- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of B Cell Migration by Intravital Microscopy


(*contributed equally to this work) Published: Vol 10, Iss 23, Dec 5, 2020 DOI: 10.21769/BioProtoc.3842 Views: 5039
Reviewed by: Luis Alberto Sánchez VargasPaurvi ShindeJulie Weidner
Original research article
The authors used this protocol in:

Mar 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
During immune responses, B cells home to lymph nodes (LN), where they encounter antigens. Homing starts with capture and L-selectin-dependent rolling on the activated endothelium of high endothelial venules (HEV). After recognition of chemokines presented on HEV, activation of B cell integrins occurs mediating firm arrest. Subsequently, B cells crawl to the spot of extravasation to enter the LN. Extravasation can be visualized and quantified in vivo by intravital microscopy (IVM) of the inguinal LN. Here, we describe an established protocol that permits detailed in vivo analysis of B cell recruitment to LN under sterile inflammatory conditions. We describe data acquisition, exportation, quantification, and statistical analysis using specialized software. IVM of LN is a powerful technique that can provide a better understanding of B cell migratory behavior during inflammation in vivo.
Keywords: B cellsBackground
The secondary LN play a fundamental role in immune responses at different locations in the body as they are tactically localized to drain antigens via the lymphatic system. This implies in situ, the recruitment of immune cells, antigen encountering, and the mounting of immune responses (Tan and Watanabe, 2010; Tavares et al., 2014). B cell recirculation through the LN begins with its capture and L-selectin-dependent rolling, along the high endothelial venules (HEV); subsequently, B cells firmly arrest through G-protein coupled receptors (GPCR)-mediated recognition of optimal concentrations of chemokines presented on the luminal surface of the HEV. B cell arrest critically depends on the acquisition of the high-affinity conformation of the integrins LFA-1, VLA-4 (Park et al., 2016). After arrest, B cells crawl to search for a proper site for extravasation into the parenchyma of the LN, where they can recognize antigens and mount an immune response (Pereira et al., 2010; Mesin et al., 2016). The release of B cells from the LN also requires chemokine gradient sensing and interaction with the endothelium of the lymphatic sinuses; however, their exit through the efferent lymph is still poorly understood (Park et al., 2016).
The migration of B cells relies on actin cytoskeletal remodeling and is tightly controlled by signaling molecules such as kinases, GTPases, and motor proteins. Morphological changes resulting from this process are indispensable for rolling, adhesion, migration, motility, filipodia formation, and transmigration (De Pascalis and Etienne-Manneville, 2017). To study these events in vivo, IVM of the LN is a powerful technique that allows in real-time, the visualization of the earliest steps of B-cell extravasation, as well as their behavior and morphological changes during migration (Miller et al., 2003). Although, variations of this technique have been used before to visualize lymphocyte trafficking to Peyer patches or spleen, the inguinal LN has the advantage of being easily accessible for IVM without much surgical effort. Less surgical manipulation allows for better preservation of vascular integrity and blood flow, and better visibility during video recording for the quantification of migratory parameters (von Andrian, 1996; Lafouresse et al., 2015).
In previous work, we focused on the evaluation of the long-tailed class-I myosin, Myo1e, as a regulator of migration of activated B cells in the inguinal LN. We found that the absence of Myo1e causes defective B cell recruitment to the LN (Giron-Perez et al., 2020). In particular, the number of rolling B cells and their rolling velocity were increased in the different HEVs of types I and II, whereas slow-rolling and adhesion were reduced in the HEVs of types III and IV. Moreover, the absence of Myo1e diminished B cell spreading and transmigration (Giron-Perez et al., 2020).
