- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Advances in Proximity Ligation in situ Hybridization (PLISH)
(*contributed equally to this work) Published: Vol 10, Iss 21, Nov 5, 2020 DOI: 10.21769/BioProtoc.3808 Views: 7587
Reviewed by: Hui ZhuMansi BabbarCarlos Ignacio Lescano
Original research article
The authors used this protocol in:

Jan 2018


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Understanding tissues in the context of development, maintenance and disease requires determining the molecular profiles of individual cells within their native in vivo spatial context. We developed a Proximity Ligation in situ Hybridization technology (PLISH) that enables quantitative measurement of single cell gene expression in intact tissues, which we have now updated. By recording spatial information for every profiled cell, PLISH enables retrospective mapping of distinct cell classes and inference of their in vivo interactions. PLISH has high sensitivity, specificity and signal to noise ratio. It is also rapid, scalable, and does not require expertise in molecular biology so it can be easily adopted by basic and clinical researchers.
Keywords: In situ hybridizationBackground
We recently developed a multiplexed in situ hybridization technique called PLISH (Proximity Ligation in situ Hybridization) (Nagendran et al., 2018). PLISH is distinct from other existing spatial transcriptomic technologies because it combines high performance, rapid multiplexing, low cost, and technical simplicity (Wilbrey-Clark et al., 2020). PLISH results can be analyzed by automated calculation of single cell expression profiles in intact cryo- or paraffin-embedded tissue and it is compatible with concurrent immunostaining.
PLISH integrates several molecular biology techniques to provide sensitive and quantitative localization of single RNA molecules in cells and intact tissues. It is broadly accessible to the research community for small- and large-scale applications because it does not employ costly reagents or require up-front investment in specialized equipment. The technology involves rolling circle amplification (RCA) at sites of RNA-catalyzed proximity ligation, which ensures high specificity and signal-to-background with easy discrimination of bright punctal signal from autofluorescence. PLISH has several features that make it adaptable to different experimental goals. First is the ability to ‘tile’ multiple probes along the length of a target RNA. The extent of tiling can be tuned based on the expression level of each transcript, allowing for co-localization of high and low abundance RNAs in a single stain. Second, PLISH's gentle signal 'erasure' step confers a simple multiplexing capability without damaging the tissue sample, allowing for iteration of the fluorescence labeling and imaging steps. Thus, for example, 12 transcripts can be co-localized in a sample with three label-image-erase rounds of four transcripts per cycle following the initial hybridization and RCA. Third, the exceptional signal-to-background of PLISH puncta facilitates automated quantitation of transcripts in each cell using freely available image-processing software packages like CellProfiler. Fourth, the simplicity of PLISH data processing enables researchers without programming expertise to generate and interrogate largescale single cell datasets, and to quantify and spatially resolve known and novel cell classes. Last, PLISH is compatible with fluorescence immunohistochemistry, which enables co-detection of proteins and transcripts in individual cells. This feature broadens the experimental scope of PLISH and on a practical level may be useful for validation of staining patterns.
A PLISH probeset consists of a pair of DNA Hybridization probes (H probes) that target a pre-selected region of an RNA transcript. The right probe (Hybridization Right probe, HR) and left probe (Hybridization Left probe, HL) recognize adjacent sequences in the targeted region (Figure 1). Each H probe comprises a ~20 bp region complementary to the target RNA, and a single stranded overhang at the 3' (HR) or 5' (HL) end. The overhangs are complementary to two oligonucleotides, a Variable Bridge (VB) oligonucleotide and a Common Connector Circle 2.1 (CCC2.1) oligonucleotide. VB and CCC2.1 are designed to hybridize cooperatively with two Hprobe overhangs to form a stable Holliday junction structure, but only at sites where the HR and HL probe have been brought into close proximity by their interaction with the target RNA (Figure 1). A successfully assembled Holliday junction catalyzes ligation of VB and CCC2.1 into a closed circle, which then serves as a substrate for rolling circle amplification (RCA, Figure 1). The RCA amplicons are ~60 kilobase long stretches of single-stranded DNA, consisting of tandem repeats that are complementary to the VB and CCC2.1 sequences. The amplicons are detected with fluorescently tagged VB oligonucleotides (Label probes, LPs). This labeling strategy produces localized fluorescence at the site of the target transcript, which is easily distinguishable from background because of its high intensity and punctal shape (Figure 1). The detection sensitivity can be increased as desired by ‘tiling’ one or more additional H probesets along the length of a given target RNA.
For multiplexed PLISH, a unique VB barcode sequence is inserted into the probesets targeting each different transcript. We have designed and tested 24 distinguishable VB barcodes, supporting up to 24-plex PLISH experiments. Typically, three to five target RNAs are imaged per round, depending on the number of fluorophores the investigator's microscope can resolve. Selective detection of a subset of RNAs is accomplished by 'lighting up' only their VB barcodes with the corresponding label probes. Importantly, the label probes contain deoxyuracil bases, enabling gentle 'erasure' of their fluorescence by enzymatic degradation with uracil DNA glycosylase. New label probes are then applied to detect a second subset of the VB barcodes. The process of label probe hybridization, photo-documentation, and enzymatic erasure is repeated until all the transcripts have been localized, after which expression profiles for every imaged cell can be compiled.
Advantages and Adaptation
PLISH is a high-performance, rapid, technically simple and low-cost technology for transcriptional profiling of single cells in intact tissue that exploits RNA-templated proximity ligation at Holliday junctions. Our adaptation significantly improves the specificity, sensitivity, and ease of multiplexing with PLISH. Since the initial publication of PLISH (Nagendran et al., 2018) we have made three important modifications to the protocol (see “Modifications from original PLISH protocol”). In its current iteration, PLISH remains technically simple and adoptable without having to invest in specialized equipment or purchase expensive reagents. It should be straightforward for any researcher to identify optimal conditions for the tissue(s) of interest, for which we include guidelines below, after which they may embark on data collection.
- Applications
Molecular profiling of single cells in intact tissues by PLISH has applications that span from co-staining several transcripts and proteins in a single tissue section to high-level multiplexing in a massive number of cells. The former approach facilitates high-resolution mapping of cell-cell interactions, for instance between stem cells and the niche cells that support them, using a small set of curated transcripts (Nabhan et al., 2018). The latter approach enables unbiased identification and in vivo mapping of molecularly distinct cell populations, which can reveal new and unexpected biology (Nagendran et al., 2018). PLISH also allows for the incorporation of cytologic features of the profiled cells along with the molecular and spatial characteristics, which may be important in disease states like cancer.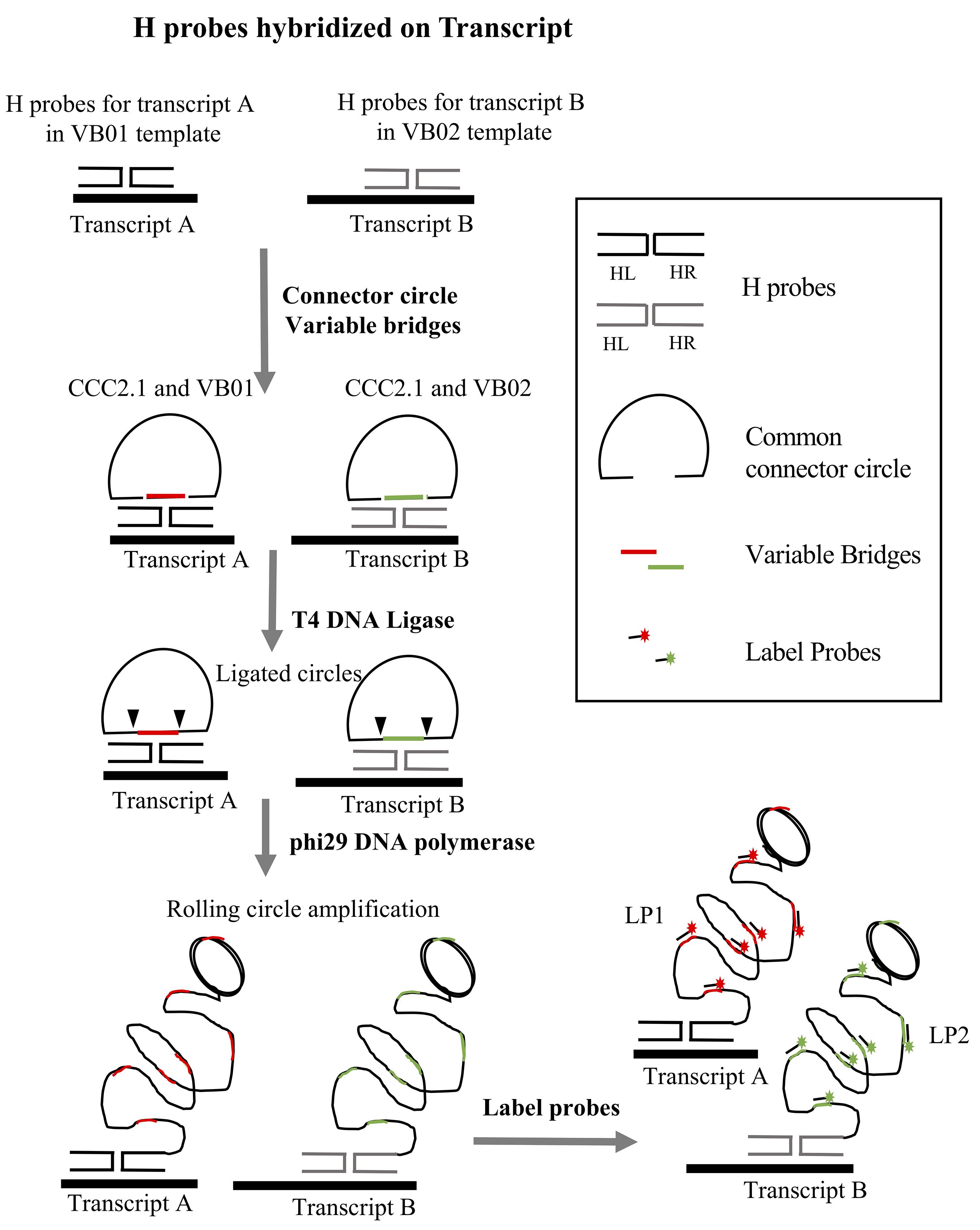
Figure 1. Overview of the PLISH protocol for two targets (Transcript A and Transcript B). Each pair of H probes in a probeset hybridize at adjacent sites on the target transcript. H probes for Transcript A are designed to work with variable bridge VB01, and probes for Transcript B with variable bridge VB02. CCC2.1 is the common connector circle used for both transcripts. CCC2.1 and the variable bridge hybridize to the single-stranded overhang regions of the H probes. T4 DNA ligase seals the nicks between the variable bridge and CCC2.1 to form a closed circle, and phi29 DNA polymerase copies the circle by rolling circle amplification. Label probes conjugated to fluorophores (LP1 for Transcript A and LP2 for Transcript B) are hybridized to complementary barcodes (colored segments) in the single-stranded RCA amplicons.
