- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Charging State Analysis of Transfer RNA from an α-proteobacterium

Published: Vol 10, Iss 23, Dec 5, 2020 DOI: 10.21769/BioProtoc.3834 Views: 3730
Reviewed by: Alka MehraSridevi Ranganathan
Original research article
The authors used this protocol in:

Oct 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Transfer RNA (tRNA) is an essential link between the genetic code and proteins. During the process of translation, tRNA is charged with its cognate amino acid and delivers it to the ribosome, thus serving as a substrate of protein synthesis. To analyze the charging state of a particular tRNA, total RNA is purified and analyzed on an acid-urea gel. Separated RNA is then transferred to a membrane and detected with a probe for the tRNA of interest. Here, we present an improved protocol to analyze the tRNA charging state in the α-proteobacterium Rhodopseudomonas palustris. Compared to the classical method, the RNA isolation step is optimized to suit this organism. Additionally, a non-radioactive platform is used for electrophoresis and Northern blots. This significantly reduces the time and the effort required for this protocol.
Keywords: TranslationBackground
The primary function of tRNA is, with the help of other translation factors, to ensure the accurate translation of mRNA to protein. Aminoacyl-tRNAs (charged) bring amino acids to the ribosome for peptide elongation, and then the uncharged tRNAs are released. The charging state of tRNA is largely determined by the available resource (i.e., amino acids) and their consumption by the ribosome. To analyze the charging state of cellular tRNA, methods have been developed using acid-urea gels to separate the total RNA and to detect the tRNA of interest by Northern blot (Janssen et al., 2012; Bullwinkle and Ibba, 2016). Recently, we explored the relationship between tRNA charging states and survival of starving bacteria (Yin et al., 2019). The method used in that study and elaborated here, has optimized the RNA purification step to better capture the tRNA charging state in bacteria other than E. coli. It also utilizes a commercially available system to perform the analysis without using radioactive materials, which could be helpful for some research groups.
Materials and Reagents
Notes:
It is crucial to keep all the materials and reagents RNase-free for this protocol. To this end, the reagents and materials are all RNase-free, and are purchased pre-made or ready-to-dissolve in order to simplify the procedure as much as possible. Disposable RNase-free centrifuge tubes are used to make reagents whenever applicable.
Multiple kits are available for DIG reagents. Items 19-23 are listed here for readers to choose from to suit the scale required.
Centrifuge tubes, RNase free (1.5 ml, 2 ml, 15 ml and 50 ml, as needed)
QIAzol Lysis Reagent (Qiagen, catalog number: 79306 )
Chloroform, molecular biology grade
Isopropanol, molecular biology grade
Ethanol, molecular biology grade
UltraPure DNase/RNase-Free Distilled Water (Invitrogen, catalog number: 10977-023 )
TEMED (National Diagnostics, catalog number: EC-503 )
Ammonium persulfate (National Diagnostics, catalog number: EC-504 )
Urea (National Diagnostics, catalog number: EC-605 )
AccuGel 29:1 (30%) (National Diagnostics, catalog number: EC-851 )
Sodium acetate, 3 M solution, pH 4.5 (National Diagnostics, catalog number: EC-905 )
Sodium acetate, 3 M solution, pH 5.2 (National Diagnostics, catalog number: EC-906 )
EDTA, 0.5 M, pH 8.0 (Thermo Fisher Scientific, catalog number: AM9260G )
Xylene cyanol, molecular biology grade
Bromophenol blue, molecular biology grade
Tris-HCl solution, 1 M, pH 9.0 (Sigma-Aldrich, catalog number: T2819-100ML )
Amersham Hybond-XL membrane (GE Life Sciences, catalog number: 45-001-148 )
Tris-broate-EDTA (TBE) buffer, 10x (Thermo Fisher Scientific, catalog number: AM9863 )
DIG RNA labeling Mix (Sigma-Aldrich, catalog number: 11277073910 )
DIG RNA labeling kit (Roche, catalog number: 11175025910 )
DIG system (Roche, catalog number: 12039672910 )
DIG Easy Hyb (Sigma-Aldrich, catalog number: 11603558001 )
DIG Wash and Block Buffer Set (Sigma-Aldrich, catalog number: 11585762001 )
Anti-Digoxigenin-AP, Fab fragments (Sigma-Aldrich, catalog number: 11093274910 )
CDP-Star (Sigma-Aldrich, catalog number: 11685627001 )
RNase Zap (Thermo Fisher, catalog number: AM9780 )
Growth medium for R. palustris (PM medium) (see Recipes)
Acid-urea gel (see Recipes)
Sodium acetate-EDTA running buffer (see Recipes)
Sodium acetate-EDTA loading buffer (see Recipes)
Equipment
Note: The working bench as well as electrophoresis, transfer and Northern blot systems should be cleaned with RNaseZap or equivalent to decontaminate RNase before use.