Here we describe a protocol to analyze B cell migration into the inguinal LN by IVM. We provide a detailed description from the purification of B cells to data analysis. This protocol may be adapted to explore the migration of other cell types and the effect of different treatments in this context. Our method can be easily established and multiple adaptations are feasible including application of blocking antibodies or pharmaceuticals to interfere with B cell migration in vivo.
Materials and Reagents
B cell isolation and preparation
Cell culture plate, 6 wells (Sigma-Aldrich, catalog number: SIAL0516-50EA )
Petri dish polystyrene (Sigma Aldrich, catalog number: P5481 )
Syringes 27 gauge (G), 1 ml (BD Plastipak, catalog number 301629 )
Falcon cell strainer 40 µm (Thermo Fischer Scientific, catalog number: 08-771-19 )
Corning 50 ml centrifuge tubes (Merck, catalog number: CLS430828 )
Female C57BL6/J Mice from 8 to 12 weeks
Ficoll-Paque Plus (GE Healthcare, catalog number: 17-1440-02 )
RPMI 1640 medium (without antibiotics) (Life Technologies, catalog number: 21870076 )
Heat inactivated Fetal Bovine Serum (Thermo Fischer Scientific, catalog number: 10100139 )
LPS from Escherichia coli O55: B5 (Sigma-Aldrich, catalog number: L 2637-5MG )
Absolute ethanol (Sigma-Aldrich, catalog number 64-17-5 )
Murine IL-4 (R&D System, catalog number: 404-ML-010 )
Anti-B220-Alexa fluor 488 (Biolegend, Clone RA3-6B2, catalog number: 103228 )
Murine CXCL12 (Preprotech, catalog number: 25020-A )
α-Thy1 (NIM-R1) (ascites)
Hoechst 33342 (Thermo Fisher Scientific, catalog number: H3570 )
Rat anti-Mouse CD44 APC-Cy7 (BD Biosciences, catalog number 560568 )
70% Ethanol
PBS 1x (see Recipes)
31 G x 8 mm Insulin syringes (BD Ultra-fine, catalog number: 305106 )
27 G x 13 mm Insulin syringes (BD Plastipak, catalog number: 301629 )
PE10 polyethylene tubing (Clay Adams, catalog number: 427401 )
Sodium Heparin 5,000 UI/ml (PISA. Mexico)
Ketamine hydrochloride 125 mg/kg (Aranda. Mexico)
Xylazine 12.5 mg/kg (Aranda. Mexico)
Sodium Chloride 0.9% (w/v) (PISA. Mexico)
PBS 1x (see Recipes)
IVM of the LN
Equipment
B cell isolation and preparation
Dissection Kit-8 (Fisherbrand, catalog number: 08-855 )
Tissue culture hood
Dissecting board
Polyester thread (any brand)
2 curved forceps (Moria MC40/B. 11 cm) (Fine Science Tools)
Straight Clamp (Micro Serrefine. 13 mm) (Fine Science Tools)
Clip applicator forceps (14 cm) (Fine Science Tools)
Spring scissors (3 mm) (Fine Science Tools)
Extra fine scissors (13 mm) (Fine Science Tools)
Customized animal stage made of Plexiglas (Figure 4)
Infrared lamp
Superfusion system (composed of a 2 L glass bottle with an adapted flow valve and a PE10 tube)
Stereo-microscope (Stemi 305, Carl Zeiss)
Equipped with a gooseneck light guide (CL4500, Carl Zeiss)
Upright microscope (Axioscope A1, Carl Zeiss)
Equipped with a 10x, and a 20x water immersion objective
Mercury lamp (HBO100, Carl Zeiss)
Filters (430-465 nm Blue)
Axiocam (Camara, Carl Zeiss)
IVM of the LN
Software
Zen Blue Edition (https://www.zeiss.com, Carl Zeiss, Germany)
ImageJ (www.imagej.nih.gov, National Institutes of Health, USA)
Procedure
B cell purification
Note: Before starting the Petri dish needs to be coated for 1 h with α-Thy1 monoclonal antibody ascites (NIM-R1) (1:10,000) (Chayen and Parkhouse, 1982).
Euthanize female mice of 8-12 weeks by cervical dislocation. Place the mouse on its back on a dissecting board.
Thoroughly soak the fur of the mouse with 70% ethanol. Transfer the board with the mouse to the tissue culture hood.
Make an incision in the skin with scissors on the left side (from the perspective of the mouse, right when looking onto the mouse, compare Figure 1), then grab the skin with both hands and pull it away to expose the left side of the abdominal wall (the spleen is located between the stomach and the left side of the diaphragm of the mouse) (Figure 1). Use forceps to lift out the spleen and carefully resect it with scissors. The spleen should be removed intact.
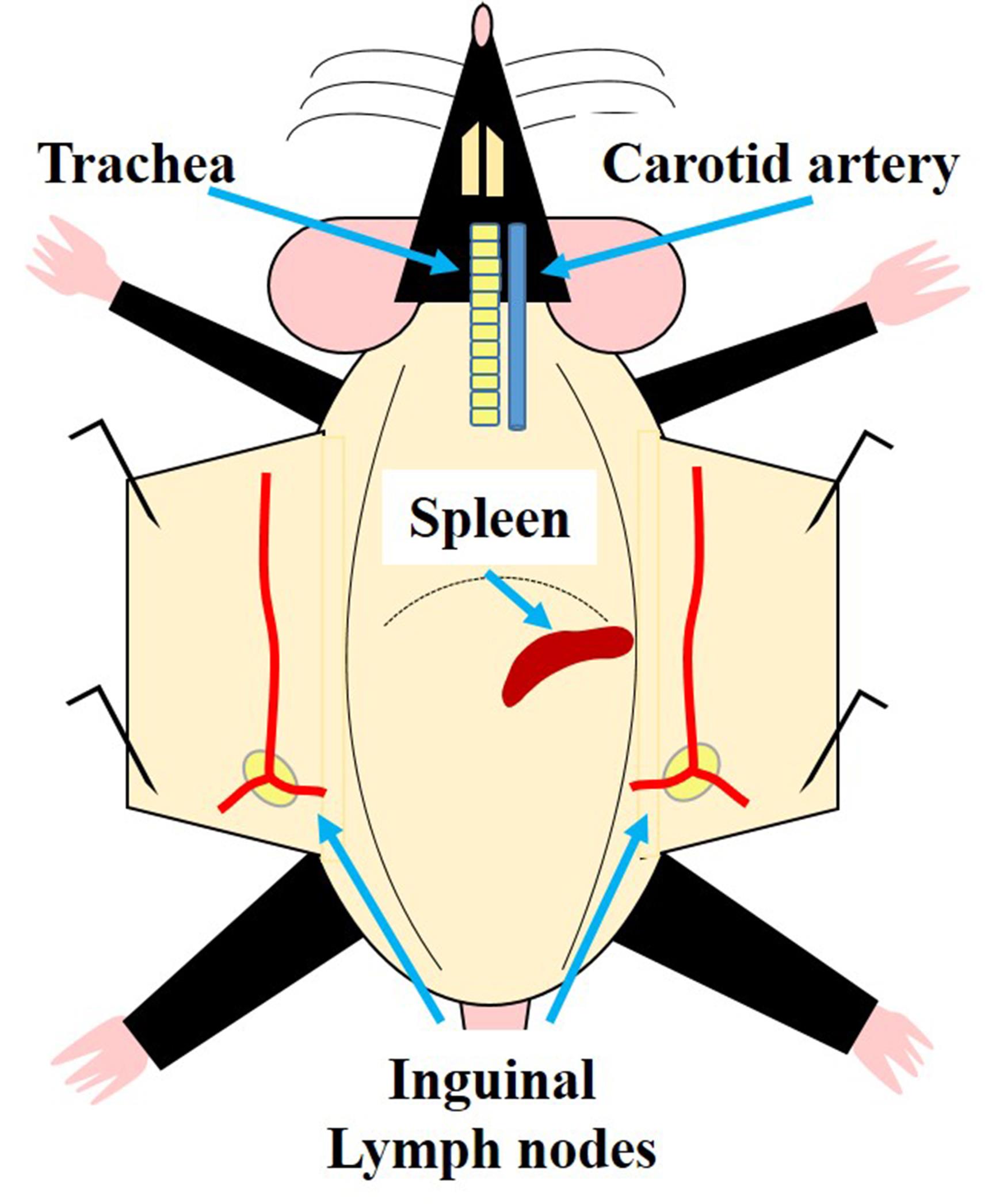
Figure 1. Schematic representation of the localization of relevant organs involved in the IVM of the LN. The trachea is shown here as a reference for the localization of the carotid artery, which is used for posterior catheterization. The localization of the spleen is shown for its resection in a donor mouse to isolate B cells. The two blue arrows in the lower part of the figure point to the right and left inguinal LN which are locally activated by injection of CXCL12 close to it. Both LN can be used for IVM.Submerge the spleen in 10 ml of PBS 1x in a Petri dish.