Materials and Reagents
Consumables
Sterile microcentrifuge tubes, 1.5 ml (Fisher Scientific, catalog number: 05-408-130 )
0.2 µm syringe filter (Thermo Scientific, Nalgene, catalog number: 194-2520 )
Microscope slides (Thermo Fisher Superfrost Plus, catalog number: 4951PLUS4 )
Kimwipes (Fisher Scientific, catalog number: 06-666 )
Rectangular coverslips (Thermo Fisher Scientific, catalog number: 22X60-1 )
Petri dish (Fisher Scientific, catalog number: 08-757-099 )
Reagents
Diethyl Pyrocarbonate (DEPC) (Sigma-Aldrich, catalog number: D5758 ); DEPC is a suspected carcinogen. Use a fume hood and wear gloves while working with DEPC
Milli Q water
Nuclease-free water (Thermo Fisher Scientific, catalog number: AM9920 ); can be used instead of preparing DEPC-treated water in the lab (Recipe below)
Paraformaldehyde (Sigma-Aldrich, catalog number: P6148 )
Caution: Paraformaldehyde fumes are toxic. Wear gloves and use a fume hood while working with paraformaldehyde.
10% Neutral Buffered Formalin (Thermo Fisher Scientific, catalog number: 5705 )
Caution: Wear gloves and use a fume hood while working with formalin.
Sodium Chloride (NaCl) (Fisher Scientific, catalog number: BP3581 )
Sodium Phosphate Dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S7907 )
Sodium Phosphate Monobasic (NaH2PO4) (Sigma-Aldrich, catalog number: S8282 )
Dimethyl Sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418 )
Sodium Trichloroacetic acid (Fisher Scientific, catalog number: AC184155000 )
Ethanol (Sigma-Aldrich, catalog number: E7023 )
Tween 20 (Sigma-Aldrich, catalog number: P1379 )
Formamide (Sigma-Aldrich, catalog number: F9037 )
Caution: Formamide is a skin, eye, nose and throat irritant and possible teratogen. Wear gloves and use a fume hood while working with formamide.
Sodium Citrate Dehydrate (Fisher Scientific, catalog number: S279 )
Heparin Sodium salt (Sigma-Aldrich, catalog number: H3393 )
Tris Base (Fisher Scientific, catalog number: BP152-1 )
Glycerol (Sigma-Aldrich, catalog number: 5516 )
DAPI (Sigma-Aldrich, catalog number: D9542 )
Caution: DAPI is a potential carcinogen. Wear gloves while working with DAPI.
Triton X-100 (Fisher Scientific, Acros organics, catalog number: 215680010 )
Xylene (Fisher Scientific, catalog number: XS-1 )
Caution: Xylenes affect the central nervous system. Vapors of xylenes are flammable. Use a fume hood and wear gloves while working with this reagent.
Ethylenediaminetetraacetic Acid Disodium Salt Dehydrate (EDTA) (Fisher Scientific, catalog number: S312-500 )
Sodium Hydroxide (NaOH) (Fisher Scientific, catalog number: S318-500 )
Hydrochloric Acid (Fisher Scientific, catalog number: A144S-500 )
Caution: Work in a fume hood, wear gloves, and exercise extreme caution when handling concentrated hydrochloric acid.
OCT or Optimal Cutting Temperature (Fisher Scientific, catalog number: 23-730-571 )
dNTP mix, 10 mM each (Thermo Fisher Scientific, catalog number: R0192 )
ELIMINase (Deacon labs, catalog number: 1101 )
Poly-L-Lysine solution (Sigma Aldrich, catalog number: P4707 )
Sterile filtered water for cell culture (Sigma Aldrich, catalog number: W3500 )
Sucrose (Fisher Scientific, catalog number: ICN821713 )
Enzymes
T4 Poly Nucleotide Kinase (T4 PNK) (New England Biolabs catalog number: M0201L ), store at -20 °C
Pepsin (Sigma-Aldrich-Roche, catalog number: 10108057001 ), store at 4 °C
T4 DNA Ligase (New England Biolabs, catalog number: M0202T ), store at -20 °C
RNaseIN, Recombinant (Promega, catalog number: N2511 ), store at -20 °C
phi29 DNA polymerase, Nxgen (Lucigen, catalog number: 30221-2 ), store at -20 °C
USER enzyme (New England Biolabs, catalog number: M5508 ), store at -20 °C
Uracil Glycosylase Inhibitor (UGI) (New England Biolabs, catalog number: M0281 ), store at -20 °C
Reagent set up–see corresponding recipe for each reagent in the Recipes section below
- Stock Solution Recipes
Nuclease-free water
3 M Sodium Chloride (NaCl)
1 M Disodium Hydrogen Phosphate (Na2HPO4)
1 M Sodium Dihydrogen Phosphate (NaH2PO4)
10x Phosphate Buffered Saline (PBS)
1x Phosphate Buffered Saline (PBS) pH 7.4
30% Sucrose solution
5% Tween-20
0.5% Tween-20
4% Paraformaldehyde
1x Phosphate Buffered Saline with 0.05% Tween-20 (PBST)
0.01 M Citric Acid
0.01 M Sodium Citrate
10% Lithium Dodecyl Sulfate (LDS)
0.1 M Hydrocholoric acid (HCl)
Pepsin stock solution from powdered enzyme
2 M Sodium Trichloroacetate
0.5 M Ethylenediaminetetraacetic Acid Disodium Salt Dehydrate (EDTA)
1 M Tris pH 7.4
1 M Tris pH 8.0
20 mg/ml Heparin
50% and 80% Glycerol
20 mg/ml Bovine Serum Albumin (BSA)
20x Saline Sodium Citrate (SSC) pH 7.0
DAPI
Blocking serum
- Buffer Recipes
10 mM Citrate buffer with 0.05% LDS
TE buffer pH 8.0 contains 100 mM Tris pH 8.0 and 10 mM EDTA pH 8.0
Hybridization buffer
High Salt Buffer
Circle hybridization buffer
1x Ligase Buffer
1x phi29 DNA polymerase buffer
Label probe buffer
Equipment
Heat block set at 37 °C
Water bath set at 100 °C
Seal chambers (Grace Biolabs, Sigma, catalog number: GBL621505 )
Laser confocal microscope (we acquire the images using a Leica Sp8 confocal microscope)
P-2, P-10, P-20, P-200, and P-1000 pipettors
Rotating platform (rocker)
4 °C refrigerator
-20 °C and -80 °C freezer
Water bath set at 100 °C
Hybridization oven or humidified chamber set at 37 °C
Software
Fiji (https://fiji.sc/)
Cell Profiler (https://cellprofiler.org/)
Procedure
Probe design, sequences and templates
Label probes (oligonucleotides conjugated to fluorophores) (Table 1)
Label probes (LPs) bind to the VB barcodes within the single-stranded RCA amplicon. Each LP consists of 15-20 bases that are identical to the barcode region of the corresponding VB (and are therefore complementary to the RCA amplicon).
Table 1. Label probe sequences for a 4 channel PLISH

Order LPs from a vendor (e.g., IDT) on the 100 nanomole scale, with 5’ conjugation of the fluorophore and HPLC purification. Resuspend the LPs in TE buffer to a stock concentration of 100 µM and store at -20 °C. Prepare 10 µM working solutions in nuclease-free water and store at 4 °C.
Plan your experiment so that you will use Cy5 for the most important target RNA. The simultaneous use of Cy3 and Texas red readouts may require linear unmixing to maximize the accurate discrimination of these two fluorophores, given their overlapping emission spectra. Consult your microscope software manual for instructions on linear unmixing, if available.
If you plan to perform PLISH for more than 4 transcripts in a specimen, order LPs with the ‘T’ nucleotides substituted with deoxyuracil (Table 2). This enables the enzymatic erasure of LPs by USER (Uracil specific excision reagent) after photo-documentation (Refer to detailed protocol in Step C5b, in situ hybridization).
- Some LPs have additional nucleotides on the 5′ or 3′ ends to facilitate fluorophore conjugation and/or compatibility with the USER digestion protocol.
Table 2. Label probe sequences for iterative PLISH

- Circle components
CCC2.1 is the Common Connector Circle
CCC2.15′ ATTCCTGACCTAACAAACATGCGTCTATAGTGGAGCCACATAATTAAACCTGGCTAT 3'
(CCC2.1 is compatible with all Variable Bridges).VB barcodes that are identical to the corresponding label probe are in bold (Table 3).
Table 3. Variable bridge sequences for PLISH

Order CCC2.1 and VB on a 100 nanomole scale as 5′ phosphorylated oligonucleotides with HPLC purification. Prepare 10 µM stock aliquots in nuclease-free water and store at -20 °C.
Hybridization probe design overview
Stringent probe design is critical for optimizing transcript detection efficiency and minimizing the risk of false positive signal. A single PLISH probeset consists of one right probe (HR) and one left probe (HL). Each H probe is ~46 bases long, with an ~20 base region that is the reverse complement of the target RNA. The other ~26 base pairs comprise an “overhang” sequence that is the reverse complement to the variable bridge (VB) and common connector circle (CCC, see Figure 1) sequences. The design process starts by choosing an RNA transcript of interest from the National Center for Biotechnology Information (NCBI) Nucleotide database. We generally name the probeset using the HUGO (Human Genome Organization) Gene Nomenclature Committee (HGNC) approved gene symbol found at https://www.genenames.org.
Dinucleotides in the target transcript that can serve as the center of a Holliday junction are first identified, and the surrounding ~40 base RNA sequence is nominated as a candidate probe site. The candidate probe site sequence is then fed to the NCBI Basic Local Alignment Search Tool (BLAST) both to quantify homology to the target RNA and its transcript variants, and to determine if the degree of homology to off-target RNAs is below a minimum threshold (see “Criteria for Off-Target Homology” below). When the specificity criteria are met, the reverse complement of the RNA sequence on the 5' side of the junction center (the HL homology region) and the reverse complement of the RNA sequence on the 3' side of the junction center (the HR homology region) are catenated to an appropriate overhang sequence to produce complete H probes. The H probeset is analyzed using an online oligo calculator (refer to Step A3b.xvi) to determine whether the melting temperature of each arm of the prospective junction is acceptable, and to rule out the possibility of hairpin or dimer formation. Between five and ten H probeset pairs are designed per transcript. Cognate VB, CCC2.1 and LP oligonucleotides are ordered along with the H probes.
The RNA dinucleotide sequence at the junction center is intended to force the Holliday junction into a conformational isomer that is favorable for PLISH (Miick et al., 1997; Eichman et al., 2000; Liu et al., 2005; Hays et. al., 2006; Khuu et al., 2006). In general, we recommend using RNA dinucleotides 5′-AG-3′ or 5′-UA-3′ at base pairs 20 and 21 of candidate probe sites. However, requiring the optimal central dinucleotide sequence can overly restrict the number of candidate probe sites for short transcripts, for which the criterion may be relaxed.
