanding exactly what is being done.
Data analysis
Analyze data as described in Tosar et al. (2020). Briefly, identify and clip 3’ adapter sequences, map clipped reads to the human genome and count features corresponding to microRNAs, tRNAs, rRNAs, snRNAs, snoRNAs, YRNAs and Vault RNAs. Low-quality reads can be eliminated, but do not trim low quality bases at the end of the reads because doing so generates artificial 3’ variants that complicate interpretation. A comparison of the P1 peak between RI-treated and control samples is shown in Figure 3. Note the identification of a putative dimer between an internal fragment of the 28S rRNA and the full-length U49 snoRNA in the RI-treated sample, which is completely absent in the control. This is an example of how RI-SEC-seq enables to identify extracellular nonvesicular RNA complexes that are lost in conventional exRNA profiling due to their low extracellular stability (compared to highly stable dimers formed by glycine 5’ tRNA halves of exactly 30-31 nucleotides) (Tosar et al., 2018 and 2020).
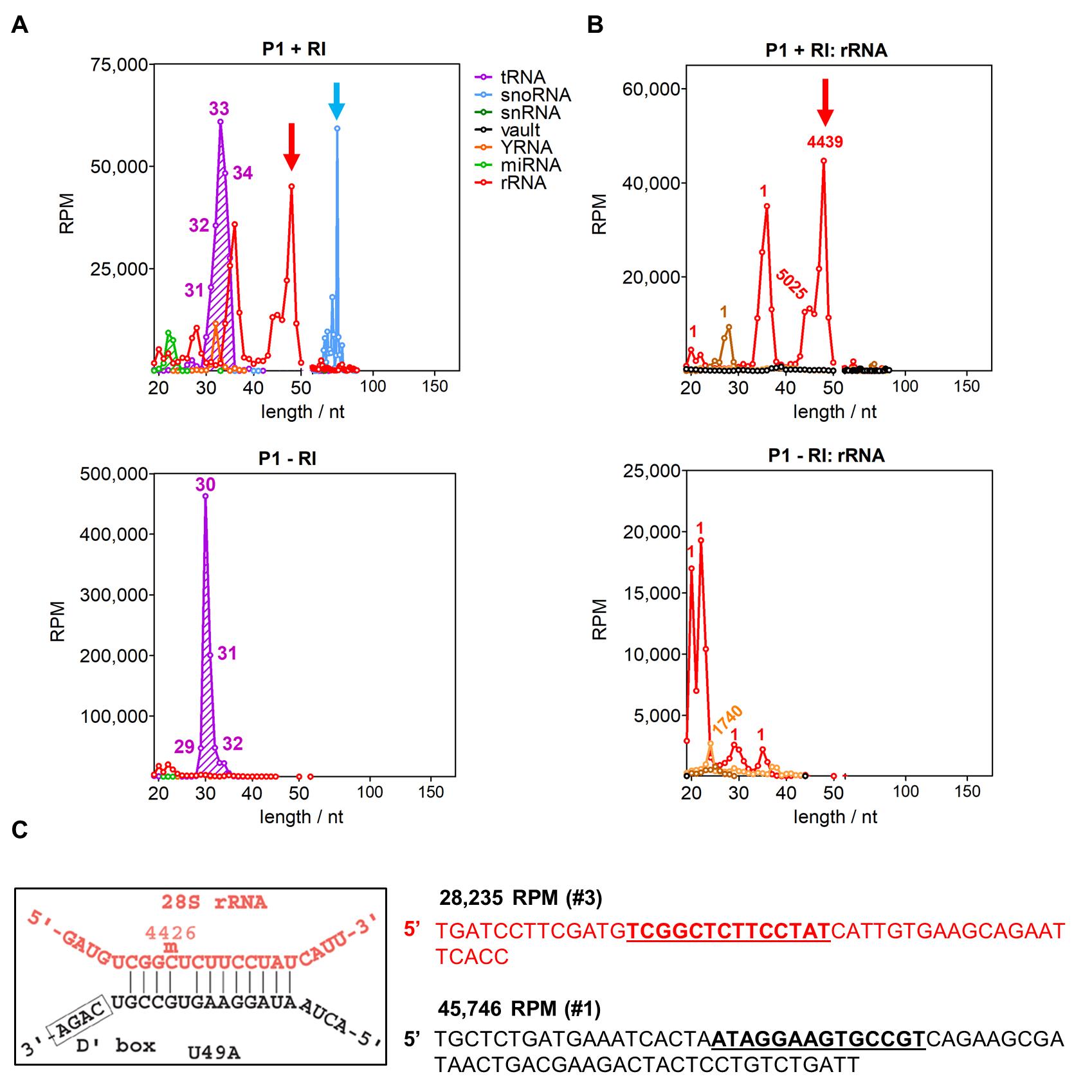
Figure 3. overview of RI-SEC-seq results. A. Size distribution of small RNA sequencing reads mapping to different ncRNAs (see legend) in the P1 peak of RI-treated (top) or control sample (bottom). RPM: reads per million mapped reads. Numbers indicate the length of 5’ tRNA-derived reads. B. Same, but showing only the reads aligning to rRNAs (28S rRNA in red, 5.8S rRNA in orange, 5S in black). In this case, numbers indicate starting position of most reads. C. Representation of the predicted SNORD49A (U49A; black)/28S rRNA (red) interaction, as depicted in snoRNABase (www-snorna.biotoul.fr). Below is the sequence with the highest number of reads. Its relative abundance is expressed as reads per million mapped reads (RPM). Its ranking in the “P1 + RI” dataset is also shown. Modified from Tosar et al. (2020).
