- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Serial Cryomicrotomy of Saccharomyces cerevisiae for Serial Electron Cryotomography
Published: Vol 10, Iss 22, Nov 20, 2020 DOI: 10.21769/BioProtoc.3831 Views: 4476
Reviewed by: Ashraf Al-AmoudiKrishna SaharanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics









Abstract
Electron cryotomography (cryo-ET) is an increasingly popular technique to study cellular structures and macromolecules in situ. Due to poor penetration of electrons through thick biological samples, the vitreously frozen samples for cryo-ET need to be thin. For frozen-hydrated cells, such samples can be produced either by cryomicrotomy or cryo-FIB-milling. As a result, a tomogram of such a sample contains information of a small fraction of the entire cell volume, making it challenging to image rare structures in the cell or to determine the distribution of scattered structures. Here, we describe the tools and workflow that we designed to facilitate serial cryomicrotomy, which makes possible the exploration of a larger volume of individual cells at molecular resolution. We successfully used serial cryomicrotomy to locate and image the Dam1/DASH complex located at microtubule plus ends inside mitotic Saccharomyces cerevisiae cells.
Keywords: Serial electron cryotomographyBackground
Chromosomes have to be coupled to kinetochore microtubules for successful segregation during cell division. The kinetochore is the macromolecular complex responsible for coupling the chromosomes to the kinetochore microtubules’ dynamic plus ends. In the budding yeast Saccharomyces cerevisiae, the Dam1/DASH complex is a part of the kinetochore that interfaces with kinetochore microtubules. There have been many studies to reconstitute kinetochore subcomplexes in vitro to understand its structure and function (Musacchio and Desai, 2017; Hinshaw and Harrison, 2018). These attempts have led to multiple models that suggest how the kinetochore functions. To test some of these outer-kinetochore models in S. cerevisiae, we use electron cryotomography (cryo-ET) to visualize the S. cerevisiae outer kinetochore at molecular resolution (4-6 nm) in situ.
Cryo-ET is an increasingly popular technique used to study cellular structures and macromolecules in situ at molecular resolution (Ng and Gan, 2020). Many of the earlier cryo-ET studies involve using biological samples that are thick (> 500 nm), resulting in reconstructions with low contrast. To increase the contrast of the reconstructions while preserving cellular context of the samples, the samples have to be much thinner, but not too thin. Therefore, a reasonable compromise is ~100 nm thickness. The two main techniques used to obtain thin samples for cryo-ET are cryo-FIB-milling and cryomicrotomy. Cryo-FIB-milling involves using gallium ions to sputter away biological material until a thin plank-like cryo-lamella of biological sample remains (Marko et al., 2006; Hayles et al., 2007; Rigort et al., 2010; Villa et al., 2013; Mahamid et al., 2015; Medeiros et al., 2018). Cryomicrotomy involves cutting a frozen-hydrated sample, such as a cell paste, into thin cryosections using a diamond knife (Al-Amoudi et al., 2004a and 2004b). Each cryo-lamella or cryosection contains only a small fraction of a cell volume. It is worth noting, however, that serial cryomicrotomy would allow us to explore a larger fraction of a cell at molecular resolution compared to cryo-FIB-milling. This can be done by imaging multiple consecutive cryosections of the same cell. Here, we describe both the tools and workflow that we designed to facilitate serial cryomicrotomy and data collection. We successfully used serial cryomicrotomy and serial cryo-ET to locate and image the Dam1/DASH complex in S. cerevisiae. We also explore the cryomicrotomy parameters that minimize artefacts in yeast cryosections. This approach has also been used to study mitotic fission yeast and meiotic budding yeast (Cai et al., 2018; Ma et al., 2019).
Materials and Reagents
Parafilm
50 ml Falcon tube
1 ml pipette tip
S. cerevisiae cells
0.1 mg/ml bovine serum albumin (Sigma-Aldrich)
10-nm gold nanoparticles (BBI Solutions, EM.GC10 )
50% (v/v) ethanol in distilled H2O, 0.2 µm filtered (500 ml)
18 MΩ H2O, 0.2 µm filtered (1 L)
Ensure that there are no debris in both H2O and ethanol before using as debris can damage the diamond knife.
Dextran (Mr ~6000) (Sigma Aldrich, catalog number: 31388 )
Pressurized ethane gas
Liquid nitrogen
Equipment
Tools (Standard)
Parallel-bar EM grids coated with continuous carbon film (Electron Microscopy Sciences, catalog number: G150PB-CU )
EM grid boxes
K100X Glow Discharge (Quorum Emitech)
Note: Product has been discontinued in 2010. The current equivalent is the SC7620 Mini Sputter Coater/Glow Discharge System.