In our experience, the major challenges in PLISH probe design relate to off-target homology and RNA access, since the tertiary and quaternary structure of endogenous target RNAs likely limits the accessibly of some candidate probe sites more than others. Probe dimerization and hairpin formation are less significant concerns, possibly due to the use of strong denaturants in the hybridization buffer.
We include a MATLAB script that was written to automate the above steps (see Appendix for the MATLAB file–PLISH_Probe_Design_BLAST210.m and accompanying instructions “How_to_use_the_MATLAB_Probe_Design_Script.pdf”), allowing the user to quickly identify candidate H-probe binding regions that do not have a high level of off-target homology. It also generates lists of H probes with overhang sequences corresponding to a specified VB barcode.
Please note that the MATLAB script is not required to perform PLISH. It is included as a potential convenience for some users. We recommend first designing probes by hand and optimizing the entire PLISH protocol prior to moving on to an automated design script.
Additional considerations are required prior to using the MATLAB script. First, if PLISH will be used for a low number of genes (e.g., less than 10) then it is likely more efficient to design the probes by hand rather than investing in the required ancillary software needed for the MATLAB script. Second, the MATLAB script requires that the user have a stand-alone, or “local” installation of the NCBI BLAST search tool and Refseq database, which may necessitate additional computing resources. It also requires the user to set up the local BLAST search tool and related Refseq databases such that they can be “called” by MATLAB (and other programs) in the user’s operating system (usually Windows). Although we provide basic guidelines on how to install these NCBI tools, it is beyond the scope of this protocol to guide the user through this process, especially as updates and newer versions of BLAST are regularly released by the NCBI. Instead, the user is directed to the NCBI’s current online instructions and help resources for installation of both the search tool and the databases. Finally, updates in MATLAB or the NCBI tools could render the current script obsolete over time.
Our MATLAB script determines the specificity of a candidate probe to its probesite based on the number of significant BLAST alignments to genes other than the one from which the probesite is derived. The design of oligonucleotide probes specific for a transcript of interest at a genome-scale is an active area of research, with new tools emerging. One solution, OligoMiner (Beliveau et al., 2018), provides a python-based pipeline which leverages both next-generation sequencing (NGS) alignment tools (such as Bowtie2) and linear discriminate analysis machine learning to determine target specificity.
If electing to use the MATLAB script, it is still recommended that the user manually verify (by entering the region of the RNA sequence targeted by the H-probe into NCBI BLAST) that off-target homology is acceptably low. The script will provide a FASTA file of these sequences which can be entered directly into the online NCBI BLAST search query for this purpose.
Criteria for off-target homology
When performing a BLAST query, the settings outlined in Table 4 should be applied. Based on our experience with the hybridization conditions detailed in the new protocol, an alignment with a BlastN bit score of greater than 30 should be considered to have significant homology, irrespective of the number of base pair matches or gaps in the alignment. Note that when parsing the search results, only alignments to RNA (and not genomic DNA) are considered. Also, because the target RNA transcript sequence is used as input for the BLAST query, only "Plus/Plus” (and not “Plus/Minus”) strand alignments are relevant. For example, a candidate sequence that has an alignment to an off-target transcript with a "Plus/Plus” bit score of 35 would be deemed to have unacceptable off-target homology.
Note: Plus/Plus vs. Plus/Minus Convention:
For every Blast search, the result report will include 2 strands: The query (~40 bp candidate RNA sequence) and the subject (the RNA transcript pool database). Metrics describe how good the alignment is between the two strands.
The Query is always presented as a “Plus” strand with a 5’-3’ orientation.
Although the subject is also written in 5′-3′ orientation, BLAST will consider alignments to both the sense mRNA strand (“Plus”) and the anti-sense mRNA strand (“Minus”). The BLAST algorithm compares strands for similarity in a base-for-base fashion.
After performing a BLAST query for a 40 bp query sequence, clicking on the ‘Alignment’ tab in the results page will show the pairwise alignment of sequences. The first few BLAST hits generally align to the sequence of the RNA of interest and its isoforms, with identities–40/40 (100%) and Strand–Plus/Plus indicating a base-for-base, query-to-subject alignment to the sense strand. In some of the subsequent hits (possible off-target RNA sequences) the strand alignments may be Plus/Plus (i.e., the query sequence aligns entirely or partly to the sense mRNA subject strand) or Plus/Minus (i.e., the query sequence aligns to the reverse complement of the sense mRNA subject strand). You may ignore hits that have Plus/Minus, because they are reverse complementary to your candidate RNA sequence (i.e., the aligned subject sequence is the same as the H probe sequence so there will be no off-target binding).
Table 4. Settings for performing NCBI Blast search
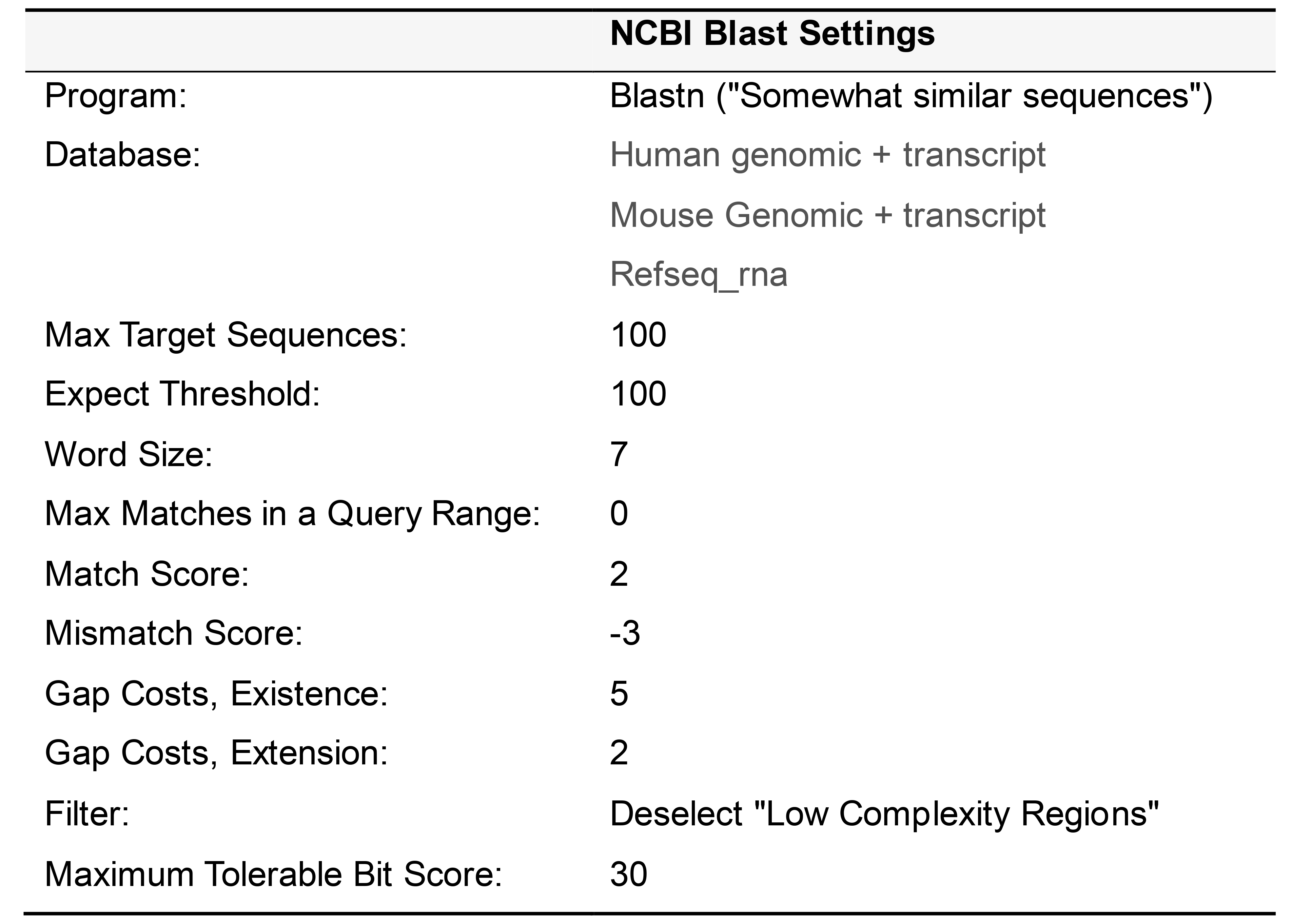
Detailed H Probe Design Protocol for mRNA
Navigate to the NCBI Nucleotide Database (https://www.ncbi.nlm.nih.gov/nucleotide?cmd=search).
Search for the transcript of interest. On the results page, first filter the results by the taxon of interest using the menu in the top right corner (e.g., Homo sapiens or Mus musculus) and then by the molecule type using the menu in the left sidebar (RNA).
Record the number of transcript variants, predicted or otherwise. Choose the search result for the primary transcript entry or for the lowest numbered transcript variant.
Navigate to the bottom of the web page and examine the sequence (denoted by ORIGIN). Paste the full sequence into a word processor document such as Microsoft Word.
Use the Control-F ‘find’ function to identify suitable central dinucleotides for a prospective Holliday junction. As noted above, these sites are 5′-AG-3′ or 5′-UA-3′.
Once a Holliday junction site is found, use the Control-C function to ‘copy’ the 40 base pair sequence.
Open the NCBI Basic Local Alignment Search Tool website (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Click on the “Nucleotide Blast” button.
Paste the candidate sequence in the “Enter Query Sequence” Box at the top of the BlastN suite webpage.
Next, choose the appropriate database. For Homo sapiens or Mus musculus the curated species-specific genome + transcript databases can be selected from the appropriate dropdown menu located to the right of “Database”. For other species, or for performing local blast queries, we recommend using the Referenced RNA sequences (Refseq_rna) database. This option will require the user to enter the organism name or Taxid (https://www.ncbi.nlm.nih.gov/taxonomy) in the box next to Organism (e.g., “Zebrafish” Taxid “7955”).
Under “Program Selection” to the left of “Optimize for”, choose the “Somewhat similar sequences” or “blastn” option.
Click on the “+” next to Algorithm Parameters.
Change the Expect Threshold to 100 and the Word Size to 7. Match and Mismatch scores should be set to 2 and -3, and gap costs should be +5 and +2 for existence and extension, respectively. De-select the check box for Low complexity regions. Finally, click on the bottom BLAST button to run the search.
Parse the Blast search results page. First ensure that the transcript of interest and all of its variants appear in the search results. Next, examine the off-target alignments. If the bit score of the alignment is over 30 and the strand result is “Plus/Plus”, then the candidate sequence has unacceptable off-target homology.
1)If unacceptable off-target homology exists, the candidate sequence should be abandoned in favor of an alternative sequence. Return to Step A3bv. and try a new candidate sequence.
2)If it is known with high confidence that an off-target transcript is not expressed in the tissue or cells that you plan to stain, the probe can still be considered for use in this context.Continue with Steps A3bv. through A3Bxiii. until a pool of 5-10 candidate RNA sequences have been identified.
