- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Reference-free Association Mapping from Sequencing Reads Using k-mers
Published: Vol 10, Iss 21, Nov 5, 2020 DOI: 10.21769/BioProtoc.3815 Views: 5491
Reviewed by: Avinash Chandra PandeyRAMESH KUDIRAAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Association mapping is the process of linking phenotypes with genotypes. In genome wide association studies (GWAS), individuals are first genotyped using microarrays or by aligning sequenced reads to reference genomes. However, both these approaches rely on reference genomes which limits their application to organisms with no or incomplete reference genomes. To address this, reference free association mapping methods have been developed. Here we present the protocol of an alignment free method for association studies which is based on counting k-mers in sequenced reads, testing for associations between k-mers and the phenotype of interest, and local assembly of the k-mers of statistical significance. The method can map associations of categorical phenotypes to sequence and structural variations without requiring prior sequencing of reference genomes.
Keywords: Association mappingBackground
Association mapping, i.e., the process of associating genotypes to phenotypes is most frequently performed in the form of genome wide association studies (GWAS) with single nucleotide polymorphisms (SNP). Microarrays are used to genotype individuals at a large number of known SNP locations and each SNP is tested for association with the phenotype of interest. But this approach requires prior sequencing of a reference genome and determining the locations of the SNPs. Moreover, this precludes mapping associations to structural variations such as insertion-deletions (indels) and copy number variations, and to variations outside of the reference genome.
With advances in sequencing technologies, the use of whole genome sequenced reads for association mapping is increasingly becoming more widespread. This is most commonly done by mapping the reads to a reference genome, calling variants, and testing for association between the variants and the phenotype. However, this approach also requires a reference genome and regions missing from the reference are not included in the study.
To address these issues, a number of reference free methods for association mapping have been developed. They are based on testing for association between k-mers, i.e., contiguous sequence of length k in sequenced reads and the phenotype. Sheppard et al., 2013, Earle et al., 2016, Lees et al., 2016 and Jaillard et al., 2018 presented methods for association mapping in bacterial genomes where high plasticity makes application of reference based methods difficult. Rahman et al., 2018 introduced a method for mapping associations to categorical phenotypes applicable to organisms with large genomes and more recently Voichek et al., 2020 presented a method for both categorical and quantitative phenotypes.
Here we present the protocol of the reference free association mapping tool HAWK, which was developed by Rahman et al., 2018 and extended by Mehrab et al., 2020. It works by first counting k-mers in reads from each individual using Jellyfish (Marçais and Kingsford, 2011). Then likelihood ratio test is used to find k-mers with significantly different counts in case and control samples. Next, population structure is determined using Eigenstrat (Patterson et al., 2006, Price et al., 2006). Finally, k-mers associated with the phenotype are identified and the k-mers are locally assembled to get a sequence for each associated loci. The results found by HAWK were found to be largely in agreement with reference based methods. Moreover, HAWK was able to map associations to structural variants and to variants in regions not present in the reference. It is worth reiterating that the method is applicable for any genetic diseases or traits. However, it currently only supports categorical, i.e., binary phenotypes although work is ongoing to extend it to quantitative phenotypes.
Equipment
Computer (We recommend at least 16GB RAM and multiple cores)
Software
HAWK ( Rahman et al., 2018 ; Mehrab et al., 2020 )
The primary software for association mapping from sequencing reads using k-mers. The software is available for download at https://github.com/atifrahman/HAWK/releases and installation instructions are at https://github.com/atifrahman/HAWK.
Modified version of Jellyfish/Jellyfish 2 (Marçais and Kingsford, 2011)
For k-mer counting. Modified versions available for download at https://github.com/atifrahman/HAWK/tree/master/supplements and installation instructions are in the README.md file.
Modified version of EIGENSTRAT ( Patterson et al., 2006 , Price et al., 2006 )
For population structure determination. A modified version can be downloaded from https://github.com/atifrahman/HAWK/tree/master/supplements and installation instructions are available in the README file.
ABySS ( Simpson et al., 2009 )
To assemble k-mers of statistical significance. Download and installation instructions are at https://github.com/bcgsc/abyss.