Notes
RI-SEC-seq incorporates the concept of differential extracellular stability in exRNA profiling. However, it suffers from the limitations of the sequencing method that is applied. For instance, we know that the P1 peak in RI-treated samples contains mainly full-length tRNAs, and this is evident in Figure 2. However, sequencing of P1 in RI-treated samples did not show a significant number of reads corresponding to full-length tRNAs. On the contrary, most of the tRNA-derived reads corresponded to 5’ tRNA halves (Figure 3). Thus, RI-SEC-seq should be combined with orthogonal techniques such as Northern blot and density gradient centrifugation to get a more representative picture of the extracellular RNAome. Alternatively, the small RNA sequencing technique described here can be substituted for alternative methods that are able to sequence both full-length tRNAs and their fragments, such as TIGRT-seq (Qin et al., 2016; Shurtleff et al., 2017).
There is a change in the length of tRNA-derived fragments between P1 + RI and P1 – RI. Fragments of 33-34 nt are observed in RI-treated samples but they are lost in the controls (Figure 3A). A similar pattern is observed by Northern blot (Tosar et al., 2020). The reason is that the main cleavage products of samples containing full-length tRNAs (like PI + RI) are 5’ tRNA halves of 33-35 nt (and their cognate 3’ halves, usually harder to sequence). However, shorter 5’ halves of 30-31 tend to be much more stable. This could be due to the formation of dimers (Tosar et al., 2018) or interactions with extracellular RNA-binding proteins with size-dependent preferences. As a consequence, 5’ halves of 30-31 nt (mainly derived from tRNAGly) tend to accumulate when RNase activities are higher. Thus, RI-SEC-seq can identify variations in the stability of different RNAs within a single RNA class (e.g., between shorter and longer 5’ tRNA halves) as well as between different RNA classes (Figure 3A).
Small RNA sequencing in P0 (RI-treated samples) showed a variety of rRNA-derived fragments, mainly derived from the 28S rRNA. However, Figure 2 clearly shows that the RNAs eluting in P0 have a high molecular weight. It should be noted that when applying small RNA sequencing to samples containing mainly full-length long non-coding RNAs (such as 28S and 18S rRNAs), only their fragments will be retrieved. This can lead to the misconception that exRNAs are mainly fragmented in situations where they are clearly not. This is further supported by our finding of a significant number of reads corresponding to the full-length 5.8S rRNA, which is sufficiently short to fit our sequencing window. By combining RI-SEQ-seq with mass spectrometry, Western blot and velocity gradients, we showed that the P0 peak contains ribonucleoprotein complexes composed of rRNAs and ribosomal proteins. Thus, development of the RI-SEQ-seq method was a key step leading to the discovery of extracellular ribosomes (Khamsi, 2020; Tosar et al., 2020).
Applications of this protocol extend to any situation where it is important to capture the whole set of RNAs released to the extracellular space, rather than only the most stable ones (i.e., glycine or glutamic acid 5’ tRNA halves and EV-associated RNAs). For instance, testing how intracellular changes in gene expression between infected and noninfected cells, or between stressed and nonstressed cells, are reflected in the extracellular space. Additionally, RI-SEC-seq is a valuable tool to dissect extracellular RNA processing mechanisms and exRNA biogenesis.