Divide each candidate RNA sequence in half. Base pairs 1-19 form the target site of HL and base pairs 20-40 form the target site of HR. Determine the reverse complements of the two 20 base target site sequences, which will constitute the target homology regions of HL and HR, respectively. Online tools such as “Sequence Massager” can be used.
Analyze the HL and HR homology region sequences for Tm, hairpin formation, self-annealing, and 3′ complementarity. A Tm range of 45-65 °C is recommended. Acceptable deltaG values for hairpin formation should be greater than -3 kcal/mole, and for self-dimerization greater than -10 kcal/mole. The calculations can be performed with the online “Oligo Calculator”, with proprietary packages such as “Oligocalc” (MATLAB) or with the online IDT “OligoAnalyzer” tool. Since H probe hybridization is conducted in a denaturing high salt buffer, weak self-dimerization or hairpin formation should not necessarily rule out a candidate H probe.
An appropriate left overhang sequence is catenated to the 3′ end of the HL homology region sequence, and a corresponding right overhang sequence is catenated to the 5′ end of the HR homology region sequence.
Notes:
1)When designing probes for transcripts that will be imaged together, it is essential to choose VR barcodes that are read out in distinct color channels.
2)Pick at least 5 pairs of probes for abundantly expressed transcripts and 10 pairs for low or moderately expressed transcripts.
3)In some cases, we have observed specificity issues when using probes with partial homology to an off-target RNA. The off-target signal is usually weaker than the on-target signal. Some probes with partial off-target homology may not be problematic, for instance when the off-target RNA is very lowly expressed or absent in the stained tissue.
4)It is not essential that the H probes have exactly 20 bases of homology to the target RNA. Depending on the sequence, 19-25 bases of homology can provide an acceptable melting temperature. Melting temperatures should be at least 9 °C above the temperature at which the probes are incubated with the tissue sample, which is 37 °C in this protocol.
5)For an initial test experiment, select one abundant transcript (5 pairs of H probes) and one or two RNAs that are expressed at a lower level (10 pairs of H probes) in the tissue you will be staining. It is most useful to select RNAs for which you already know the proper expression pattern, for instance cell type markers, to help gauge which condition provides the optimal sensitivity and specificity in the target tissue.