Fine-tipped and self-locking tweezers for handling EM grids (Ted Pella, Inc.)
Metal chuck (included with UC7/FC7)
Chuck adaptor (0.45 mm) (included with UC7/FC7)
Plastic FC7 cover (included with UC7/FC7)
UC7/FC7 (Leica Microsystems)
Note: An alternative cryomicrotome by RMC Products is commercially available. However, we do not have any experience with that product.
Diamond trimming and cutting knives (Diatome)
Crion (Leica Microsystems)
Centrifuge
Diamond knife
Forceps
Tools (Non-standard)
Fibre tool (Figure 1A)
Guinea pig hair (available from pet shops or animal husbandry facilities)
Micromanipulator (Narishige Scientific Instrument Lab, catalog number: MN-151S [6 mm joystick travel]) (Figure 1B) (Inspired by Ladinsky et al., 2006 ; Ladinsky, 2010; Studer et al., 2014 )
Note: Any commercially available micromanipulator can be adapted for use in cryomicrotomy.
Copper tubes (0.45 mm inner diameter, 0.65 mm outer diameter) and wire tool for drawing samples into copper tubes (Part 733-1, Engineering Office M Wohlwend GmbH) (Figure 1C)
Flat-nosed pliers (Figure 1D)
Copper tube cutting device (Part. 732, Engineering Office M Wohlwend GmbH) (Figure 1E)
Steel bucket for storing copper tubes (Figure 1F) (design based on Zuber lab’s cryosectioning tools)
Aluminium mount (base and insert pieces) (Figure 1G)
Slotted plastic FC7 covers [modified from (Studer et al., 2014) to minimize ice contamination] (Figure 1H)
Air dust blower

Figure 1. Tools for enabling serial cryomicrotomy. A. Custom-made fibre tool for manipulating the cryoribbon during cryomicrotomy. B. Custom micromanipulator for fine control of the fibre tool. During cryomicrotomy, the micromanipulator is secured to a heavy steel block with a magnetic stand. C. Copper tubes (0.45 mm inner diameter) and wire tool (upper) for drawing samples into the copper tubes for self-pressurized freezing. D. Pliers for clamping shut the end of the copper tubes before freezing. E. Device for cutting off flat ends of copper tube along the cyan dotted lines. Note that the rust, which cannot be completely removed, does not affect the performance of the cutting device. Users can also consider a cutter made from a rust-prone material. F. Custom steel bucket for storing copper tubes containing frozen samples. Steel bucket can easily fit into a 50 ml Falcon tube. G. Base and insert pieces of the aluminium mount that can be assembled on a diamond knife. H. Slotted plastic FC7 cover with cutouts for the (i) forceps, (ii) Crion, (iii) fibre tool, and (iv) for observing during cryomicrotomy. The translucency of the cover does not affect cryomicrotomy workflow.
Software
IMOD (Mastronarde, 1997) (https://bio3d.colorado.edu/imod/)
Procedure
EM grid preparation
Plasma clean the EM grids using the K100X Glow Discharge (15 mA, 45 s) to increase the hydrophilicity of the grids.
Secure the grids using self-locking tweezers.
Cover the carbon side of the grids with 4 µl of 10-nm gold nanoparticles in 46 µl of 0.1 mg/ml bovine serum albumin.
Note: Gold nanoparticles are used as fiducials for tilt series alignment for tomogram reconstruction.
Air-dry the grids overnight and then store them in a dry box until use.
Self-pressurized freezing (Leunissen and Yi, 2009)
Pellet 50 ml of S. cerevisiae cells (OD600 ~1) using a centrifuge (5,000 x g, 4 °C for 5 min).
Discard the supernatant.
Resuspend the cell pellet in an equal volume of 50% dextran using a cut 1 ml pipette tip.
Note: Using a cut 1 ml pipette tip will help resuspend the cell pellet in the viscous 50% dextran.
Pipette ~20 μl of the mixture onto a piece of parafilm.
Insert the wire tool through the copper tube, then dip one end of the copper tube into the droplet and draw the mixture into the tube by pulling out the wire tool.
Notes:
To prevent drawing air into the copper tube, ensure that the end of the copper tube is fully submerged in the droplet before drawing the sample.