GNU sort with parallel support. Usually included with Linux distribution
R. Available for download at https://cran.r-project.org/
Procedure
The overview of the procedure of association mapping from sequencing reads with k-mers using HAWK is shown in Figure 1. The steps are described in more details below. More information on the process is available at https://github.com/atifrahman/HAWK/blob/master/README.md.
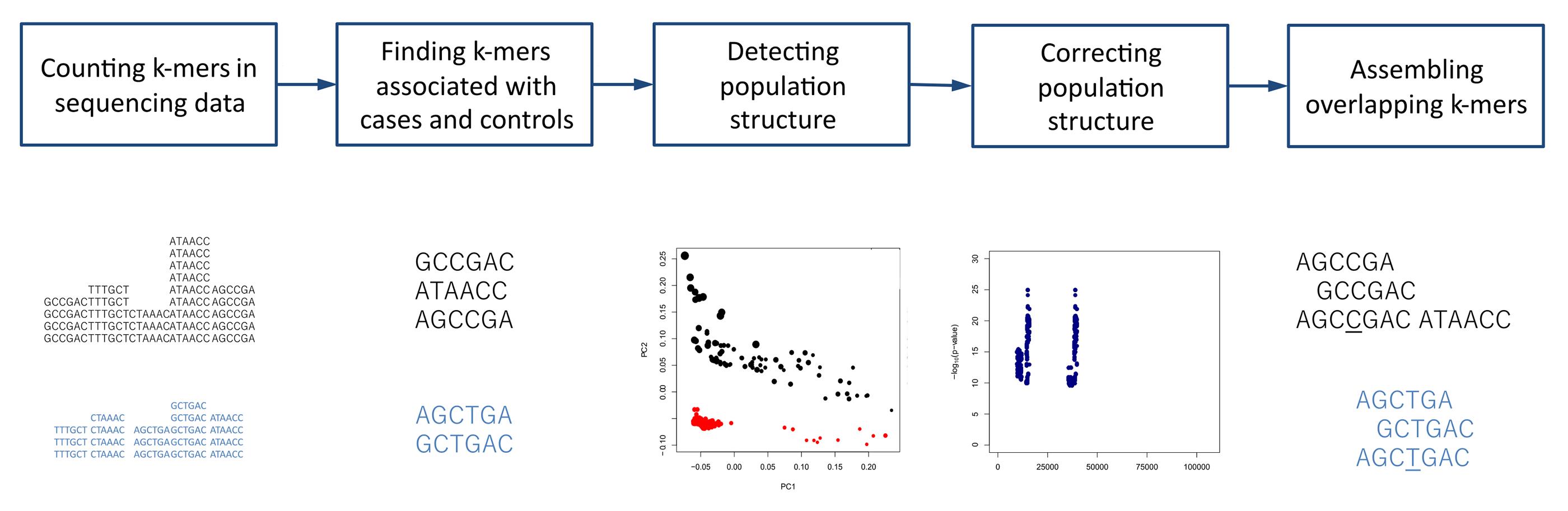
Figure 1. Overview of association mapping from sequencing reads with k-mers using HAWK
Counting and sorting k-mers
Count k-mers in each sample by running Jellyfish or Jellyfish 2. To do this, modify the example scripts available at https://github.com/atifrahman/HAWK/tree/master/supplements.
countKmers_jf2 or countKmers_jf1 can be used to count and sort k-mers using Jellyfish 2 or Jellyfish respectively when the reads from each sample are in a separate folder whereas countKmers_jf2_sra or countKmers_jf1_sra can be used when reads first need to be downloaded from the sequence read archive (SRA).
The scripts will write the names of the sorted k-mer count files in 'sorted_files.txt' and the total k-mer counts in each sample in 'total_kmer_counts.txt'.
Running HAWK
Create a file named 'gwas_info.txt' containing three columns separated by tabs in each row giving a sample ID, male/female/unknown denoted by M/F/U and Case/Control status of the sample for each sample.