Preparing individual and pooled stocks of H probes
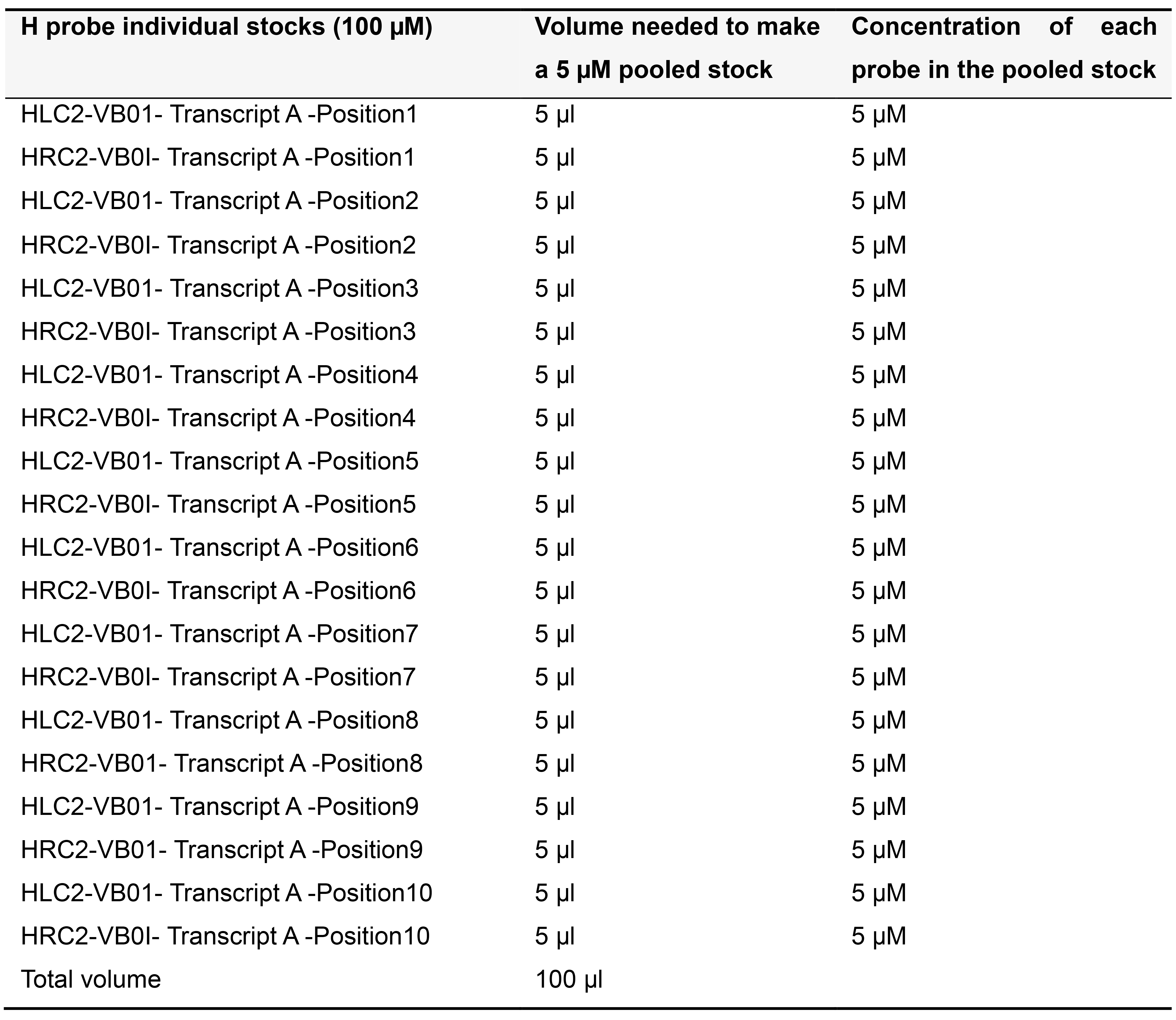
Order H probes on the 25 nanomole scale as standard desalted oligonucleotides. Dissolve each H probe in TE buffer (pH 8.0) to make a 100 µM stock solution and store at -20 °C. Depending on the number of probe pairs designed for your transcript of interest, prepare pooled stocks as described below (Tables 5-7) and store at -20 °C.
Table 5. Preparing 10 µM pooled stock for 5 pairs of H probes targeting Transcript A in a template for variable bridge VB01
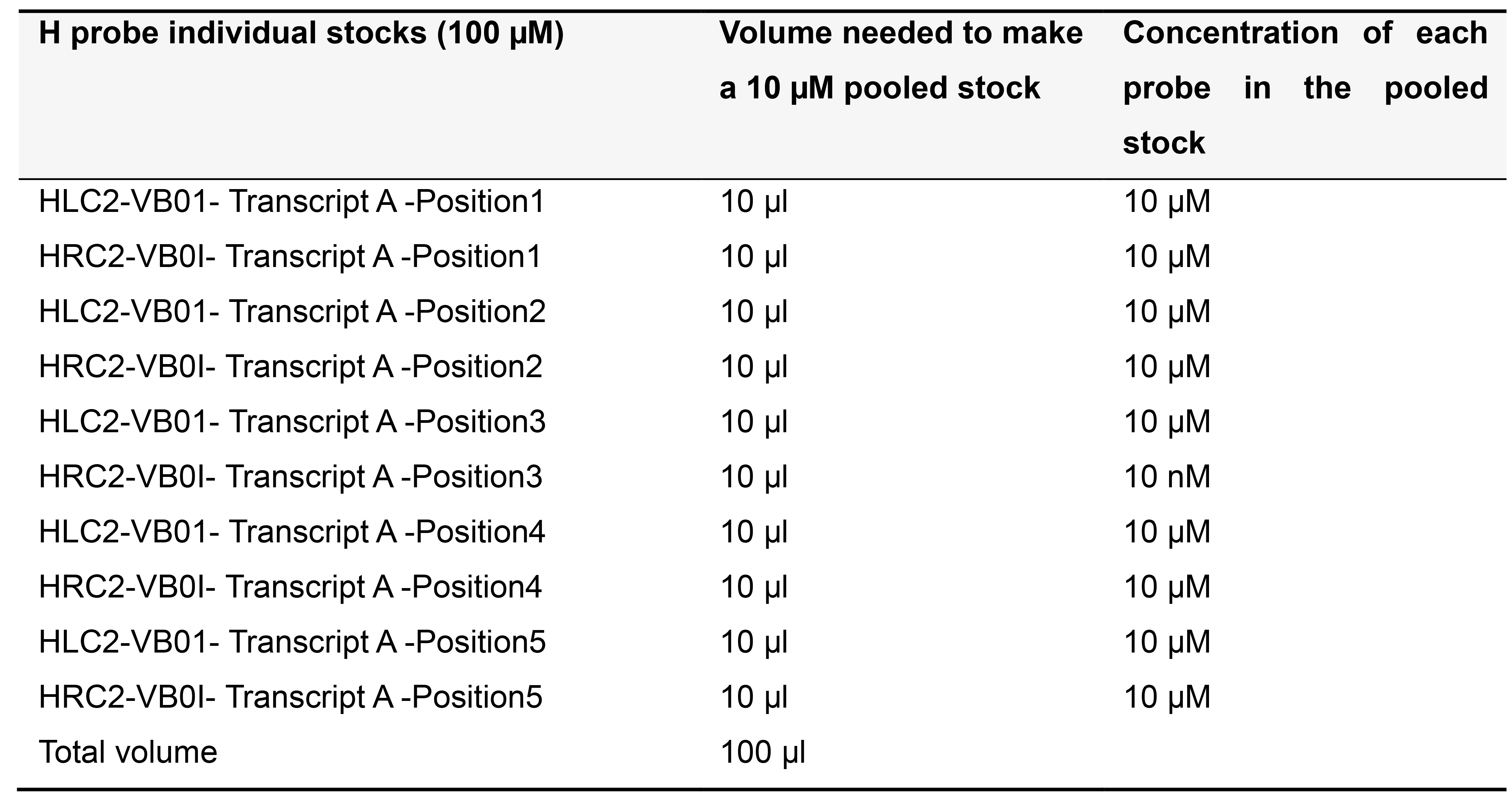
Table 6. Preparing 5 µM pooled stock for more than 5 pairs of H probes targeting Transcript A in variable bridge VB01
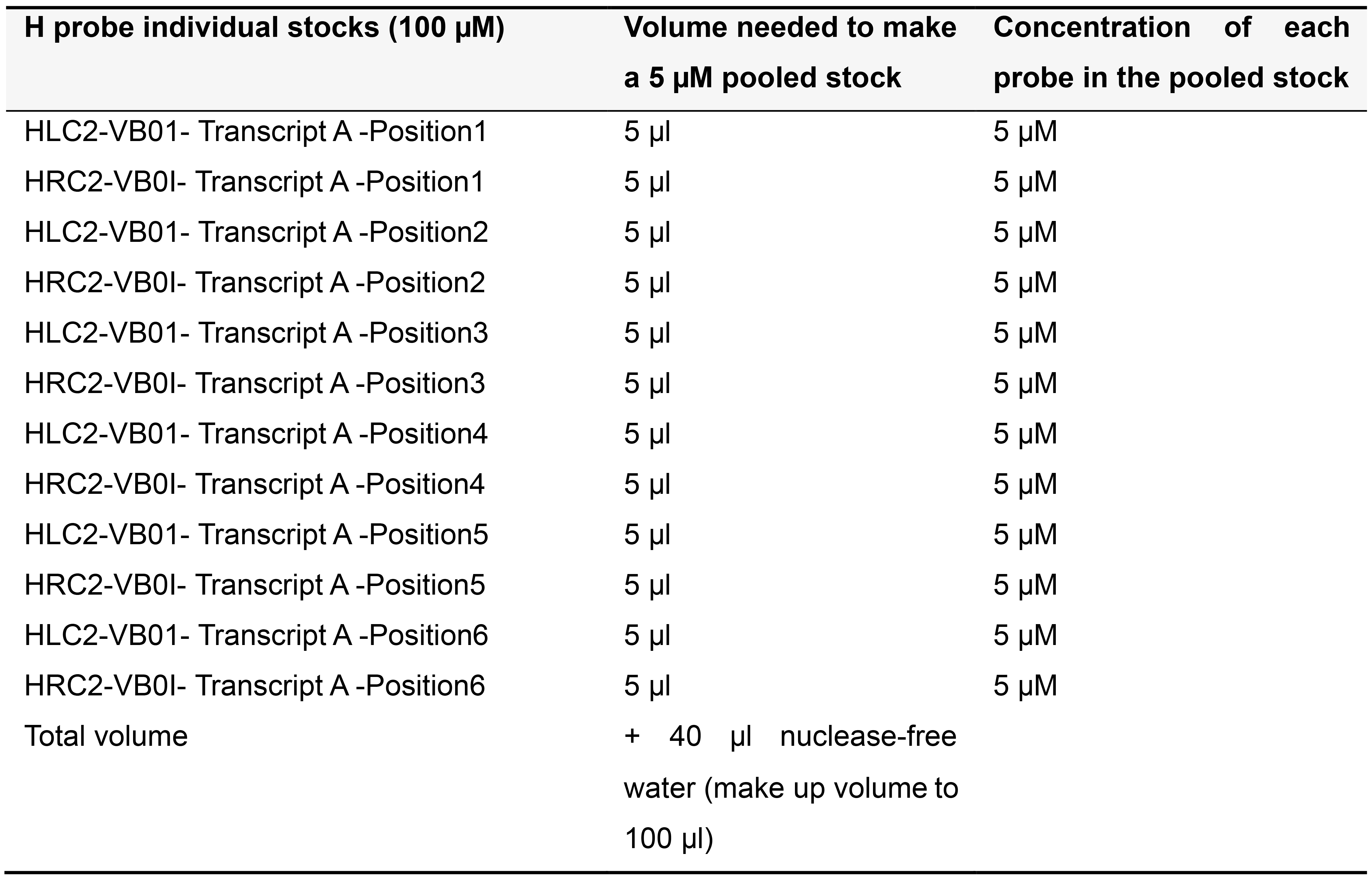
Table 7. Preparing 5 µM pooled stock for 10 pairs of H probes targeting Transcript A in variable bridge VB01
Template for designing H probes
Every H probe contains an ~20 base homology region that is complementary to the target RNA. The overhang on H probes consists of a 16-base region (highlighted in red) that binds to the VB oligonucleotide, and an 11-base region (highlighted in blue) that binds to the common connector circle oligonucleotide (Tables 8-10). Table 9 shows a 40 nucleotide region on the mouse Nkx 2.1 transcript that was used to design a pair H probes (Table 10).When designing H probes for one round of PLISH (1-4 genes), refer to Table 8 for templates. For iterative PLISH (> 4 genes), refer to Table 11.
Table 8. Template for designing H probes for simultaneous four-plex PLISH
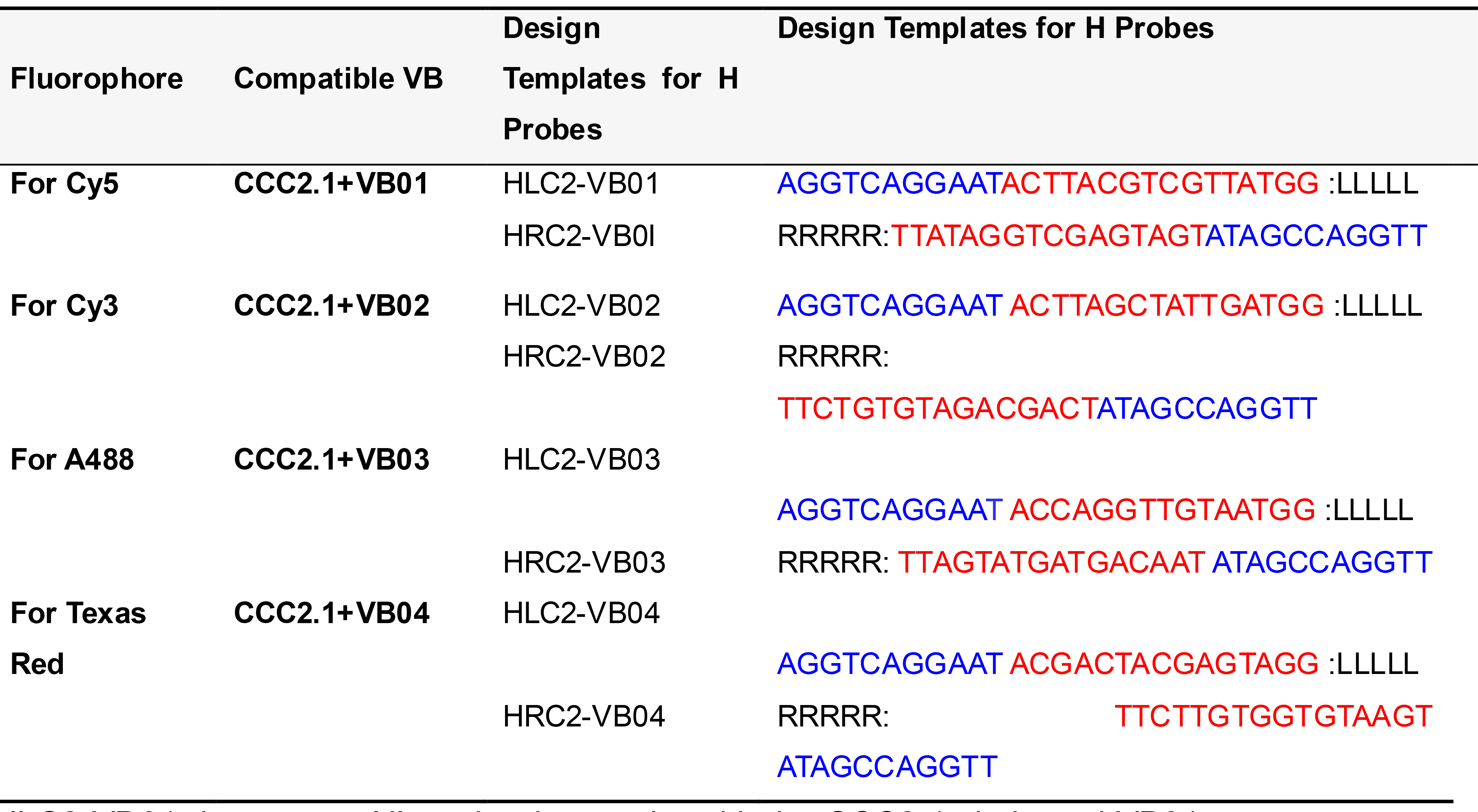
HLC2-VB01 denotes an HL probe that works with the CCC2.1 circle and VB01.
LLLLL and RRRRR designate the regions where the ~20 base target homology sequences are placed.
Table 9. Identifying a candidate probe site and prospective Holliday junction for the mouse Nkx2.1 transcript (NCBI reference number NM- 009385.3)

The dinucleotide at the junction center (AG or UA in the RNA sequence) is highlighted in green.
Table 10. Designing a pair of H probes targeting the mouse Nkx2.1 transcript (NCBI reference number NM- 009385.3)

The two nucleotides complementary to the junction center (AG or UA in the RNA sequence) are highlighted in green. mm, Mus Musculus.
Table 11. H probe template sequences for iterative PLISH
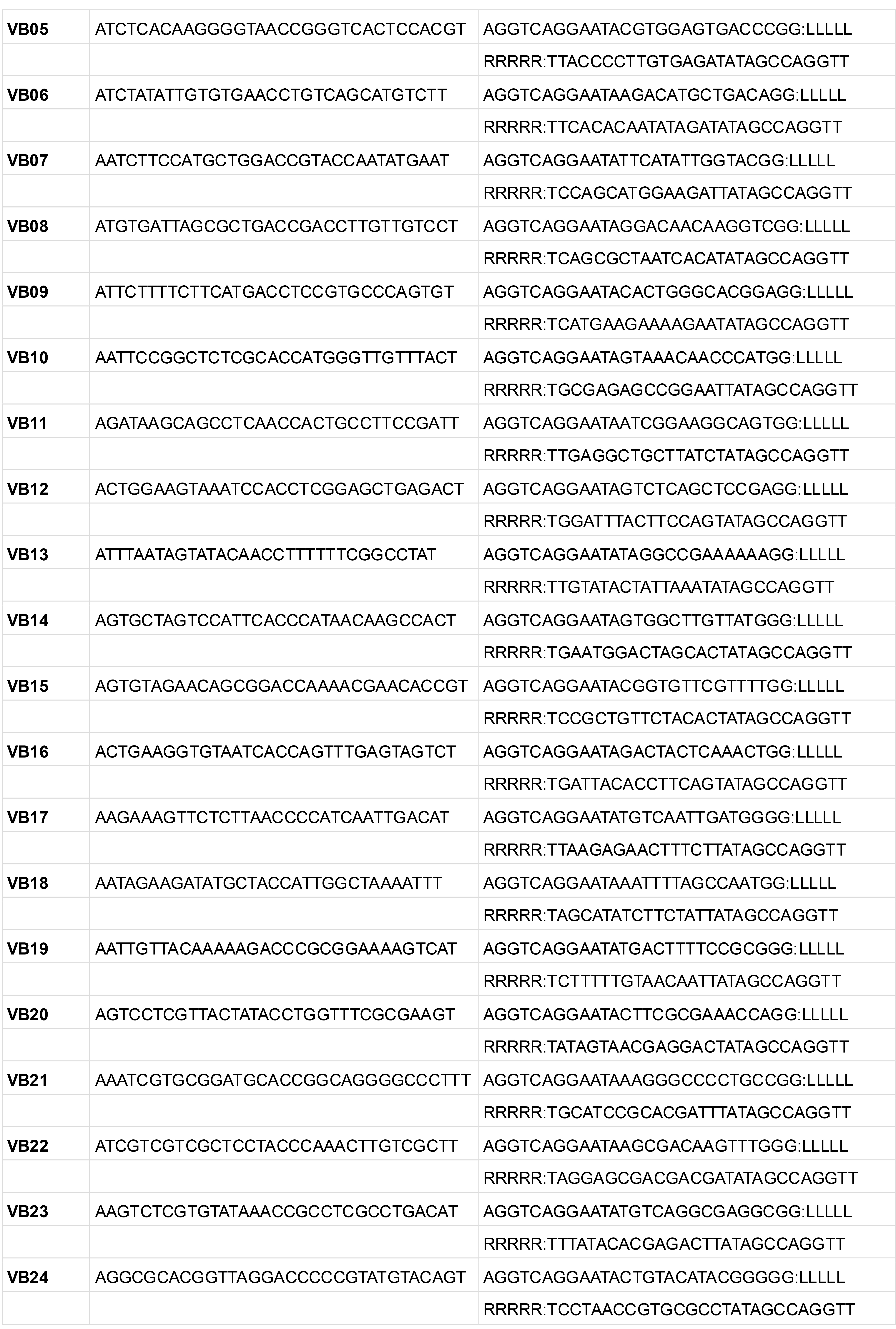
LLLLL and RRRRR indicate the regions where the ~20 bp probe sequences are inserted.