Zirconia/Silica Beads, 0.1 mm dia (Bio Spec Products Inc, manufacture number: 11079101z )
Mini-Beadbeater-24 (Bio Spec Products Inc, catalog number: 112011 )
Electrophoresis, transfer and Northern blot system that fits a gel 20 cm in height
Temperature-controlled tabletop centrifuge
PCR machine for temperature-controlled incubation or equivalent incubator
Software
ImageJ
Procedure
Isolation of total RNA from R. palustris
Notes:
It is crucial to choose the proper cell lysis method for the organism of interest in order to capture the native charging state of tRNA. The method described here, based on micro beads and a beadbeater, is routinely used in our group to isolate RNA from R. palustris and related α-proteobacteria. We strongly advise the reader to test different lysis methods for their organism of interest (e.g., the ratio between bead volume and medium volume, the number of cell beating cycles, etc.).
Another note about tRNA charging states is that, as elaborated elsewhere (Janssen et al., 2012; Bullwinkle and Ibba, 2016), the sample needs to be kept in acidic conditions all the time. Acidic buffers and gel systems are used throughout this protocol.
RNA purified from Procedure A can be used directly in the following steps in this protocol. This is because the charging state of tRNA is calculated based on the ratio of charged vs uncharged tRNA. Slight contamination of DNA and/or protein, therefore, will not affect the end result after the purification described here. If the purified RNA is to be used in other procedure such as RNAseq or in vitro translation, further purification would be required.
Before the experiment, prepare one screw-capped 2 ml centrifuge tube for each sample. Add ~500 µl of Zirconia/Silica Beads into each tube and autoclave all of the tubes.
Grow R. palustris cultures as required by the experimental question. Typical growth conditions that we use are given in Kim and Harwood (1991). However, the composition of the cultivation medium is not important for the protocol we describe here. The typical sample contains 3-5 ml of culture at Abs660 ~1.2. Collect the cells at 4 °C by centrifugation at 4,000 rpm and store the pellets at -80 °C.
Resuspend each frozen cell pellet in 1 ml of QIAzol Lysis Reagent. Mix gently with pipetting and transfer the resuspended samples into the tubes with beads. This step should be performed on ice.
Break the cells with a beadbeater at 3,500 rpm for 1 min and then place the samples on ice for another 1 min. Repeat this cycle 3 more times.
Place all the samples at room temperature for 5 min.
Add 200 µl chloroform to each sample. Shake the tube vigorously by hand for 15 s.
Place the tube at room temperature for 3 min.
Centrifuge the samples for 15 min at 12,000 x g at 4 °C. Carefully transfer 560 µl of the colorless upper aqueous phase to a new 1.5 ml centrifuge tube.
Add 0.5 ml isopropanol and mix thoroughly by vortexing.
Centrifuge for 15 min at 12,000 x g at 4 °C. Discard the supernatant.
Add 1 ml of 75% ethanol. Centrifuge for 5 min at 7,500 x g at 4 °C.
Remove the supernatant completely. Air-dry the RNA pellet in a fume hood briefly (5-10 min).
Re-dissolve each RNA pellet in 10 mM of sodium acetate, pH 4.5. The purified RNA can be stored at -80 °C for at least two weeks. A typical yield is 50 µl of 5 µg/µl total RNA. This number can vary depending on factors such as species, growth states and sampling volume.