Repeat 2-3 times to ensure that there are no air bubbles in the copper tube.
Clamp both ends of the copper tube shut using the flat-nosed pliers.
Drop the copper tube into liquid ethane for self-pressurized freezing.
Transfer the copper tube into the cutting device and cut off flat ends of the copper tube (Figure 1E).
Self-pressurized freezing works well on our yeast paste samples. An alternative method for making frozen-hydrated samples of cell paste is high-pressure freezing (Dubochet, 1995).
Cryo-microtome setup (Figures 2A-2D)
Place the diamond knives and sample holder (chuck and chuck adaptor) into FC7 and tighten securely.
Cover the FC7 with the plastic FC7 cover to prevent accumulation of ice crystals in the cryochamber.
Cool down the system to -150 °C.
Ensure that the trimming knife is at the trimming position (Figure 2E).
Transfer the steel bucket containing the copper tube samples into the FC7 and place it on the platform at the back of the FC7 (Figure 2A; also see Figure 2 for the definition of the ‘back’).

Figure 2. Assembly of tools for cryomicrotomy. A. Positioning of the forceps on the built-in micromanipulator, and the steel bucket containing copper tube samples on the platform at the back of the FC7. B. Micromanipulator and fibre tool set up for manipulating the cryoribbon. C. Positioning of the tools in the FC7 chamber to enable transfer of a cryoribbon onto a carbon-coated EM grid. D. Close-up of chuck, chuck adaptor and copper tube. Knife stage in (E) preparation position and in (F) cutting position for trimming. G-I. Assembly of the aluminium mount onto a cutting knife.Use cooled tweezers to remove a copper tube of frozen sample from the steel bucket and transfer it swiftly into the FC7 cryochamber.
Minimizing the magnitude of movement will decrease the amount of disturbances to air flow, thus limiting introduction of ice contamination into cryochamber.
Insert the copper tube into the sample holder and ensure that the tube is held securely in place.
Trimming
Position the trimming knife at cutting position (Figure 2F) by moving the knife closer to the copper tube such that the knife edge is just grazing the outer surface of the copper tube.
Notes:
Trimming should only start from the ends of the copper tube.
Trimming copper does not harm the diamond knife, but the knife can be easily damaged by side-on collision with the copper tube.
Insert the Crion and let it cool for 10 min.
Set the trimming settings on UC7 (500 nm feed, 50 mm/s cutting speed, medium return speed).
Turn on the Crion and set it to discharge mode to prevent debris from sticking to knife edge.
Note: If the Crion does not keep the knife edge clean enough, pour liquid nitrogen, drop-wise, to wash away debris that have accumulated on the knife edge.
Start trimming until the surface of the sample appears smooth and dark, indicating vitreous ice.
Notes:
If the sample does not appear smooth and dark throughout, then it is probably not vitreous.
For self-pressurized-frozen samples, the thin layer of sample that is in contact with the copper tube’s inner surface might appear to be frosted because it is not vitreous. This small amount of crystalline ice is acceptable as long as there is sufficient vitreous ice closer to the centre of the tube to create a square mesa (see following section).
Slow down the trimming speed to 25 mm/s, medium return speed, and 200 nm feed, then trim another 1 µm.
Block trimming to create a 100 x 100 x 30 µm square mesa (Figures 3A-3C)
Set the trimming settings on UC7 (200 nm feed, 25-50 mm/s cutting speed, medium return speed).
Note: Slowing down trimming speed reduces vibrations introduced to the system when the sample is being trimmed, resulting in smoother surfaces on the slopes of the mesa, which is essential for cryosection-to-cryosection attachment.
Translate the trimming knife to the right so that its left corner is ~0.1 mm from the right inner wall of the copper tube.
Translate the trimming knife forward so that the knife edge is lightly touching the surface of the sample.
Trim until ~30 µm deep.
Translate the trimming knife backwards ~50 µm.
Translate the trimming knife to the left so that the right corner of the knife blade is ~110 µm from the newly created mesa edge.
Trim until ~30 µm deep.
Translate the trimming knife backwards ~50 µm.
Unlock the metal chuck and rotate the copper tube 90°, lock the chuck and repeat Steps E2-E8.
There should now be a 100 µm square mesa, 30 µm tall.
The length of the mesa (along the cutting direction, indicated as L in Figure 3F) can be shortened to increase the number of cryosections per unit length of the cryoribbon.
Cryomicrotomy
Rotate the knife stage so that the cutting knife is facing the sample.