Copy the files 'sorted_files.txt' and 'total_kmer_counts.txt' into a folder as well as the file 'gwas_info.txt'
Copy the scripts 'runHawk' and 'runAbyss' into the folder, edit the variables hawkDir, eigenstratDir and isDiploid, and run ./runHawk
This will complete the following steps:
Identifying k-mers associated with cases and controls. This will be done by running ‘hawk’ and will generate the initial lists of k-mers associated with cases and controls in files ‘case_out_w_bonf.kmerDiff’ and ‘control_out_w_bonf.kmerDiff’ respectively as well as the files to be used by Eigenstrat to determine population structure.
Determining population structure. Population structure will be detected by running Eigenstrat. Optionally, the population structure can be investigated by running the R script pca_plot.R. This will read the ‘gwas_eigenstrat.evec’ file generated by Eigenstrat and output the principal component analysis (PCA) plot in ‘pca_plot.eps’. Adjust the variables PC1 and PC2 to select along which principal components the data will be plotted.
Correcting for population structure. The p-values for the k-mers identified in Step B3a will be adjusted for confounding factors such as population structure, total number of k-mers and sex. The k-mers found significantly associated with cases and controls will be in files 'case_kmers.fasta' and 'control_kmers.fasta'. Additional information about the k-mers will be in ‘pvals_case_top_merged.txt’ and ‘pvals_control_top_merged.txt.’
Assembling k-mers
Edit the variable ‘abyssDir’ in the script ‘runAbyss’ and run ./runAbyss
This will assemble the k-mers to generate one sequence for each associated region and the sequences associated with cases and controls will be in ‘case_abyss.25_49.fa’ and ‘control_abyss.25_49.fa’ respectively.
Downstream analysis
Once the k-mers or the assembled sequences are obtained, they can be analyzed in a number of ways.
To obtain summary stats such as average p-values, average counts of constituent k-mers and average number of times they are present in cases and controls edit the HAWK directory, input filename, and whether the sequences are from case or control in the script ‘runKmerSummary’ (at https://github.com/atifrahman/HAWK/tree/master/supplements) and run ./runKmerSummary (see https://github.com/atifrahman/HAWK for details).
If no reference genome is available, BLAST ( Altschul et al., 1990 ) the sequences to check for hits to sequences in related organisms and analyze the matched sequences.
If a reference genome is available, the k-mers can be mapped to the reference using a tool such as Bowtie 2 (Langmead and Salzberg, 2012) and their positions and p-values can be visualized using Manhattan plots. Edit the shell script ‘runBowtie2’ and the R script ‘manhattan_plasmid.R’, available at https://github.com/atifrahman/HAWK/tree/master/ecoli_analysis to align the k-mers to a reference and generate Manhattan plots respectively.
Data analysis
To identify the k-mers present significantly more times in cases or controls compared to the other, HAWK assumes that k-mer counts are Poisson distributed and performs a likelihood ratio test. Population structure is determined by running Eigenstrat to do a principal component analysis on the binary matrix denoting presence or absence of a random set of k-mers. Correction of population structure and other confounders is done by fitting logistic regression models of the phenotype against the confounders as well as a k-mer count vector and the confounders and adjusted p-values for the k-mers identified in the first step are identified. Bonferroni correction is performed to correct for multiple testing. See Rahman et al., 2018 and Mehrab et al., 2020 for a detailed description of the methods and supporting results.
Next we present an example data analysis using the pipeline. We use the E. coli ampicillin resistance dataset from Earle et al., 2016 , which was also analyzed by Rahman et al., 2018 . The dataset contains sequenced reads from 241 strains, of which 189 were ampicillin resistant and the rest were not. First, Jellyfish 2 was used to count k-mers in reads from each sample. Of the 176,284,643 distinct k-mers in total, the first step of HAWK identified 4,752,738 and 4,007,202 k-mers to be associated with cases and controls respectively before correcting for confounders. Next, Eigenstrat was run on 342,988 randomly chosen k-mers to detect population structure. Figure 2A shows the PCA plot of the samples along the first two principal components revealing population stratification.
We then adjust the p-values using the first ten principal components and total number of k-mers in each sample. After correcting for confounders, we get 4,125 k-mers associated with cases and none associated with controls. The k-mers associated with cases were then assembled with ABySS, revealing 11 sequences.