Alkaline pH treatment to produce a deacylated (uncharged) tRNA control
Take ~10 µg of purified RNA. Add stock solution of 0.5 M EDTA and 1 M Tris-HCl (pH 9.0) directly to the sample in order to adjust the final buffer to 1 mM EDTA and 100 mM Tris-HCl, pH 9.0. Adjust final volume to 50 µl. Mix well by pipetting.
Incubate the sample at 42 °C for 60 min.
Add 5 µl of 3 M sodium acetate, pH 5.2 to the sample. Reverting the buffer back to pH 5.2 will not make the sample acylated again.
Add 137.5 µl of ice-cold 95% ethanol to the sample, then incubate on ice for 10 min.
Centrifuge at 13,000 x g for 10 min at 4 °C to precipitate RNA.
Remove the supernatant and wash the RNA pellet with 1 ml of 70% ethanol.
Centrifuge at 13,000 x g for 10 min at 4 °C.
Remove the supernatant completely. Air-dry the RNA pellet as needed.
Dissolve deacylated RNA with ~30 µl 10 mM sodium acetate, pH 4.5.
Making DIG-labeled probe for tRNA-Trp
DIG RNA labeling kit was used in our project to generate labeled probe for subsequent experiments. Equivalent reagents, such as DNase I and T7 RNA polymerase, can be substituted where suitable. Other strategies such as the classical P32-labeling can also be used for this step. If so, the following experiments should be adjusted accordingly. As the detailed protocol for the DIG kit is commercially available, this section will focus on how we adapted it to detect tRNATrp in R. palustris CGA009.
A stretch of 31 nucleotides in R. palustris CGA009 tRNATrp was chosen as the target of the probe. The following dsDNA was commercially made (IDT) as the template of in vitro transcription:
TGAATTGTAATACGACTCACTATAGGGGGTTTTGGAGACCGGTGCTCTACCAATTGAG
A basic purification level, which should be molecular biology grade by default, was sufficient for dsDNA in our experience. We have also ordered two complementing ssDNA and annealed them together to form the dsDNA (see Reference 5 for more information), and it worked as well.
Within the above sequence, the shaded part is the promoter for T7 RNA polymerase and the underscored part is the reverse-complement of our target on tRNATrp. A 7-bp clamp is added before the promoter to facilitate polymerase binding. The selection of tRNA target-sequence needs to be done case by case and the readers are encouraged to try several candidate sequences.
Make the reaction mixture following the instructions for the DIG RNA labeling kit. Below is an example we used for our project (all reagents here except the template are included in the kit). The volume can be scaled up or down as needed.
9 µl RNase-free water
4 µl transcription (5x) buffer
4 µl DIG labeling mix
2 µl T7 polymerase
1 µl template of dsDNA (10 ng/µl is sufficient)
Incubate the reaction mixture at 42 °C for 1.5 h.
For each reaction, add 2 µl DNaseI and incubate at 37 °C for 15 min.
Acid-urea polyacrylamide gel electrophoresis
An electrophoresis system of at least 20 cm in height is recommended to analyze the charging states of tRNAs. The thickness of gel/spacer does not matter as long as the well can be sufficiently cleaned of urea. The difference between charged and uncharged tRNA is small in size. A smaller gel might be suitable to analyze some peptidyl-tRNAs, but in general does not provide the resolution to analyze aminoacyl-tRNAs. For each run, a sample of uncharged tRNA must be included as the negative control.
Assemble the electrophoresis system with acid-urea gel. Add the cold sodium acetate-EDTA running buffer and pre-run the gel at 20 V for 30 min at 4 °C. The purpose of pre-running is to bring up the temperature of the gel. See more information at company’s page (see Reference 6).
Incubate the sample at 72 °C for 3 min. Briefly spin the sample and mix it with the loading buffer at a 1:1 ratio. We usually loaded ~20 µl of sample, a total of 2.5-5 µg total tRNA, per well for a gel of 20 cm height. The minimum and maximum amounts of sample required need to be tested and determined case by case.
Clean the loading wells with syringe and needle. This needs to be done immediately before applying the sample to the well. For acid-urea gel, urea continues to accumulate in the well. This can prevent the sample from settling at the bottom of the well if not properly cleaned.