Place the base piece of the aluminium mount onto the cutting knife (Figures 2G and 2H).
Insert the fibre tool into the FC7 and wait 5 min for the tools to cool down.
Slowly bring the knife edge close to the sample surface.
Position the guinea pig hair close to the sample.
Notes:
Make sure that the guinea pig hair does not touch the sample surface or the knife edge.
Check that the guinea pig hair can translate at least 3 mm from the open end of the sample tube towards the back of the FC7.
Input the cutting settings on the UC7 (100 nm feed, 1 mm/s, slow return speed).
Turn on the Crion and set it to discharge mode.
Start cutting.
After the first few (approximately 3-5 cryosections for a 100 µm mesa) cryosections are produced, stop cutting.
Using the fibre tool, gently lift the cryoribbon from the second cryosection and loop it upwards and then towards the knife edge (Figure 3D).
Notes:
Ensure that the fresh edge of the cryoribbon remains attached to the knife edge.
Use the positioning knobs on the micromanipulator to move the fibre tool up/down, and the joystick to move the fibre tool front/back.
Make 2-3 cuts while holding the loop in position (Figure 3E). This will cause the cryosections to fuse at the cut site, generating a loop at the start of the cryoribbon that can be used to hold onto and manipulate the cryoribbon.
Insert the tip of the guinea pig hair through the loop.
Start cutting again while maintaining constant tension on the cryoribbon using the micromanipulator joystick (Figure 3F).
Once the cryoribbon is longer than 3 mm, stop cutting.
A 3-mm-long cryoribbon will span the diameter of an EM grid, maximizing the number of serial cryosections, and is therefore important for serial cryo-ET.
Pick up an EM grid using the forceps. Ensure that the copper bars of the EM grid are parallel with the cryoribbon.
Place the forceps onto the built-in micromanipulator to insert the EM grid into FC7, and then wait ~1 min for the grid and forceps to cool down.
Note: Do not let the EM grid touch the knife edge.
Position the EM grid under the cryoribbon. Use the fibre tool to align the cryoribbon parallel to and in between the copper bars of the EM grid (Figure 3G). Position the cryoribbon near the middle of the grid to maximize the number of imageable serial cryosections.
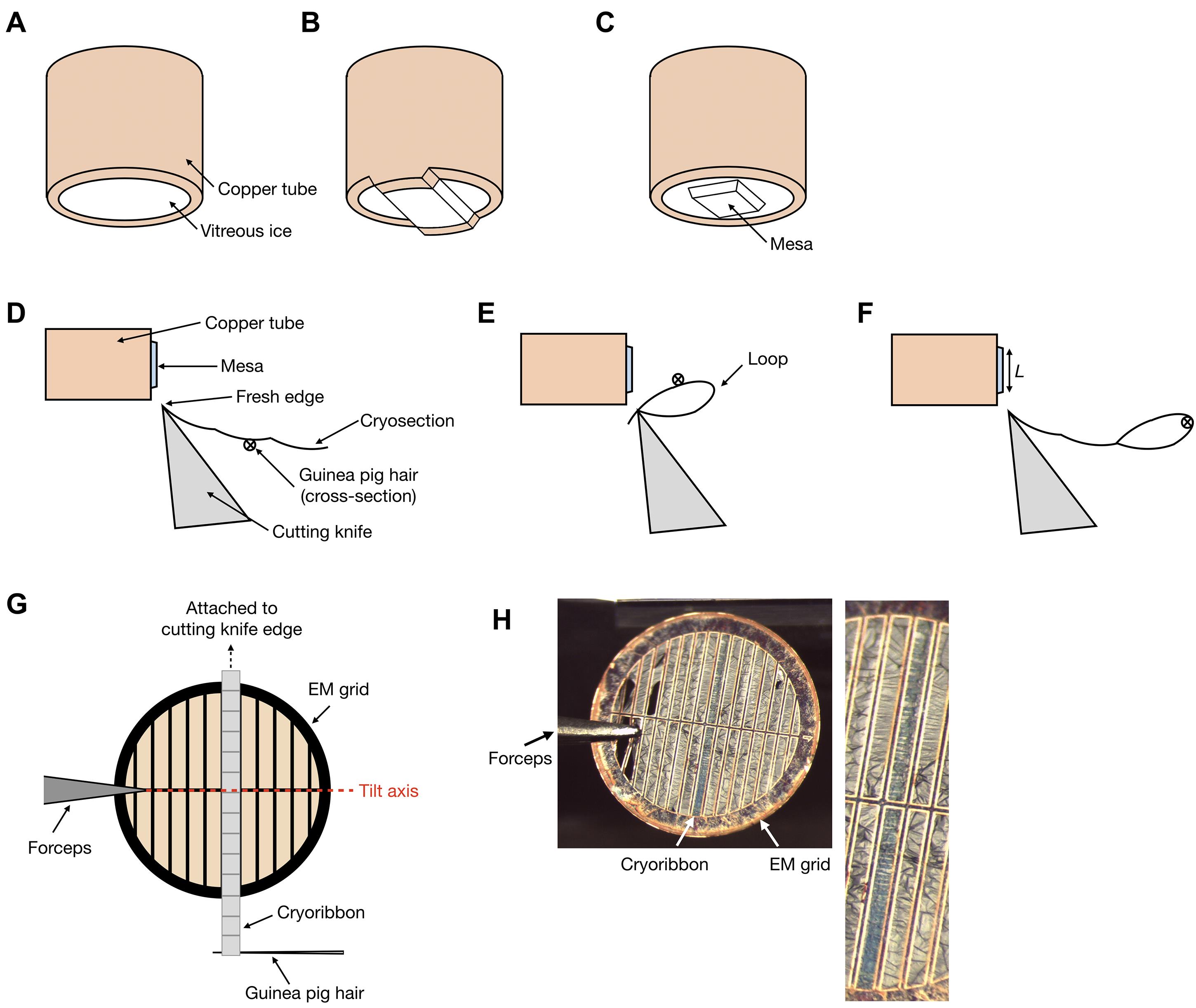