Sample preparation
Cells grown on coverslips
Preparing poly-L-Lysine coated coverslips in a tissue culture hood:
Wash coverslips in 70% ethanol briefly then transfer them into sterile plates (that have also been pre-washed with 70% ethanol).
Rinse once with tissue culture grade sterile water (Pyrogen-free).
Incubate the coverslips in the poly-L-Lysine solution for 20 min.
Rinse twice with tissue culture grade sterile water.
Aspirate off all remaining water and allow the coverslips to air dry for 10 min in the tissue culture hood.
Splitting cells growing in a flask (perform steps in a tissue culture hood):
Aspirate off the media and discard it. For a medium-sized flask, wash once in 5 ml of a solution containing 1x PBS and 1 mM EDTA. Alternatively, you may use Trypsin.
Cover the cells in 1 ml of a solution containing 1x PBS and 1 mM EDTA, then incubate the cells at 37 °C for 2-3 min (no more than 5 min). Gently tap the flask to ensure that the cells have detached into solution (cells should be visibly floating).
Pre-dilute the cells at least ten-fold into fresh, pre-warmed media. Perform a final dilution (e.g., 1:20) with fresh media then gently pipet the cells onto Petri plates containing coverslips until they are submerged. Move Petri plates into 37 °C incubator where cells will settle onto coverslips and adhere. Grow the cells until they reach the desired level of confluency.
When the cells are ready for analysis, rinse the coverslips in 1x PBS. Fix cells in 3.7% Formaldehyde (prepared in 1x PBS using 37% molecular biology grade Formaldehyde) at room temperature for 20 min.
Pre-treat the coverslips with 10 mM Citrate pH 6.0 + 0.05% LDS at 70 °C for 30 min, followed by air drying (~5 min).
Apply seal chambers. Proceed to the H probe hybridization step.
Tissue sections collected on Superfrost glass slides
Prepare sections from OCT embedded cryoblocks or from paraffin blocks of Paraformaldehyde (PFA) fixed tissue. To cryoembed adult mouse lungs, immersion fix in 4% PFA with 1x PBS, 5 mM EDTA and 10 mM Citrate at 4 °C overnight. After fixation, transfer the lungs into a 30% Sucrose solution prepared in 1x PBS with 5 mM EDTA and 10 mM Citrate, then incubate at 4 °C overnight. Embed the lungs in OCT, freeze them, and store the cryoblock at -80 °C.
For OCT blocks: Cut 14 µm sections and collect on clean Superfrost glass slides. Air dry for 5-10 min then proceed to sample treatment step.
For FFPE samples: Cut 10 µm paraffin sections and collect them on clean Superfrost glass slides. Air dry the sections for 30 min then bake them at 60 °C for 1 h. To deparaffinize, immerse in Xylene twice for 5 min each; immerse in 100% ethanol twice for 5 min each; air dry slides for 5 min. Rehydrate the sections in a graded ethanol series (80% ethanol, 50% ethanol, then 1x PBS for 5 min each). Proceed to the sample treatment step.
Tissue sections collected on poly-L-Lysine coated coverslips
Collecting tissue sections on coverslips facilitates iterated labelimage-erase cycles because the seal chambers do not have to be removed for imaging each cycle (which is the case when a slide is used).
After imaging, LPs can be erased by USER enzyme (see Step C5b for protocol) then the specimen incubated with a new set of LPs to image another subset of transcripts.
Wash the coverslips in 70% ethanol for 2 min and rinse them with nuclease-free water. Incubate the clean coverslips with poly-L-Lysine solution for 20 min with gentle rocking (the solution should uniformly coat the coverslips), remove the excess solution and air dry the coverslips for 10 min or until all of the moisture has evaporated. Store the coverslips protected from dust.
Collect tissue sections on poly-L-Lysine coated coverslips. Air dry for 5 min at room temperature. Bake coverslips at 37 °C for 15-30 min for strong adhesion of the tissue. Proceed to the sample treatment step (Figure 2).
Note: Consider the following when preparing tissue samples for PLISH.
Prior to fixation, trim each tissue piece so its size does not exceed 10 mm.
During collection, transfer tissues immediately to fixative solution to minimize RNA degradation.
For cells, only perform the sample pretreatment step 1. Skip the sample pretreatment step 2.
All reagents should be prepared using nuclease-free water. Maintain a clean work area and use ELIMINase to minimize risk of exposure of cells or tissue to RNases.
Sample treatment for adult mouse lung
Sample treatment step 1: Prepare 10 mM Citrate pH 6.0 + 0.05% LDS in nuclease-free water and pour into a slide jar. Pre-warm this buffer in a water bath at 100 °C for 5 min. Place the air-dried slides into the jar and incubate for 5 min at 100 °C. Use forceps to transfer slides into a jar containing nuclease-free water. Rinse by lifting then re-immersing the slides in the water 4 times. Air dry the slides for 10 min. Use a Kimwipe to remove excess moisture. Apply seal chambers over the tissue. Rehydrate the sections with nuclease-free water for 1 min before proceeding to the next step. Each seal chamber accommodates ~50 µl volume (Figure 2).
Sample treatment step 2: Prepare 0.025 mg/ml Pepsin (in 0.1 M HCl). Add this solution to the seal chamber wells and incubate at 37 °C in a humidified chamber for 5 min. Quickly remove the Pepsin solution and add 1x PBS to inactivate residual Pepsin (Figure 2). Proceed to hybridization of H probes.
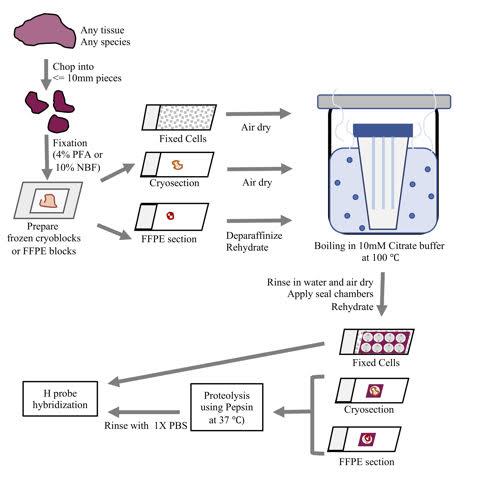
Figure 2. An overview of the sample preparation protocol for PLISH. A fresh tissue specimen is chopped into smaller pieces (for full penetration of the fixative solution) then rapidly fixed. The tissues are then used to prepare OCT embedded cryoblocks or Formalin Fixed Paraffin Embedded (FFPE) blocks. Sections from these blocks are collected on Superfrost slides, deparaffinized (if required), subjected to de-crosslinking by boiling in 10 mM Citrate buffer at 100 °C, followed by proteolysis using Pepsin and hybridization of H probes. Cells grown on coverslips or slides are fixed, subjected to de-crosslinking by boiling in 10mM Citrate buffer at 100 °C, then H probes are hybridized.
Note: The provided sample treatment is optimized for 14 µm adult mouse lung cryosections, but in our experience each tissue requires a customized treatment protocol that varies depending on the tissue under study, the fixation and embedding conditions, and the species of origin. Use the guidelines below to determine the optimal sample treatment conditions for your tissue of interest (Table 12; Figure 3). Note that tissue thickness can also impact on PLISH performance, so if the initial trials are suboptimal, you may wish to empirically test the effect of varying the section thickness.