Place the spleen on the top of a strainer of 40 µm (Falcon cell strainer) and disaggregate it by employing a syringe plunger and wash the strainer with up to 10 ml of PBS 1x. Collect the cell suspension in a 50 ml centrifuge tube.
The cell suspension is transferred on top of a 15 ml tube containing 5 ml of Ficoll-Paque. Slowly pour the cell suspension on top of the Ficoll layer without disrupting the interphase between cell suspension and Ficoll). Centrifuge at 50 x g for 30 min at room temperature (with an acceleration and deceleration rate of 1 or no brake). Collect the thin white cell layer with a 1 ml pipette.
Resuspend the cells in 5 ml of PBS 1x and centrifuge at 50 x g for 5 min at room temperature and repeat this washing step, then recollect the pellet of splenic mononuclear cells (MNC) in 10 ml of PBS 1x.
Deposit the cell suspension in the Petri dish previously coated for 1 h with an α-Thy1 monoclonal antibody ascites (NIM-R1) (1:10,000) (Chayen and Parkhouse, 1982) (as mentioned at the beginning of this procedure) and incubates for 1 h at 37 °C with 5% of CO2, to allow adhesion of other MNC and enrichment of the B220+ cell population in the supernatant.
After 1 h, carefully transfer the supernatant from the Petri dish into a 15 ml tube and centrifuge for 5 min at 50 x g twice at room temperature. Then, decant and resuspend the pellet in 2 ml of PBS 1x and analyze the purity of the population by flow cytometry using an anti-B220 monoclonal antibody. Purity should be over 90% (Figure 2).

Figure 2. B cell enrichment and stimulation. A. After B cell enrichment, purity was assessed by flow cytometry employing a monoclonal antibody against B220 coupled to AF488. Typically, a purity over 90% is obtained using α-Thy1 antibody-coated Petri dishes. B. CD44 expression on B cells increases after stimulation with LPS and IL-4 for 48 h.
Activation of B cells
Stimulate 2 x 106 B cells in 2 ml of RPMI 1640 in one well of a 6-well cell culture plate, supplemented with 10% of fetal bovine serum (the supplemented medium should be at room temperature) by adding 10 µg/ml of LPS from Escherichia coli O55: B5 and 10 U/ml of IL-4.
Incubate the cells at 37°C and 5% of CO2 for 48 h.
Recover the cells with a 1 ml pipet, resuspend them gently by pipetting up and down a few times, and wash cells with 5 ml of PBS 1x twice. (centrifugation is done at 50 x g for 5 min at room temperature). Then, resuspend the B cells in 2 ml of PBS 1x in a 15 ml tube.
B cell labeling
Place 1 x 107 activated B cells in a 50 ml tube containing 30 ml of RPMI1640 at room temperature without supplements and add 10 µl of Hoechst 33342 for 1 h.
Note: Reconstitute Hoechst 33342 according to the manufacturer’s instructions; aliquot and store at -20 °C.
Wash cells twice with RPMI 1640 without supplements and centrifuge at 50 x g for 10 min at room temperature.
Resuspend cells in 200 µl of PBS 1x in an Eppendorf tube. Store cells on ice until use in experiments (not longer than 1 h).
Note: An example of increased CD44 expression on B cells after LPS stimulation as measured by flow cytometry is shown in Figure 2B (This step is optional).
Injection of CXCL12 into the host mouse
Grab the mouse by the neck to handle it for the procedure.
Hold the tail of the mouse with the fingers and locate the inguinal LN below the inguinal ligament and the bifurcation of the epigastric vein). The injection can be performed as previously described (Andorko et al., 2014).
Inject the chemokine into the zone of the inguinal LN (prior to the administration, you can visualize the LN localization with a dissection microscope) with 200 µl of CXCL12 (25 ng/ml) using a 22 G syringe.
Note: Be careful with this process as an incorrect puncture can damage the LN and can also cause alterations in the blood flux.
Let the mouse rest for 1 h.
Preparation of the catheter and the syringes needed during the procedure
Fill a 31 G syringe with 100 μl of a heparin solution diluted to a final concentration of 500 UI/ml (Use physiological saline to dilute the heparin).
Cut an approximately 15 cm long PE tube. Cut the end of the tube in a 45° angle (like the tip of an injection needle).
Insert the other end to the needle of the syringe (Use the forceps to facilitate its insertion).
Purge the heparinized catheter (make sure no air bubbles are present in the syringe or the PE tube).
In addition to the heparinized catheter, prepare another syringe with 300 μl of anesthesia (Ketamine hydrochloride 125 mg/kg/Xylazine 12.5 mg/kg) to be used to deliver additional anesthesia systemically during the procedure.
Prepare an additional 31 G syringe. It will be employed to deliver the labeled B cells through the catheter.
Carotid artery catheterization
Clean and disinfect with 70% alcohol where the surgery is going to be performed.
Anesthetize the mouse delivering intraperitoneally (IP) 10 ml/kg weight of the previously mentioned anesthetic mixture
Example: For a 20 g mouse, inject IP 200 μl of the anesthetics mixture.