The k-mers found associated with ampicillin resistance were mapped to the E. coli strain DTU-1 genome [GenBank: CP026612.1] and the E. coli strain KBN10P04869 plasmid pKBN10P04869A sequence [GenBank: CP026474.1] using Bowtie 2. Manhattan plots in Figures 2B and 2C show -log10 (p-values) of the k-mers against their locations in E. coli strain DTU-1 genome and plasmid pKBN10P04869A sequence respectively. The vertical lines denote locations of the β-lactamase TEM-1 gene. We observe that no k-mers map to the E. coli strain DTU-1 genome. However, k-mers map to the plasmid pKBN10P04869A sequence near the β-lactamase TEM-1 gene, the presence of which is known to provide ampicillin resistance.
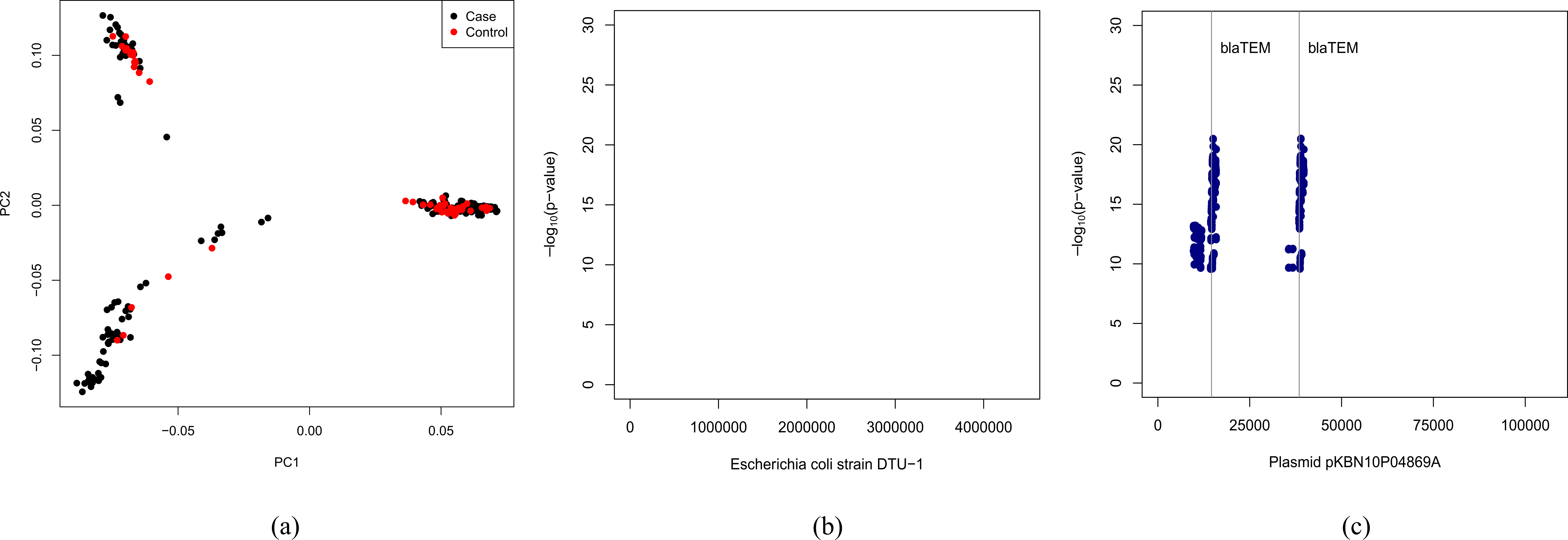
Figure 2. Association mapping of ampicillin resistance in E. coli (A) Plots of the first two principal components of the E. coli strains in the ampicillin resistance dataset. Manhattan plots showing -log10(p-values) of k-mers associated with ampicillin resistance and their locations in (B) Escherichia coli strain DTU-1 genome and (C) plasmid pKBN10P04869A sequence.
Notes
By default, HAWK uses Bonferroni correction to address the issue of multiple testing. If the study is underpowered for Bonferroni correction, the Benjamini-Hochberg procedure can be used instead. For this after executing ‘runHawk’, run ./runBHCorrection. The resulting k-mers will be in ‘case_kmers_bh_correction.fasta’ and ‘control_kmers_bh_correction.fasta’.