Table 12. Range of suggested conditions for determining optimal pretreatment for fixed frozen or FFPE samples of your tissue of interest
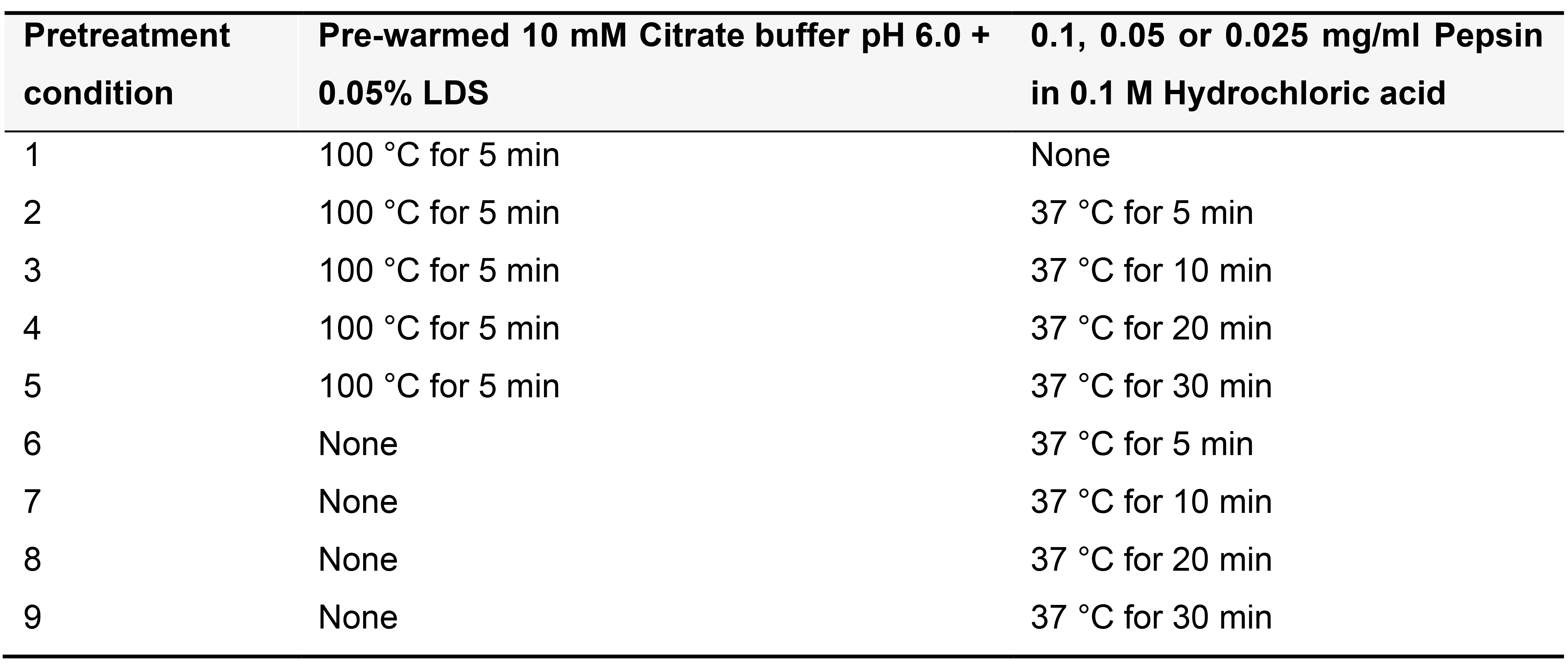
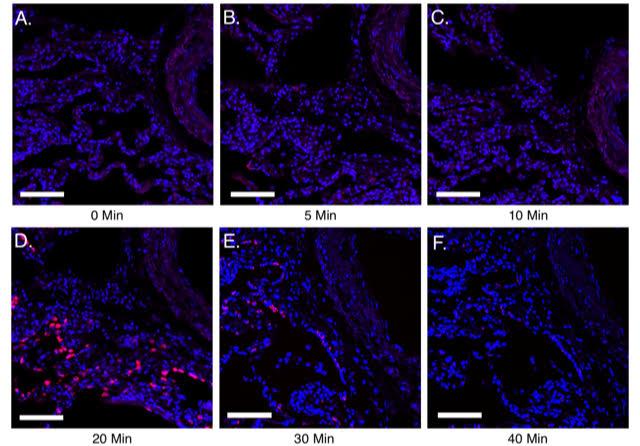
Figure 3. Utilizing guidelines to identify optimal pretreatment conditions for normal human lung FFPE sections. A-F, 10 μm FFPE sections of normal human lung were processed for PLISH. The sample pretreatment guidelines mentioned in Table 12 were followed and PLISH was performed to detect human Surfactant protein C (SFTPC, red) using Cy5-conjugated label probes. Samples (A-F) were boiled in 10mM citrate buffer with 0.05% LDS at 100 °C for 5 min. This was followed by no pepsin treatment (A) or 5, 10, 20, 30 and 40 min of pepsin (0.1 mg/ml in 0.1 M HCl) digestion at 37 °C (B-F). A-C show weak detection of SFTPC. D shows maximum SFTPC detection indicating optimal pretreatment conditions. E and F show diminished SFTPC signal due to excess proteolytic digestion leading to loss of RNA. DAPI, blue. Scale, 50 μm. Images were acquired on a Leica Sp8 confocal microscope using a 633 nm laser (with a laser power level of 10%; PMT detector gain of 750 V and offset of–0.5%) and a 25x water immersion objective.
In situ hybridization
The example below shows how to perform PLISH for 2 transcripts. Transcript A has 5 pairs of H probes designed to work with the VB01 bridge and LP1 label probe. Transcript B has 10 pairs of H probes designed to work with the VB02 bridge and LP2 label probe.
H probe Hybridization (Table 13)
If using 5 pairs of H probes to target a transcript of interest (as described in Table 5), the final concentration of each probe in the hybridization reaction should be 100 nM. If using more than 5 pairs, the final concentration of each H probe in the hybridization reaction should be 50 nM. Therefore, for Transcript A (targeted using 5 pairs of H probes or a total of 10 probes) the pooled stock is 10 µM (Table 5), the final concentration of each H probe in the hybridization reaction is 100 nM and the total final concentration of all H probes targeting Transcript A is 1,000 nM (100 nM x 10 probes = 1,000 nM). For Transcript B (targeted using 10 pairs of H probes or a total of 20 probes) the pooled stock is 5 µM (Table 7), the final concentration of each H probe in the hybridization reaction is 50 nM and the total final concentration of all H probes targeting Transcript B is 1,000 nM (50 nM x 20 probes = 1,000 nM).
Table 13. Hybridization buffer with probes targeting Transcript A and Transcript B

Add the hybridization buffer with probes to the sample and incubate at 37 °C in a humid chamber for 2 h. Wash four times for 5 min each in pre-warmed hybridization buffer at 37 °C, followed by one wash for 10 min with pre-warmed high salt buffer at 37 °C.
Assembly of circle components (Table 14)
Use each phosphorylated oligonucleotide (CCC2.1 and each VB) at a final concentration of 100 nM.
Table 14. Circle hybridization buffer with phosphorylated circle and bridge oligonucleotides

Add circle hybridization buffer containing phosphorylated VB and CCC2.1 to the sample and incubate at 37 °C in humid chamber for 60 min. Wash twice for 5 min each in pre-warmed circle hybridization buffer at 37 °C. Wash for 2 min in pre-warmed 1x ligase buffer at 37 °C.
Ligation Reaction (Table 15)
Table 15. T4 DNA Ligase reaction mix
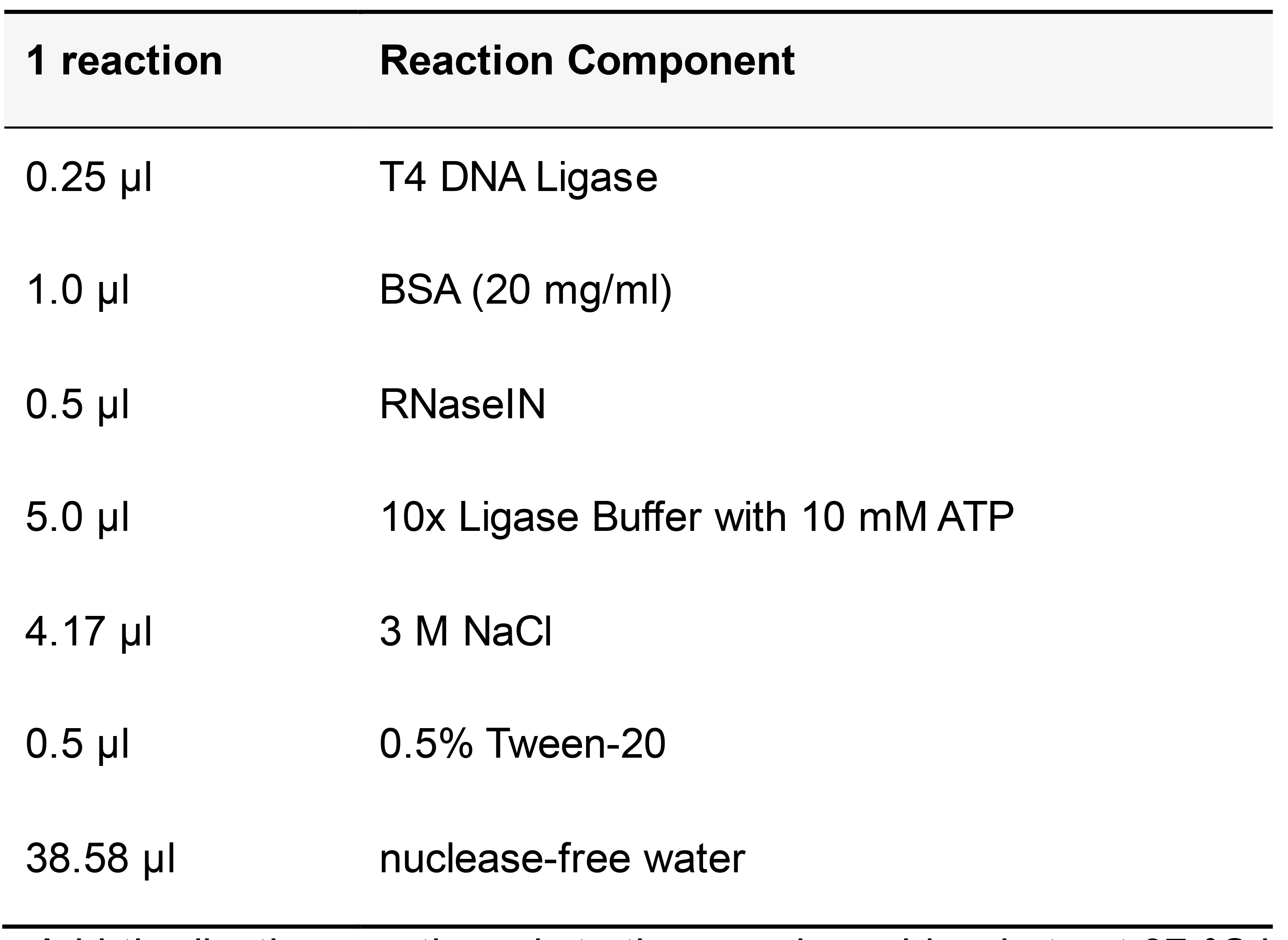
Add the ligation reaction mix to the sample and incubate at 37 °C in a humid chamber for 2 h. Wash twice for 5 min each in pre-warmed circle hybridization buffer at 37 °C. Wash for 2 min with pre-warmed 1x phi29 DNA polymerase buffer at 37 °C.
Rolling Circle Amplification (RCA) Reaction (Table 16)
Table 16. Rolling circle amplification reaction mix

Add the RCA reaction mix to the sample and incubate at 37 °C in a humid chamber overnight. Wash twice for 5 min each in pre-warmed label probe hybridization buffer at 37 °C.
Label Probe Hybridization (Table 17)
Use label probes at a final concentration of 100 nM each.
Table 17. Label probe hybridization mix

Add the label probes to the sample and incubate at 37 °C in humid chamber for 30 min. From this point, the protocol diverges to three possible steps, depending on the experimental design.
If no additional staining is planned
Wash samples twice for 5 min each in PBST, then incubate for 5 min with DAPI in PBS (DAPI stock is 1 mg/ml; dilute the stock 1:1,000 in 1x PBS to prepare a working solution). Peel off the seal chamber and mount the sample in 1x PBS if using a water objective, or 80% glycerol if using a glycerol objective.
If conducting additional round(s) of label probe hybridization for iterative PLISH
Wash samples twice for 5 min each in PBST, then incubate for 5 min with DAPI in PBS (the DAPI stock is 1 mg/ml; dilute stock 1:1,000 in 1x PBS to prepare a working solution). Image the specimen in PBS. After photo-documentation of labeled transcripts, proceed with the enzymatic erasure of label probes as detailed below.
USER digestion (Table 18)
Table 18. USER digestion mix

Incubate at 37 °C in a humidified chamber for 20 min. Wash samples twice for 5 min each in PBST.
UDG (Uracil DNA Glycosylase) Inhibition (Table 19)
Table 19. Uracil DNA Glycosylase Inhibitor reaction mix

Incubate the samples at 37 °C in a humidified chamber for 20 min then remove the digestion solution and add the next round of LPs in label probe buffer. Incubate at 37 °C in humid chamber for 30 min.