Wait 10 min; monitor pain reflexes of the mouse during induction to anesthesia by pinching the paws with forceps.
Upon anesthesia, place the mouse on its back; fix the paws with adhesive tape in a dorsal position. Also, secure the head with a 5 cm thread positioned behind the teeth and fix it with tape. Both ends of the thread should be fixed in front of the nose of the mouse (Jacobs and Hopper-Borge, 2014).
Optional: The carotid artery can be more accessible if a 1 ml pipette tip is positioned under the neck of the mouse before the catheterization.
Place the mouse under the dissection microscope. Make sure all the material needed is accessible.
Below the chin, make a small incision, introduce the scissors in a closed position between the skin and submaxillary glands and separate them by carefully opening the scissors; Make a 2 cm longitudinal cut through the skin of the neck.
Once the submaxillary glands are exposed, separate them by cutting the connective tissue between them. Separate them gently by employing 2 curved forceps. After these, use the left forceps to separate the muscle from the trachea while inserting the right forceps next to the trachea and carefully dissecting the fascia that surrounds the carotid artery. Also, dissect the vagus nerve located next to the artery (This serves to completely isolate the artery which avoids mistakes when inserting the catheter).
Cut approximately 8 cm of the polyester thread and bend it in a hairpin-like shape and with the forceps, draw it below the carotid artery and leave equal portions of the thread aside of the artery. Cut the thread so that two equal portions are located beneath the carotid artery. One portion should be towards the thorax while the other thread should be located towards the head (Figure 3A).
Close the blood flux to the brain by firmly tying a knot as close as possible to the head (Figure 3B, red arrow). Leave the two extremes of the thread free; after catheterization, they are used for securing the catheter with a double knot.
Make a loose knot in the farthest portion of the artery, in direction to the thorax (Figure 3B, black arrow). (This knot will remain loose and will serve for securing and fixing the catheter once it is inserted inside the artery).
With the left forceps, slightly lift the carotid artery by pulling out the loose knot in a way that it does not loses its shape. With the right hand, use a clip applicator to place a straight clamp to the artery. This clamp should be placed after the loose knot (in direction to the thorax) (Figure 3C). Make sure enough space (approximately 1 cm length) is left to insert the catheter to the carotid artery.
Once the clamp is securely placed, use the spring scissors to make a small incision close to the portion of the artery located towards the head of the mouse (Blood will be spilled as a consequence of this cut).
Hold the tight knot to fix the artery with the right hand while inserting the catheter into the carotid artery. (Insert the catheter with the tip in an upward direction; this will avoid damaging the artery. Once the catheter is inside, tighten the loose knot to secure in place the catheter; fix the catheter with the threads located towards the head with a double knot (Figure 3D).
Slowly release the clamp with a clip applicator. Blood will invade the inner part of the catheter. If it is correctly placed, blood pumping inside the catheter will be seen (Figure 3D) (Obstruction of the catheter can be tested by delivering a small amount of saline solution with a 31 G syringe. This is an optional step).
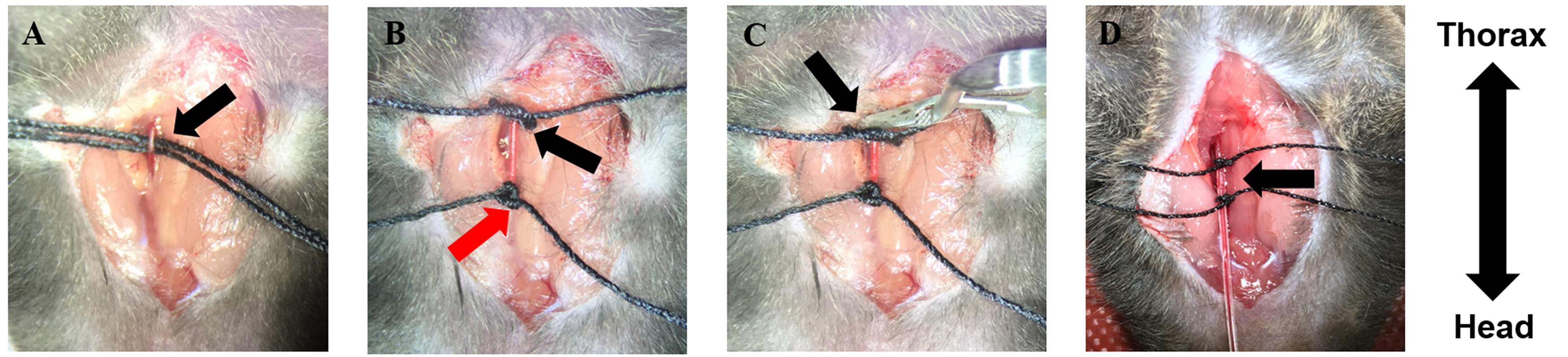
Figure 3. Carotid artery catheterization. A. A hairpin-like polyester thread is drawn below the carotid artery: one thread is placed towards the thorax; the other one towards the head. B. Close the blood flow towards the brain with a double knot (red arrow); with the other thread, make a loose knot (black arrow). C. Place a straight clamp to close the artery. D. Catheterize the carotid artery; fix the catheter with two knots and release the clamp from the artery. Blood will invade the inner part of the catheter (black arrow). The double arrow indicates the orientation of the mouse.
Labeled B cell injection
Transfer the labeled B cells (1 x 107 cells/200 μl) to a 31 G syringe. Avoid the presence of bubbles in the syringe.
Unplug the heparinized syringe from the catheterized mouse and avoid drawing blood by pinching the catheter’s tube with forceps. Immediately, plug the syringe that contains the cell suspension.
Note: Take the cells to room temperature before injection. Avoid light exposure of the cell suspension at any time.
Slowly administer the cell suspension through the catheter.