Recipes
Commercial reagents and concentrated buffers were used through this protocol, as indicated above. Nevertheless, most of these buffers are routinely used in molecular and cellular biology laboratories and can be easily made from common solid stocks. In this particular case, we have opted to purchase commercial solutions to minimize RNase contamination throughout the protocol.
Acknowledgments
This work received funding from the following sources: Agencia Nacional de Investigación e Innovación (ANII, Uruguay) [FCE_3_2018_1_148745]; Comisión Sectorial de Investigación Científica (CSIC-UdelaR, Uruguay) [MIA_PAS2019_2_99] and National Institutes of Health, USA [UG3CA241694, supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director]. J.P.T. and A.C. are researchers from PEDECIBA (Uruguay), The National System of Researchers (ANII) and the full-dedication program of UdelaR.
The rationale for this protocol was first presented in Tosar et al. (2018), but RI-SEC-seq was formally developed in Tosar et al. (2020). All figures shown in this protocol were adapted from the latter publication.
Competing interests
None declared
References
- Arroyo, J. D., Chevillet, J. R., Kroh, E. M., Ruf, I. K., Pritchard, C. C., Gibson, D. F., Mitchell, P. S., Bennett, C. F., Pogosova-Agadjanyan, E. L., Stirewalt, D. L., Tait, J. F. and Tewari, M. (2011). Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad of Sci U S A 108(12): 5003-5008.
- Chevillet, J. R., Kang, Q., Ruf, I. K., Briggs, H. A., Vojtech, L. N., Hughes, S. M., Cheng, H. H., Arroyo, J. D., Meredith, E. K., Gallichotte, E. N., Pogosova-Agadjanyan, E. L., Morrissey, C., Stirewalt, D. L., Hladik, F., Yu, E. Y., Higano, C. S. and Tewari, M. (2014). Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U S A 111(41): 14888-14893.
- Dhahbi, J. M., Spindler, S. R., Atamna, H., Yamakawa, A., Boffelli, D., Mote, P. and Martin, D. I. (2013). 5' tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genomics 14: 298.
- Hoshino, A., Costa-Silva, B., Shen, T. L., Rodrigues, G., Hashimoto, A., Tesic Mark, M., Molina, H., Kohsaka, S., Di Giannatale, A., Ceder, S., Singh, S., Williams, C., Soplop, N., Uryu, K., Pharmer, L., King, T., Bojmar, L., Davies, A. E., Ararso, Y., Zhang, T., Zhang, H., Hernandez, J., Weiss, J. M., Dumont-Cole, V. D., Kramer, K., Wexler, L. H., Narendran, A., Schwartz, G. K., Healey, J. H., Sandstrom, P., Labori, K. J., Kure, E. H., Grandgenett, P. M., Hollingsworth, M. A., de Sousa, M., Kaur, S., Jain, M., Mallya, K., Batra, S. K., Jarnagin, W. R., Brady, M. S., Fodstad, O., Muller, V., Pantel, K., Minn, A. J., Bissell, M. J., Garcia, B. A., Kang, Y., Rajasekhar, V. K., Ghajar, C. M., Matei, I., Peinado, H., Bromberg, J. and Lyden, D. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527(7578): 329-335.
- Kalluri, R. and LeBleu, V. S. (2020). The biology, function, and biomedical applications of exosomes. Science 367(6478).
- Kamerkar, S., LeBleu, V. S., Sugimoto, H., Yang, S., Ruivo, C. F., Melo, S. A., Lee, J. J. and Kalluri, R. (2017). Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546(7659): 498-503.
- Khamsi, R. (2020). The biologist on the hunt for extracellular ribosomes. Nature 582: S6-S8.
- Kojima, R., Bojar, D., Rizzi, G., Hamri, G. C.-E., El-Baba, M. D., Saxena, P., Ausländer, S., Tan, K. R. and Fussenegger, M. (2018). Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nature Commun 9(1): 1305.
- Li, K., Rodosthenous, R. S., Kashanchi, F., Gingeras, T., Gould, S. J., Kuo, L. S., Kurre, P., Lee, H., Leonard, J. N., Liu, H., Lombo, T. B., Momma, S., Nolan, J. P., Ochocinska, M. J., Pegtel, D. M., Sadovsky, Y., Sanchez-Madrid, F., Valdes, K. M., Vickers, K. C., Weaver, A. M., Witwer, K. W., Zeng, Y., Das, S., Raffai, R. L. and Howcroft, T. K. (2018). Advances, challenges, and opportunities in extracellular RNA biology: insights from the NIH exRNA Strategic Workshop. JCI Insight 3(7): e98942.