Figure 3. Cryoribbon preparation and grid positioning. A-C. After obtaining a smooth surface of vitreous ice, as shown in (A), trim the outer edges of the tube in two directions perpendicular to each other (B and C) to generate a mesa. D. After the first few cryosections have been produced, place the guinea pig hair under the cryoribbon and loop it towards the knife edge as shown in (E). Holding the loop in place, make two to three more cuts to fuse the end of the cryoribbon to the new cryosection. F. Insert the guinea pig hair through the loop and use it to manipulate the cryoribbon for the rest of the cutting process. Shorter cryosections (length of cryosection defined by L) will result in more cryosections per cryoribbon. G. After cutting, use the fibre tool to align cryoribbon parallel to the bars of the EM grid before attachment. The grid should be loaded into the cryo-electron microscope such that the parallel bars of the grid are perpendicular to the tilt axis of the microscope stage. To facilitate visualization, the cryosections and grid bars are not drawn to scale. H. Left panel: Photograph of a parallel-bar EM grid with a 3 mm-long cryoribbon attached. Right panel: Enlargement of the attached cryoribbon shown in the left panel.Turn on the Crion in charging mode for ~10 seconds to attach the cryoribbon to the EM grid (Figure 3H).
Remove the fibre tool from the FC7.
Cool the insert piece of the aluminium mount in liquid nitrogen and place it onto the base piece on the cutting knife (Figure 2I).
Adding the insert piece towards the end of the experiment provides a clean surface to store the EM grid while the EM grid box is being prepared.
Gently place the EM grid on the aluminium mount and carefully remove the fibre tool and forceps from the FC7.
Note: Placing the EM grid on the platform allows easier transfer into an EM grid box.
Cool an EM grid box in liquid nitrogen and put it into the FC7.
Note: This is done towards the end of the experiment to minimize ice contamination in the grid box.
Place the EM grid into the EM grid box and store it in liquid nitrogen until imaging.
Packing up and tool maintenance
Stop liquid nitrogen cooling of the FC7.
Immediately remove the knives and the knife stage, and wash by pouring 500 ml of 18 MΩ H2O over the knife edge, followed by 500 ml of 50% ethanol, then 500 ml of 18 MΩ H2O again.
Use an air dust blower to remove any visible debris on the knife blades, then use Kimwipe to dry the rest of the knife without touching the knife blade.
Store all the knives in a dry cabinet.
Use a hair blow dryer to dry all the other tools prior to storage.
Tilt series collection on serial cryosections
Load the EM grids in the cryo-electron microscope such that the parallel bars are perpendicular to the tilt axis (Figure 3D).
Note: This geometry ensures that the grid bars do not occlude the cryosections at high tilts.
At low magnification, select a cell of interest for imaging in one cryosection near the edge of the grid, then locate and record the stage positions of the same cell on multiple consecutive cryosections for tilt series collection.