Unplug the syringe that contained the cell suspension. As before, immediately stop the blood return with forceps and plug a 31 G syringe with the mixture of anesthetics. Deliver 10 μl of anesthesia (This step serves two purposes: to maintain anesthesia and to wash the catheter).
After administration of the B cell suspension, wait for 1 h to allow B cell interaction in the LN.
Start to warm the PBS 1x superfusion system to 45 °C. (At this temperature, while the PBS 1x is superfused, it is naturally cooled down to 37 °C. This should be optimized in every laboratory).
Inguinal LN dissection
After 30 min of B cell administration, check for pain reflexes in the pawn and if necessary, administrate more anesthesia.
Carefully place the mouse on the animal stage. Secure the mouse by fixing the paws with tape.
Note: Our animal stage (Figure 4) was designed with an area filled with silicon where one can fix the retracted skin by pins. Other options have been previously reported (Sellers and Payne, 2011).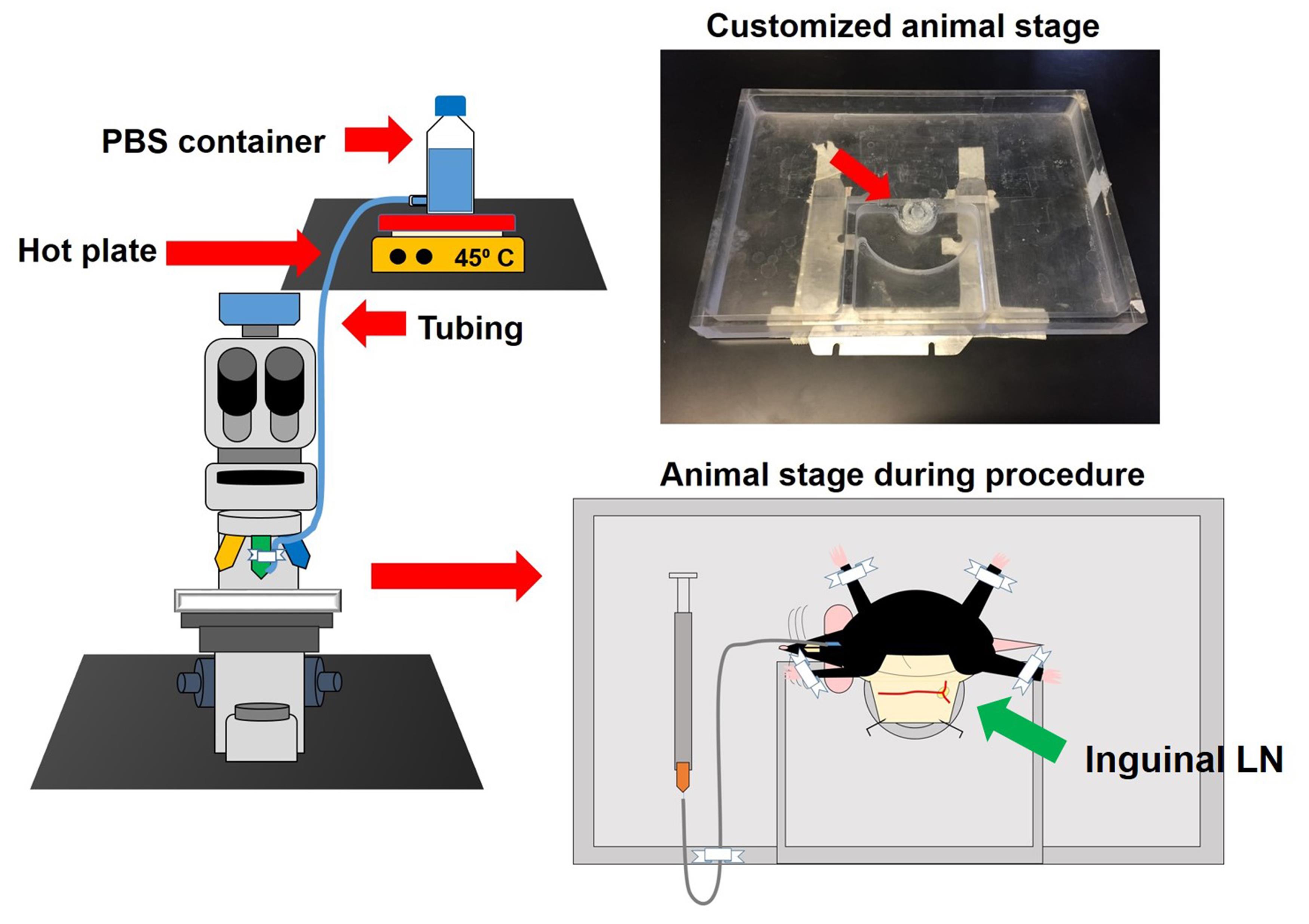
Figure 4. Microscope set-up, superfusion system and customized animal stage. The superfusion system is composed of a hot stirring plate set at 45 °C to warm the superfusion buffer (PBS or 0.9% saline). With this setting, the buffer reaches the tissue at around 37 °C (this needs to be optimized in each laboratory). The buffer flask with an exit at the bottom is connected to a plastic tube that is fixed with regular tape to the 20x objective of the microscope. The customized animal stage is 25 cm long and 15 cm wide. This animal stage has the purpose of controlling the flow of PBS used to moisturize the exposed tissue. Additionally, it contains a little ring filled with silicon where the retracted skin can be fixed with needles (red arrow). This stage can be used for IVM of the LN, the cremaster muscle, and the mesentery. In the lower right part of the figure, an example of how the mouse is fixed to the animal stage is shown. Also, it exemplifies how the catheter should be fixed and how the mouse preparation should look prior IVM.Under the dissection microscope, make a small incision in the abdominal cavity and dissect the skin from the muscle by introducing the scissors in a closed position; then open them carefully. Enlarge to 2 cm the length of the skin incision and cut diagonally 1 cm of the skin towards the limbs; dissect the connective tissue and avoid disturbing the LN.