Load the sample. There should be no change of dye color. Run the gel at 15 W for 12 h (for a gel of 20 cm height) at 4 °C. It is crucial to bring up and maintain the operating temperature of the gel during the run, hence the setting of constant power as well as the pre-run specified above (see Reference 5 for more information).
Northern blot analysis
After the electrophoresis, any standard protocol suiting the available system can be used to transfer RNA from the gel to a membrane. In our case, we used 0.5x TBE as the transfer buffer. We did a standard wet-transfer was at 4 °C, either at 20 V overnight or at 650 mA for 30 min. No obvious difference was observed between the two settings. The membrane is then ready for cross-link and following Northern blot. In our experience, exposure to a UV-light box for 10-15 min was sufficient and could serve as a start point.
The standard protocol of the DIG system (Roche, see Reference 7 for more information) was used for Northern blot blocking, washing and detection steps. Briefly, the probe for tRNATrp is used to detect tRNATrp on the membrane, followed by interaction with Anti-Digoxigenin-AP and then CDP-Star to generate chemiluminescence signals. Any suitable imaging system, such as film or digital imager, can be used to detect and quantify the results.
Data analysis
ImageJ was used for quantification of the image on films. For results obtained with a digital imager, the accompanying software was used for quantification. The charging rate of tRNA is calculated as: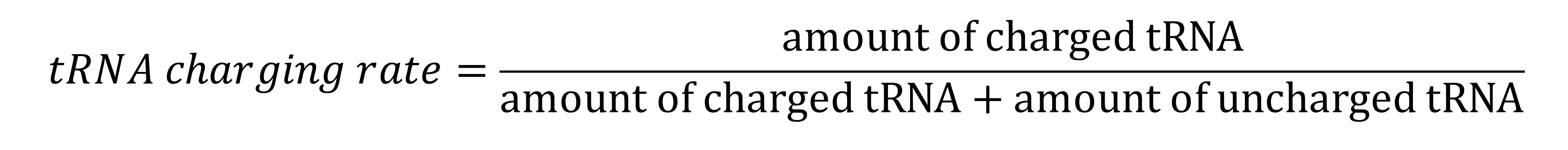
A brief walkthrough of tRNA charging gel analysis is provided here. As shown in Figure 1; lane 1 contains untreated total RNA from growing R. palustris, which mainly contains charged tRNA. The same sample is deacylated as described in this protocol and analyzed in lane 2. The sample to be analyzed is in lane 3. The bands corresponding to charged and uncharged tRNA are determined by referring to the bands in lanes 1 and 2. The intensity of the individual bands is the analyzed with ImageJ. An example can also be found in Figure 5d and 5e from Yin et al. (2019).
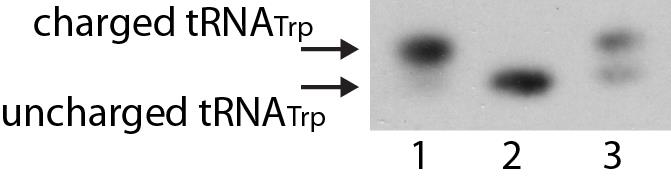
Figure 1. Northern blot showing separation of charged and unchaged tRNATrp
Recipes
Growth medium for R. palustris (PM medium)
For 1,000 ml PM
800 ml distilled water
25 ml 0.5 M Na2HPO4
25 ml 0.5 M KH2PO4
10 ml 10% (NH4)2SO4
1 ml Concentrated base (see below)
0.1 M Na2S2O3·5H2O
2 mg/ml p-aminobenzoic acid
Bring volume to 1,000 ml
For 1,000 ml Concentrated base
20 g Nitrilotriacetic acd (NTA-acid free)
28.9 g MgSO4 anhydrous
6.7 g CaCl2·2H2O
18.5 mg (NH4)6Mo7O24·4H2O
198 mg FeSO4·7H2O
100 ml Metals 44 (see below)
Dissolve NTA separately in 600 ml water and neutralize with KOH (14.6 g KOH). Add other components, and dissolve in the order given. Adjust to pH 6.8 before bringing to final volume of 1,000 ml. A precipitate forms when adjusting the pH from the acid side of 6.8 with KOH (need about 100 ml of 1M KOH) but will eventually redissolve with stirring. When the pH is near 6.8, the color of the solution changes from a deep yellow to straw color. Then, filter steriize and store in a glass bottle wrapped in aluminum fold. Store in refrigerator. It is good for at least one year.