Note: If a cell position is well attached to the carbon on the EM grid, it is likely that the same position on other cryosections of the cryoribbon will also be well attached.
Record tilt series of each cellular position, noting down each tilt series’ order along the cryoribbon.
Reconstruct tomograms and join them together as a serial tomogram using the IMOD programme Etomo (Figure 4A).
Data analysis
Results
Our modified cryomicrotomy workflow and customized tools allowed us to reproducibly create 3-mm-long cryoribbons. The quality of these cryoribbons allowed us to determine the thinnest yeast cryosection that could be produced using our setup. Our thinnest usable cryosections were produced using a 20 nm feed (Figure 4B). A 10 nm feed resulted in clumping of the cryosections and could not be imaged using cryo-electron microscopy. Cutting cryosections with a thinner feed also resulted in more severe compression of the cryosections (Figure 4C).

Figure 4. Additions to the cryomicrotomy workflow enabled production of long cryoribbons, allowing for serial cryo-electron tomography. A. Cross-section of a joined tomogram of 7 tomograms cut using 100 nm feed. The longitudinal view of a spindle microtubule can be seen spanning 6 of the 7 tomograms (black rectangle). B. A single cryoribbon produced using feeds ranging from 100 nm to 10 nm. A cutting feed of 10 nm led to clumping of cryosections. C. Cutting using a 20 nm feed results in cryosections with more severe compression artefact as shown by the increase in ellipticity of the S. cerevisiae cells (outlined in black). Compression artefacts can lead to distortion of cellular components.
We further tested our cryomicrotomy setup by characterizing the effects of cutting temperature and type of cutting knife on cryosections (Al- Amoudi et al., 2005 ; Han et al., 2008 ). We found that cutting at -140 °C instead of -150 °C resulted in less severe crevassing artefacts on the surface of cryosections while cutting with a 25° knife instead of a 35° knife led to less compression of the cryosections (Figure 5).
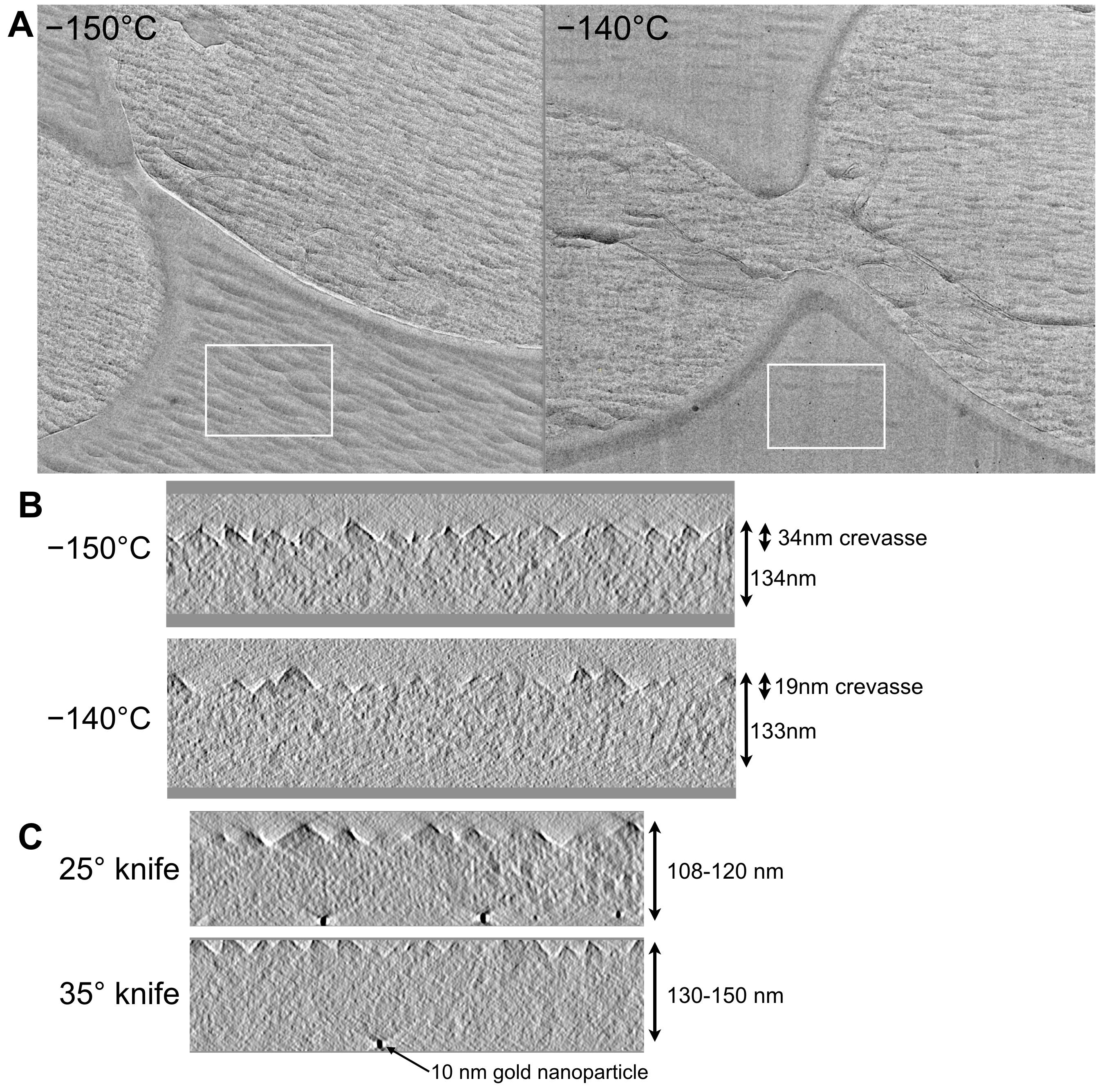
Figure 5. Comparison of yeast cryosections cut at different temperatures and by different diamond knives. A. Crevassing artefacts are diminished when cutting is done at -140 °C instead of at -150 °C. The difference is especially prominent when comparing crevassing effects on extracellular buffer (white rectangles). B. The depth of crevasses over biological material also decreases from approximately 34 nm to 19 nm as a result of cutting at -140 °C instead of -150 °C. C. The magnitude of compression of the cryosection decreases when cutting with a 25° knife, resulting in a thinner cryosection. All cryosections shown here were produced with a 100 nm feed.