Retract the skin enough to observe the inguinal LN and to make it accessible to the objectives of the microscope. Secure the skin by employing 2 pins (Figure 5A).
Moisten the tissue with PBS 1x and dissect the connective tissue surrounding the inguinal LN.
Clear the connective adipose tissue by using forceps until the microvasculature is exposed.
Note: It is important to never touch or manipulate the vasculature. Also, too much tension while fixing the skin to the stage perturbs the blood flow. Since parameters such as rolling are quantified in this technique, it is fundamental to have intact blood flow. If necessary moisten the LN again (Figures 5B and 5C).
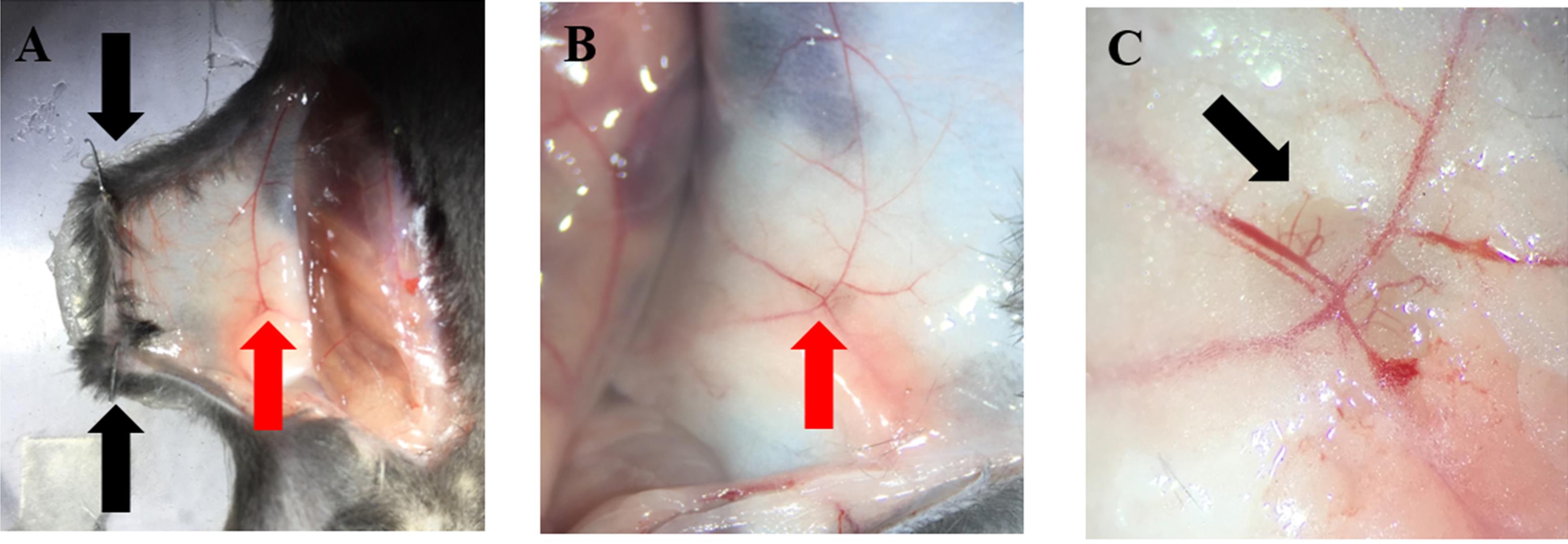
Figure 5. LN preparation. A and B. Secure the abdominal skin to the animal stage with two pins (black arrows); the inguinal LN should be visible (red arrows). C. Appearance of the LN after removal of surrounding connective and adipose tissue. HEV are clearly visible using a dissection microscope (black arrow).
IVM of the inguinal LN
Prepare the superfusion system and fix it with tape at the very end of the PE10 tube to the 20x objective.
Place the animal under the microscope and locate the inguinal LN microvasculature. Start the PBS 1x superfusion in a way that the dropping keeps constant tissue moisture and objective immersion.
Note: Constant moisture ensures correct focusing of the video.
Localize the desired area of the microvasculature to record. Employ the 430-465 nm blue filter of the microscope. The desired area to record should contain HEV of 4 different calibers (Figure 6).