- Nechooshtan, G., Yunusov, D., Chang, K. and Gingeras, T. R. (2020). Processing by RNase 1 forms tRNA halves and distinct Y RNA fragments in the extracellular environment. Nucleic Acids Research 48(14): 8035-8049.
- O’Brien, K., Breyne, K., Ughetto, S., Laurent, L. C. and Breakefield, X. O. (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol 21(10): 585-606.
- Qin, Y., Yao, J., Wu, D. C., Nottingham, R. M., Mohr, S., Hunicke-Smith, S. and Lambowitz, A. M. (2016). High-throughput sequencing of human plasma RNA by using thermostable group II intron reverse transcriptases. RNA 22(1): 111-128.
- Shurtleff, M.J., Yao, J., Qin, Y., Nottingham, R.M., Temoche-Diaz, M.M., Schekman, R. and Lambowitz, A.M. (2017). Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci U S A 114: E8987-E8995.
- Srinivasan, S., Yeri, A., Cheah, P. S., Chung, A., Danielson, K., De Hoff, P., Filant, J., Laurent, C. D., Laurent, L. D., Magee, R., Moeller, C., Murthy, V. L., Nejad, P., Paul, A., Rigoutsos, I., Rodosthenous, R., Shah, R. V., Simonson, B., To, C., Wong, D., Yan, I. K., Zhang, X., Balaj, L., Breakefield, X. O., Daaboul, G., Gandhi, R., Lapidus, J., Londin, E., Patel, T., Raffai, R. L., Sood, A. K., Alexander, R. P., Das, S. and Laurent, L. C. (2019). Small RNA Sequencing across Diverse Biofluids Identifies Optimal Methods for exRNA Isolation. Cell 177(2): 446-462 e416.
- Thomou, T., Mori, M. A., Dreyfuss, J. M., Konishi, M., Sakaguchi, M., Wolfrum, C., Rao, T. N., Winnay, J. N., Garcia-Martin, R., Grinspoon, S. K., Gorden, P. and Kahn, C. R. (2017). Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542(7642): 450-455.
- Tosar, J. P., Gámbaro, F., Sanguinetti, J., Bonilla, B., Witwer, K. W. and Cayota, A. (2015). Assessment of small RNA sorting into different extracellular fractions revealed by high-throughput sequencing of breast cell lines. Nucleic Acids Res 43(11): 5601-5616.
- Tosar, J. P., Gámbaro, F., Darre, L., Pantano, S., Westhof, E. and Cayota, A. (2018). Dimerization confers increased stability to nucleases in 5' halves from glycine and glutamic acid tRNAs. Nucleic Acids Res 46(17): 9081-9093.
- Tosar, J. P., Segovia, M., Castellano, M., Gambaro, F., Akiyama, Y., Fagundez, P., Olivera, A., Costa, B., Possi, T., Hill, M., Ivanov, P. and Cayota, A. (2020). Fragmentation of extracellular ribosomes and tRNAs shapes the extracellular RNAome. Nucleic Acids Res 48(22): 12874-12888.
- Tosar, J. P., Witwer, K.W. and Cayota, A. (2021). Revisiting Extracellular RNA Release, Processing, and Function. Trends Biochem Sci, online ahead of print. Doi: 10.1016/j.tibs.2020.12.008.
- Turchinovich, A., Weiz, L., Langheinz, A. and Burwinkel, B. (2011). Characterization of extracellular circulating microRNA. Nucleic Acids Res 39(16): 7223-7233.
- Yamasaki, S., Ivanov, P., Hu, G. F. and Anderson, P. (2009). Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol 185(1): 35-42.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tosar, J. P., Gámbaro, F., Castellano, M. and Cayota, A. (2021). RI-SEC-seq: Comprehensive Profiling of Nonvesicular Extracellular RNAs with Different Stabilities. Bio-protocol 11(4): e3918. DOI: 10.21769/BioProtoc.3918.
Category
Molecular Biology > RNA > RNA detection
Cancer Biology > General technique > Molecular biology technique
Cancer Biology > Cancer biochemistry > Cancer metabolism
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.