Notes
General tips:
To avoid injury, do not touch the knife blades. Contact with the knife edge may also damage the knife edge and compromise quality of cryosections.
Ice contamination can easily hinder collection of serial tilt series. Hence, it is important to take precautions at all steps of the experiment to minimize introduction of vapour into the FC7 cryochamber. The precautions are as follows:
Move slowly and with deliberation to avoid disturbance to air flow within the FC7. Reducing disturbance to the air flow will minimize ice contamination.
Minimize exposure of the FC7 cryochamber to external environment using plastic covers.
Wear a surgical mask during cryomicrotomy to minimize water vapour contamination from breathing.
After trimming or cryomicrotomy, move the knife away from the sample towards the back of the FC7 before translating the knife stage sideways in order to prevent damaging the surface of the sample.
Cool all tools down before bringing them close to the sample.
Acknowledgments
We thank Chen Chen for the initial cryomicrotomy set up in the lab, Gemma An for suggesting the use of parallel-bar EM grids, and Olivia Ma for her help in preparation of figures and feedback on the manuscript. We also thank members of the Gan lab for feedback and tips on cryomicrotomy. We thank Satoru Chomabayashi (Narishige) for help in modifying the MN-151 to increase the joystick travel and Eddie Lee (JEG Engineering Supplies) for fabricating the other custom tools. This protocol was used in Ng et al., 2019.
C.T. Ng and L. Gan were funded by Singapore Ministry of Education grant MOE2018-T2-2-146. M.S. Ladinsky was funded by National Institutes of Health grant (2 P50 GM082545-08) to Pamela J. Bjorkman.
Competing interests
The authors declare no competing financial interests.
References
- Al-Amoudi, A., Chang, J. J., Leforestier, A., McDowall, A., Salamin, L. M., Norlén, L. P., Richter, K., Blanc, N. S., Studer, D. and Dubochet, J. (2004a). Cryo-electron microscopy of vitreous sections. EMBO J 23(18): 3583-3588.
- Al-Amoudi, A., Norlen, L. P. and Dubochet, J. (2004b). Cryo-electron microscopy of vitreous sections of native biological cells and tissues. J Struct Biol 148(1): 131-135.
- Al-Amoudi, A., Studer, D. and Dubochet, J. (2005). Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. J Struct Biol 150(1): 109-121.
- Cai, S., Chen, C., Tan, Z. Y., Huang, Y., Shi, J. and Gan, L. (2018). Cryo-ET reveals the macromolecular reorganization of S. pombe mitotic chromosomes in vivo. Proc Natl Acad Sci U S A 115(43): 10977-10982.