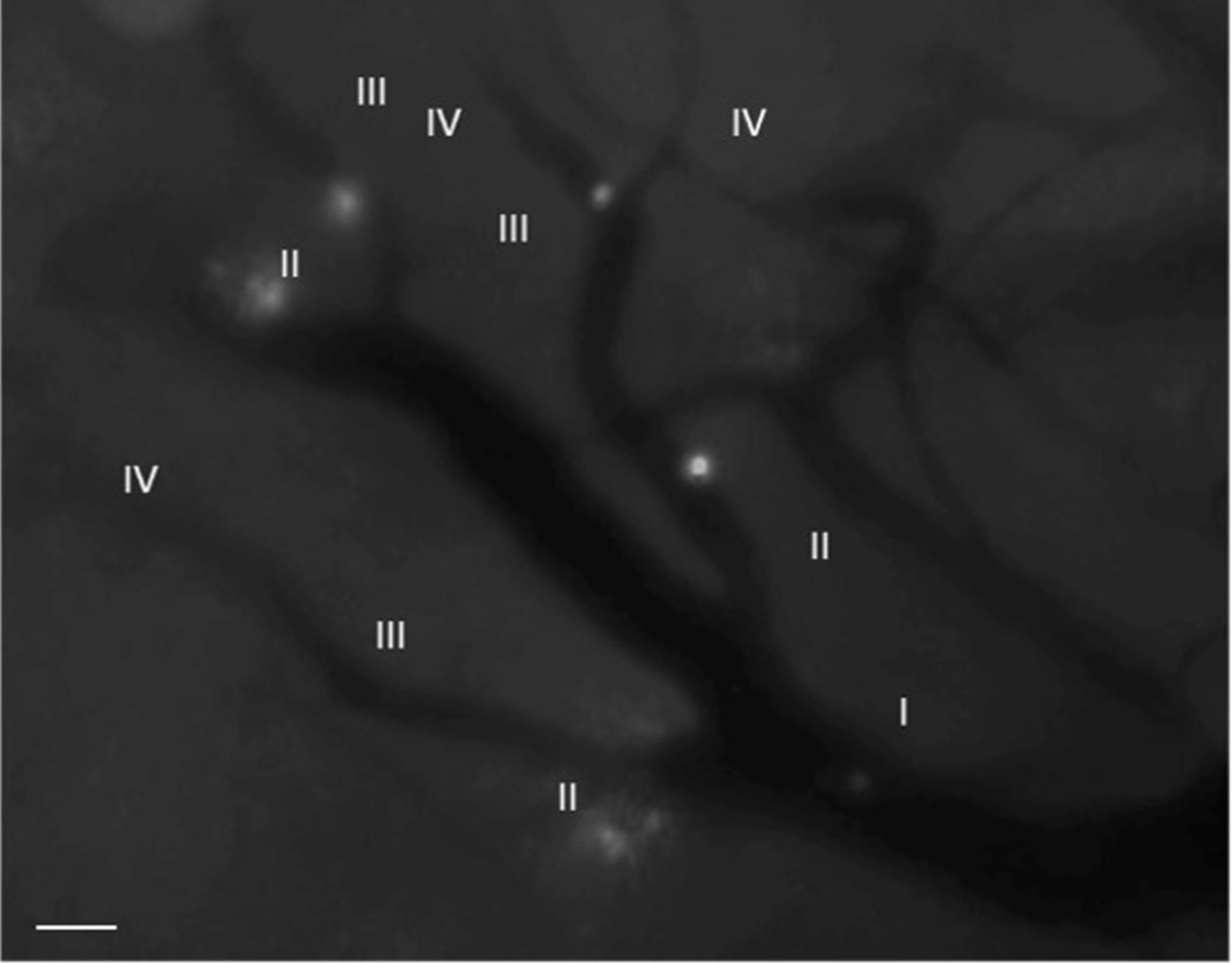
Figure 6. Identification of HEVs. The number IV refers to the thinnest HEV, while the number I denotes the thickest HEV in the inguinal LN. The labeled B cells can be observed interacting with the HEV. (Objective 20x) Scale bar 50 µm.Record 4 or more videos of 5 min each at different time points
Note: Maintain anesthesia during the whole procedure.
Take a picture of the recorded area to locate the HEV for posterior quantification (Optional).
After these steps, export the videos in AVI format and images in TIFF format.
Data analysis
Data exportation and analysis in ImageJ
From the work station of the microscope, export the videos in AVI format to an external hard disk.
Transfer the videos to another computer for further analysis (This is a desirable step since analysis directly from the hard disk can cause slow system performance).
Start ImageJ software. The program can be freely downloaded from the following link: https://imagej.nih.gov/ij/.
Import the videos to ImageJ. Change the videos to 8 bits and set the scale according to the distance covered by the objective and the camera of the microscope. For example, in our conditions, when using the 20x objective and our camera, 480 pixels correspond to 712 μm (For this step, a scale can be set simply by acquiring an image of a known scale). This is fundamental for further quantification.
Multiple data can be obtained from this technique. In our manuscript (Giron-Perez et al., 2020), we quantified the following parameters:
HEV diameter. This is achieved by tracing a line across the venule and measuring (Ctl-M).
Recruited cells (at different time points):
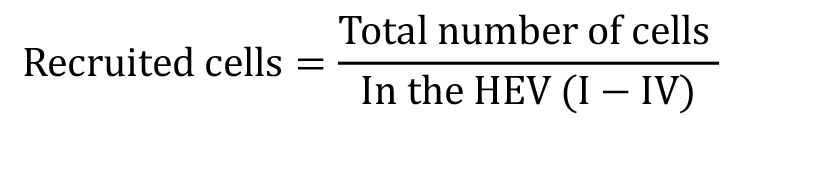
Cell flux (Number of cells passing through a fixed point in 1 min): This parameter can be quantified in each of the different HEV (I-IV)
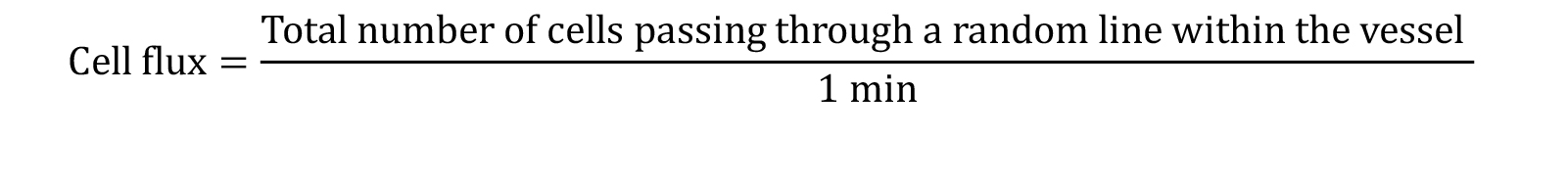
Adherent cells: Cells adherent for more than 30 s in one fixed position
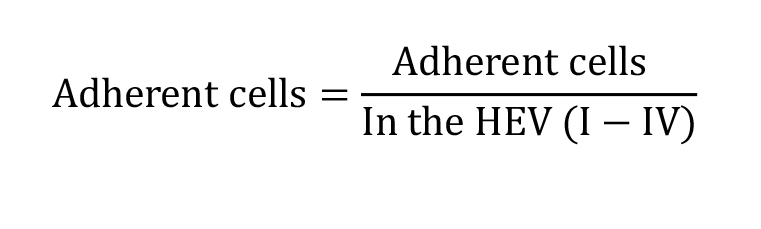
Rolling Velocity:
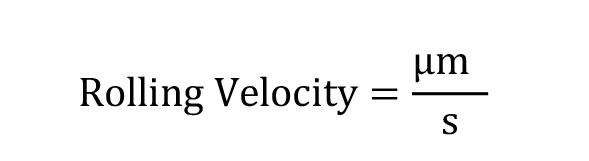
The rolling velocity can be categorized as rolling and slow rolling by establishing a threshold of ≤ 5 μm/s to be considered slow rolling.