For 1,000 ml Metals 44
2.5 g (free acid, not sodium salt)
10.95 g ZnSO4·7H2O
5 g FeSO4·7H2O
1.54 g MnSO4·H2O
392 mg CuSO4·5H2O
250 mg Co(NO3)2·6H2O
177 mg Na2B4O7·10H2O
Add EDTA to 800 ml distilled water with stirring and adjust pH to about 5.0 with 10 M NaOH to get EDTA dissoved. Add the other metals in the order given. Do not add components until the previous one has dissolved completely. Then bring to a final volume of 1,000 ml (the final pH is around 2.4, a clear and lime green solution). Then filter sterilize and store in a glass bottle wrapped in aluminum foil. Can store in refiigerator indefinitely.
Acid-urea gel
Make acid-urea gel solution in a 50 ml centrifuge tube. A recipe for 50 ml of 12% acid-urea gel solution is as follows:
22.5 g of urea
1.5 ml of 3 M sodium acetate, pH 5.2
93.8 µl of 0.5M EDTA, pH 8.0
18 ml of 30% acrylamide (29:1)
Add RNase-free water and bring the total volume to 45 ml
Add 1 ml of 10% APS
Invert the tube gently to mix
Then add 20 µl of TEMED and invert the tube gently to mix
Pour the solution immediately into the gel cassette
Sodium acetate-EDTA running buffer
Prepare sodium acetate-EDTA running buffer as follows. The buffer should be pre-chilled to 4 °C before the run.
100 mM sodium acetate, pH 5.2
1 mM EDTA
Sodium acetate-EDTA loading buffer
Prepare sodium acetate-EDTA loading buffer with the following recipe:
10 ml of RNase-free water
4.8 g of urea
20 μl of 0.5 M EDTA, pH 8.0
33 μl of sodium acetate, pH 5.2
Pinch of xylene cyanol and bromophenol blue
Acknowledgments
This work was supported by grant W911NF-15-1-0150 to C.S.H. from the U.S. Army Research Office. We thank Dr. Yasuhiro Oda, University of Washington, for his extensive RNA work in R. palustris. We also thank Prof. Michael Ibba, Ohio State University for his excellent advice about tRNA. This protocol is derived from our previous work published in 2019 (Yin et al., 2019).
Competing interests
The authors claim no financial or non-financial competing interest.
References
- Bullwinkle, T. J. and Ibba, M. (2016). Translation quality control is critical for bacterial responses to amino acid stress. Proc Natl Acad Sci U S A 113(8): 2252-2257.
- Janssen, B. D., Diner, E. J. and Hayes, C. S. (2012). Analysis of aminoacyl- and peptidyl-tRNAs by gel electrophoresis. Methods Mol Biol 905291-309.
- Yin, L., Ma, H., Nakayasu, E. S., Payne, S. H., Morris, D. R. and Harwood, C. S. (2019). Bacterial longevity requires protein synthesis and a stringent response. mBio 10(5): .
- Kim, M. K. and Harwood, C. S. (1991). Regulation of benzoate-CoA ligase in. Rhodopseudomonas palustris. FEMS Microbiology Letters 83199-203.
- https://www.idtdna.com/pages/education/decoded/article/annealing-oligonucleotides.
- https://www.nationaldiagnostics.com/electrophoresis/article/run-conditions-denaturing-page.
- 12039672910 bul.pdf" target="_blank">https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Roche/Bulletin/1/ 12039672910 bul.pdf.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yin, L. and Harwood, C. S. (2020). Charging State Analysis of Transfer RNA from an α-proteobacterium. Bio-protocol 10(23): e3834. DOI: 10.21769/BioProtoc.3834.
Category
Molecular Biology > RNA
Microbiology > Microbial biochemistry > RNA
Microbiology > Microbial physiology > Adaptation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.