- Dubochet, J., (1995). High-pressure freezing for cryoelectron microscopy. Cell 5: 366-368.
- Han, H. M., Zuber, B. and Dubochet, J. (2008). Compression and crevasses in vitreous sections under different cutting conditions. J Microsc 230(Pt 2): 167-171.
- Hayles, M. F., Stokes, D. J., Phifer, D. and Findlay, K. C. (2007). A technique for improved focused ion beam milling of cryo-prepared life science specimens. J Microsc (3): 263-269.
- Hinshaw, S. M. and Harrison, S. C. (2018). Kinetochore function from the bottom up. Trends Cell Biol 28(1): 22-33.
- Ladinsky, M. S., (2010). Chapter Eight- Micromanipulator-Assisted Vitreous Cryosectioning and Sample Preparation by High-Pressure Freezing. In: Methods in Enzymology. Jensen, G. J. (Ed.). Academic Press. 481: 165-194.
- Ladinsky, M. S., Pierson, J. M. and McIntosh, J. R. (2006). Vitreous cryo-sectioning of cells facilitated by a micromanipulator. J Micro 224(2): 129-134.
- Leunissen, J. L. and Yi, H. (2009). Self-pressurized rapid freezing(SPRF): a novel cryofixation method for specimen preparation in electron microscopy. J Microsc 235(1): 25-35.
- Ma, O. X., Cai, S., Shi, J. and Gan, L. (2019). Cryo-ET analysis of budding yeast synaptonemal complexes in situ. bioRxiv: 746982.
- Mahamid, J., Schampers, R., Persoon, H., Hyman, A. A., Baumeister, W. and Plitzko, J. M. (2015). A focused ion beam milling and lift-out approach for site-specific preparation of frozen-hydrated lamellas from multicellular organisms. J Struct Biol 192(2): 262-269.
- Marko, M., Hsieh, C., Moberlychan, W., Mannella, C. A. and Frank, J. (2006). Focused ion beam milling of vitreous water: prospects for an alternative to cryo-ultramicrotomy of frozen-hydrated biological samples. J Microsc 222(Pt 1): 42-47.
- Mastronarde, D. N., (1997). Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120(3): 343-352.
- Medeiros, J. M., Bock, D., Weiss, G. L., Kooger, R., Wepf, R. A. and Pilhofer, M. (2018). Robust workflow and instrumentation for cryo-focused ion beam milling of samples for electron cryotomography. Ultramicroscopy 190: 1-11.
- Musacchio, A. and Desai, A. (2017). A molecular view of kinetochore assembly and function. Biology(Basel) 6(1).
- Ng, C. T., Deng, L., Chen, C., Lim, H. H., Shi, J., Surana, U. and Gan, L. (2019). Electron cryotomography analysis of Dam1C/DASH at the kinetochore-spindle interface in situ. J Cell Biol 218(2): 455-473.
- Ng, C. T. and Gan, L. (2020). Investigating eukaryotic cells with cryo-ET. Mol Biol Cell 31(2): 87-100.
- Rigort, A., Bäuerlein, F. J., Leis, A., Gruska, M., Hoffmann, C., Laugks, T., Bohm, U., Eibauer, M., Gnaegi, H., Baumeister, W. and Plitzko, J. M. (2010). Micromachining tools and correlative approaches for cellular cryo-electron tomography. J Struct Biol 172(2): 169-179.
- Studer, D., Klein, A., Iacovache, I., Gnaegi, H. and Zuber, B. (2014). A new tool based on two micromanipulators facilitates the handling of ultrathin cryosection ribbons. J Struct Biol 185(1): 125-128.
- Villa, E., Schaffer, M., Plitzko, J. M. and Baumeister, W. (2013). Opening windows into the cell: focused-ion-beam milling for cryo-electron tomography. Curr Opin Struct Biol 23(5): 771-777.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ng, C. T., Ladinsky, M. and Gan, L. (2020). Serial Cryomicrotomy of Saccharomyces cerevisiae for Serial Electron Cryotomography. Bio-protocol 10(22): e3831. DOI: 10.21769/BioProtoc.3831.
- Ng, C. T., Deng, L., Chen, C., Lim, H. H., Shi, J., Surana, U. and Gan, L. (2019). Electron cryotomography analysis of Dam1C/DASH at the kinetochore-spindle interface in situ. J Cell Biol 218(2): 455-473.
Category
Biophysics > Electron cryotomography
Cell Biology > Cell imaging > Cryosection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.