Other IVM parameters can also be quantified using these experimental settings (Gavins and Chatterjee, 2004; Sperandio et al., 2006; Xu et al., 2011; Lafouresse et al., 2015; Vadillo et al., 2019).
Recipes
PBS 1x (1 L)
Dissolve in 800 ml distilled water:
8 g of NaCl (0.137 M)
200 mg of KCl (0.0027 M)
1.44 g of Na2HPO4 (0.01 M)
240 mg of KH2PO4 (0.0018 M)
Adjust solution to pH = 7.4 and distilled water to a final volume of 1 L
Acknowledgements
We thank Dr. Hilda Vargas Robles, Lenin Estudillo for expert technical assistance; and Ricardo Gaxiola-Centeno from the animal facility at CINVESTAV for taking care of mice.
Competing interests
The authors declare no competing or financial interests.
Ethics
This study was carried out in strict accordance with ARRIVE (Animal Research: Reporting of in vivo experiments) rules. The Animal Care and Use Committee of CINVESTAV approved all protocols and experiments.References
- Andorko, J. I., Tostanoski, L. H., Solano, E., Mukhamedova, M. and Jewell, C. M. (2014). Intra-lymph node injection of biodegradable polymer particles. J Vis Exp (83): e50984.
- Chayen, A. and Parkhouse, R. M. (1982). B cell subpopulations in the mouse: analysis with monoclonal antibodies NIM-R2 and NIM-R. Eur J Immunol 12(9): 725-732.
- De Pascalis, C. and Etienne-Manneville, S. (2017). Single and collective cell migration: the mechanics of adhesions. Mol Biol Cell 28(14): 1833-1846.
- Gavins, F. N. and Chatterjee, B. E. (2004). Intravital microscopy for the study of mouse microcirculation in anti-inflammatory drug research: focus on the mesentery and cremaster preparations. J Pharmacol Toxicol Methods 49(1): 1-14.
- Giron-Perez, D. A., Vadillo, E., Schnoor, M. and Santos-Argumedo, L. (2020). Myo1e modulates the recruitment of activated B cells to inguinal lymph nodes. J Cell Sci 133(5).
- Jacobs, J. D. and Hopper-Borge, E. A. (2014). Carotid artery infusions for pharmacokinetic and pharmacodynamic analysis of taxanes in mice. J Vis Exp (92): e51917.
- Lafouresse, F., Bellard, E., Laurent, C., Moussion, C., Fournie, J. J., Ysebaert, L. and Girard, J. P. (2015). L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood 126(11): 1336-1345.
- Mesin, L., Ersching, J. and Victora, G. D. (2016). Germinal Center B Cell Dynamics. Immunity 45(3): 471-482.
- Miller, M. J., Wei, S. H., Cahalan, M. D. and Parker, I. (2003). Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc Natl Acad Sci U S A 100(5): 2604-2609.
- Park, C., Hwang, I. Y. and Kehrl, J. H. (2016). Intravital Two-Photon Imaging of Lymphocytes Crossing High Endothelial Venules and Cortical Lymphatics in the Inguinal Lymph Node. Methods Mol Biol 1407: 195-206.
- Pereira, J. P., Kelly, L. M. and Cyster, J. G. (2010). Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int Immunol 22(6): 413-419.
- Sellers, S. L. and Payne, G. W. (2011). Intravital microscopy of the inguinal lymph node. J Vis Exp 4(50): 2551.
- Sperandio, M., Pickard, J., Unnikrishnan, S., Acton, S. T. and Ley, K. (2006). Analysis of leukocyte rolling in vivo and in vitro. Methods Enzymol 416: 346-371.
- Tan, J. K. and Watanabe, T. (2010). Artificial engineering of secondary lymphoid organs. Adv Immunol 105: 131-157.
- Tavares, M. R., Cruz, J. A., Waisberg, D. R., Toledo, S. P., Takeda, F. R., Cernea, C. R., Capelozzi, V. L. and Brandao, L. G. (2014). Lymph node distribution in the central compartment of the neck: an anatomic study. Head Neck 36(10): 1425-1430.
- Vadillo, E., Chanez-Paredes, S., Vargas-Robles, H., Guerrero-Fonseca, I. M., Castellanos-Martinez, R., Garcia-Ponce, A., Nava, P., Giron-Perez, D. A., Santos-Argumedo, L. and Schnoor, M. (2019). Intermittent rolling is a defect of the extravasation cascade caused by Myosin1e-deficiency in neutrophils. Proc Natl Acad Sci U S A 116(52): 26752-26758.
- von Andrian, U. H., (1996). Intravital microscopy of the peripheral lymph node microcirculation in mice. Microcirculation 3(3): 287-300.
- Xu, N., Lei, X. and Liu, L. (2011). Tracking neutrophil intraluminal crawling, transendothelial migration and chemotaxis in tissue by intravital video microscopy. J Vis Exp (55): 3296.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schnoor, M., Santos-Argumedo, L., Girón-Pérez, D. A. and Vadillo, E. (2020). Analysis of B Cell Migration by Intravital Microscopy. Bio-protocol 10(23): e3842. DOI: 10.21769/BioProtoc.3842.
Category
Immunology > Immune cell function > Lymphocyte
Immunology > Immune cell imaging > Epifluorescence Microscopy
Cell Biology > Cell movement > Cell adhesion
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.