HAWK uses first two principal components found using Eigenstrat, sex of samples and sequencing depth in the form of total k-mer counts as confounders, by default. To change the default settings, edit the variables ‘noPC’ and ‘useSexConfounder’ in the script ‘runHawk’. In order to provide additional confounders, create a file with the number of lines equal to the number of samples and in each line specify the covariates, given by numbers and separated by spaces or tabs. Edit the variable ‘covFile’ in ‘runHawk’ with the name of the confounder file.
Recipes
The HAWK pipeline can be used to find sex specific k-mers. In order to do this, in the file 'gwas_info.txt', provide the sample IDs in the first column, write U's in the second column and specify contain Case/Control status depending on whether the sample is Male/Female in the third column. For example if SRR3050845 and SRR3050847 are female and SRR3050846 is male, the file will be:
SRR3050845 U Control
SRR3050846 U Case
SRR3050847 U Control
Acknowledgments
Lior Pachter, and Atif Rahman were funded in part by NIH R21 HG006583. This paper describes protocol of a method originally presented in the paper “Association mapping from sequencing reads using k-mers” by Atif Rahman, Ingileif Hallgrímsdóttir, Michael Eisen and Lior Pachter, and extended in “A faster implementation of association mapping from k-mers” by Zakaria Mehrab, Jaiaid Mobin, Ibrahim Asadullah Tahmid and Atif Rahman.
Competing interests
The authors declare no competing interests.
References
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D. J. (1990). Basic local alignment search tool. J Mol Biol 215(3): 403-410.
- Earle, S. G., Wu, C. H., Charlesworth, J., Stoesser, N., Gordon, N. C., Walker, T. M., Spencer, C. C., Iqbal, Z., Clifton, D. A., Hopkins, K. L. and Woodford, N. (2016). Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nature Microbiol 1(5): 1-8.
- Jaillard, M., Lima, L., Tournoud, M., Mahe, P., van Belkum, A., Lacroix, V. and Jacob, L. (2018). A fast and agnostic method for bacterial genome-wide association studies: Bridging the gap between k-mers and genetic events. PLoS Genet 14(11): e1007758.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie. Nat Methods 9(4): 357-359.
- Lees, J. A., Vehkala, M., Valimaki, N., Harris, S. R., Chewapreecha, C., Croucher, N. J., Marttinen, P., Davies, M. R., Steer, A. C., Tong, S. Y., Honkela, A., Parkhill, J., Bentley, S. D. and Corander, J. (2016). Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat Commun 712797.
- Marçais, G. and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6): 764-770.
- Mehrab, Z., Mobin, J., Tahmid, I. A. and Rahman, A. (2020). A faster implementation of association mapping from k-mers. bioRxiv. .
- Patterson, N., Price, A. L. and Reich, D. (2006). Population structure and eigenanalysis. PLoS Genet 2(12): e190.
- Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A. and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38(8): 904-909.
- Rahman, A., Hallgrimsdottir, I., Eisen, M. and Pachter, L. (2018). Association mapping from sequencing reads using k-mers. Elife 7: e32920.
- Sheppard, S. K., Didelot, X., Meric, G., Torralbo, A., Jolley, K. A., Kelly, D. J., Bentley, S. D., Maiden, M. C., Parkhill, J. and Falush, D. (2013). Genome-wide association study identifies vitamin B5 biosynthesis as a host specificity factor in Campylobacter. Proc Natl Acad Sci U S A 110(29): 11923-11927.
- Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J. and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res 19(6): 1117-1123.
- Voichek, Y. and Weigel, D. (2020). Identifying genetic variants underlying phenotypic variation in plants without complete genomes. Nat Genet 52(5): 534-540.
Article Information
Copyright
![]() Mehrab et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Mehrab et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mehrab, Z., Mobin, J., Tahmid, I. A., Pachter, L. and Rahman, A. (2020). Reference-free Association Mapping from Sequencing Reads Using k-mers. Bio-protocol 10(21): e3815. DOI: 10.21769/BioProtoc.3815.
- Rahman, A., Hallgrimsdottir, I., Eisen, M. and Pachter, L. (2018). Association mapping from sequencing reads using k-mers. Elife 7: e32920.
Category
Systems Biology > Genomics > Sequencing
Molecular Biology > DNA > Genotyping
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.