Note: If performing more than one label-image-erase cycle, we recommend fixing the samples after the RCA reaction step using 4% Paraformaldehyde for 10 min at room temperature, followed by washing twice for 5 min each in 1x PBS.
If conducting combined PLISH and immunohistochemistry:
Add 5 mM EDTA to all of the solutions used for the immunohistochemistry steps. Following label probe hybridization, wash samples twice for 5 min each in at room temperature. Add blocking solution (50 µl/ml serum, 1 µl/ml Triton X-100 and 0.03 g/ml BSA in PBS) and incubate for 1 h at room temperature with gentle rocking. Remove blocking solution and add primary antibody (diluted in blocking solution). Incubate samples for 2 h at room temperature or overnight at 4 °C with gentle rocking.
Wash samples four times for 5 min each in PBST at room temperature. Add secondary antibody (diluted in blocking solution) and incubate samples for 1 h at room temperature with gentle rocking. Wash samples four times for 5 min each in PBST at room temperature, then incubate for 5 min with DAPI in PBS at room temperature. Peel off the seal chamber and mount the sample in 1x PBS if using a water objective, or 80% glycerol if using a glycerol objective.
If you plan to store samples for imaging later, seal the coverslips with a thin coat of clear sealant along the edges and allow to dry completely in a dark chamber at room temperature. Store slides at 4 °C in a slide box protected from light.
Data analysis
For detailed information on data analyses, refer the original article (Nagendran et al., 2018).
Modifications from original PLISH protocol
1)Buffers: Hybridization buffer contains the detergent Lithium Dodecyl Sulfate (LDS). All washes are carried out at 37 °C and a high salt buffer wash is included in the steps after H probe hybridization to increase the stringency. Circle and bridge oligonucleotides are added in 20% formamide and 2x SSC buffer with LDS.
2)Due to variability in the performance of circles used in the published version (CC06 performed very well while CC03 and CC01 performed relatively poorly), we implemented a modified version of CC06 for use as a common connector circle (referred to here as CCC2.1) and incorporated the barcode into the bridge oligonucleotides (referred to here as VB). As a result, all oligonucleotide sequences in the new protocol differ from the ones in the original publication. The new protocol utilizes 24 unique variable bridges for iterative (label-image-erase) multiplexing experiments. This modification eliminates the need for up-front testing of each connector circle, which are more prone to variation in performance than bridges.
3)We increased the stringency of the criteria used to select probe sites in order to minimize the risk of cross hybridization to off-target transcripts with partial sequence homology.
Recipes
- Stock Solution Recipes
Nuclease-free water
Add 1 ml of Diethyl pyrocarbonate (DEPC) to 1 liter of Milli-Q water in a glass bottle
Close lid tightly and mix well by inverting or shaking the bottle several times
Keep the bottle in a dark room and leave overnight
Autoclave on liquid cycle for 30 min to inactivate DEPC
Store at room temperature
3 M Sodium Chloride (NaCl)
Dissolve 17.53 g of Sodium Chloride in 100 ml nuclease-free water
Mix well
Autoclave on liquid cycle for 30 min
Store at room temperature
1 M Disodium Hydrogen Phosphate (Na2HPO4)
Dissolve 141.96 g of Na2HPO4 in 1,000 ml of nuclease-free water
Mix well
Store at room temperature
1 M Sodium Dihydrogen Phosphate (Na2HPO4)
Dissolve 119.98 g of Na2HPO4 in 1,000 ml of nuclease-free water
Mix well
Store at room temperature
10x Phosphate Buffered Saline (PBS)
10x PBS contains 1.5 M NaCl and 100 mM NaPO4 (pH 6.8)
To make 1,000 of 10x PBS:
Mix 500 ml of 3 M NaCl
46.3 ml of 1 M Na2HPO4
53.7 ml of 1 M Na2HPO4
Make up the volume to 1,000 ml with nuclease-free water
Autoclave on liquid cycle for 30 min
When diluted to 1x, the pH of the solution will be 7.4
Store at room temperature
1x Phosphate Buffered Saline (PBS) pH 7.4
Dilute 10 ml of 10x PBS in 90 ml of nuclease-free water
Store at room temperature
30% Sucrose solution
Dissolve 30 g of Sucrose in 100 ml of 1x PBS.
Mix well.
Make 10 ml aliquots and store at -20 °C
5% Tween-20
Add 500 µl of Tween-20 in 10 ml nuclease-free water
Mix thoroughly until completely dissolved
Store at room temperature
0. 5% Tween-20
Add 1,000 µl of 5% Tween-20 in 10 ml nuclease-free water
Mix thoroughly until completely dissolved
Store at room temperature
4% Paraformaldehyde
Add 40 g of Paraformaldehyde to 500 ml of nuclease-free water
Add NaOH pellets and stir gently on a heating block at ~60 °C until the Paraformaldehyde is dissolved
Add 100 ml of 10x PBS and allow the mixture to cool to room temperature
Adjust the pH to 7.4 with HCl, then adjust the final volume to 1,000 ml with nuclease-free water
Store in aliquots at -20 °C for several months. Avoid repeated freeze/thawing
Caution: Wear gloves, work in a fume hood and exercise extreme caution when preparing paraformaldehyde.
1x Phosphate Buffered Saline with 0.05% Tween-20 (PBST)
Dissolve 100 µl of 5% Tween-20 in 10 ml 1x PBS
Store at room temperature
0.01 M Citric Acid
Dissolve 1.92 g of Citric Acid in 1,000 ml of nuclease-free water
Autoclave on liquid cycle for 30 min
Store at room temperature
0.01 M Sodium Citrate
Dissolve 2.58 g of Sodium Citrate in 1,000 ml of nuclease-free water
Autoclave on liquid cycle for 30 min
Store at room temperature
10% Lithium Dodecyl Sulfate (LDS)
Dissolve 1 g of Lithium Dodecyl Sulfate in 10 ml nuclease-free water
Filter sterilize using a 0.2 µm syringe filter
Store at room temperature
0.1 M Hydrocholoric acid (HCl)
Add 817 µl of concentrated Hydrochloric Acid (10 N) to 100 ml nuclease-free water
Store at room temperature
Caution: Wear gloves, work in a fume hood and exercise extreme caution when handling concentrated Hydrocholoric acid.
Pepsin stock solution from powdered enzyme
Weigh 1 mg of Pepsin in a 1.5 ml microcentrifuge tube
Add 1 ml of 0.1 M HCl, mix thoroughly
This is a 1 mg/ml Pepsin stock solution
Dilute it to the required concentration in 0.1 M HCl, for example 1:10 (100 µl stock solution in 900 µl 0.1M HCl) for making 0.1 mg/ml Pepsin working solution. Use immediately then discard leftover working and stock solutions
Alternatively, you can prepare a 10 mg/ml stock of Pepsin in glycerol and store at -20 °C. When needed, dilute this stock to the required concentration in 0.1 M HCl.
Note: Pepsin activity may differ between manufactured batches, so it is recommended to test the enzyme performance when you receive a new batch. Some PLISH users have also used Proteinase K successfully.
2 M Sodium Trichloroacetate
Dissolve 185.37 g of Sodium Trichloroacetate in 500 ml nuclease-free water
Autoclave on liquid cycle for 30 min
Store at room temperature
0.5 M Ethylenediaminetetraacetic Acid Disodium Salt Dehydrate (EDTA)
Reagent Amount needed EDTA, disodium salt 9.3 g Sodium hydroxide pellets Approximately 1 g nuclease-free water Approximately 50 ml Add EDTA to approximately 45 ml of nuclease-free water with stirring. Monitor pH and slowly add 1 or 2 Sodium Hydroxide pellets with stirring until the pH reaches 8.0. Make up the volume to 50 ml. Filter sterilize using a 0.2 µm syringe filter. Store at room temperature.
1 M Tris pH 7.4
Reagent Amount needed Tris Base 12.114 g Concentrated Hydrochloric acid 7-8 ml Nuclease-free water Approximately 100 ml Add Tris Base to approximately 85 ml of nuclease-free water with stirring. Monitor pH and slowly add concentrated Hydrochloric acid with stirring until the pH reaches 7.4. Make up the volume to 100 ml. Mix well. Autoclave on liquid cycle for 30 min. Store at room temperature.
1 M Tris pH 8.0
Reagent Amount needed Tris Base 12.114 g Concentrated Hydrochloric acid 7-8 ml Nuclease-free water Approximately 100 ml Add Tris Base to approximately 85 ml of nuclease-free water with stirring. Monitor pH and slowly add concentrated Hydrochloric acid with stirring until the pH reaches 8.0. Make up the volume to 100 ml. Mix well. Autoclave on liquid cycle for 30 min. Store at room temperature.
20 mg/ml Heparin
Dissolve 0.2 g of Heparin Sodium salt in 10 ml nuclease-free water
Mix well
Filter sterilize using a 0.2 µm syringe filter
Make aliquots and store at -20 °C
50% and 80% Glycerol
Add 25 ml Glycerol to 25 ml nuclease-free water (for 50%) or 8 ml Glycerol to 2 ml 1x PBS (for 80%)
Mix well until dissolved
Store at room temperature
20 mg/ml Bovine Serum Albumin (BSA)
Dissolve 0.2 g of bovine serum albumin in 10 ml nuclease-free water
Mix well
Filter sterilize using a 0.2 µm syringe filter
Make aliquots and store at -20 °C
20x Saline Sodium Citrate (SSC) pH 7.0
20x SSC contains 3 M Sodium Chloride in 0.3 M Sodium Citrate
Reagent Amount needed Sodium Chloride 17.53 g Sodium Citrate 8.82 g nuclease-free water Approximately 100 ml Add Sodium Chloride and Sodium Citrate to 80 ml nuclease-free water. Monitor pH and slowly add concentrated Hydrochloric acid with stirring until the pH reaches 7.0. Make up the volume to 100 ml. Mix well. Autoclave on liquid cycle for 30 min. Store at room temperature
DAPI
Stock solution:
Dissolve 1 mg of DAPI in 1 ml of Dimethyl Sulfoxide (DMSO). Store protected from light at -20 °C
Working solution:
Dissolve 10 µl of the stock solution in 10 ml of 1x PBS. Mix well. Make aliquots and store protected from light at 4 °C
Precaution
Handle DMSO with care in a fume hood while wearing gloves
Blocking serum
Blocking serum contains 5% Serum, 0.1% Triton X-100, 5 mM EDTA and 3% BSA in 1x PBS
To make 10 ml of blocking serum, place 0.3 g of BSA in a 15 ml tube. Add 10 ml of nuclease-free water. Mix thoroughly. Add 10 µl of Triton X-100, 100 µl of 0.5 M EDTA and 500 µl of serum. Mix well. Always prepare fresh
- Buffer Recipes
10 mM Citrate buffer with 0.05% LDS
Mix 0.01 M Sodium Citrate with 0.01 M Citric acid at a 1:4 ratio by volume. Mix well and adjust the pH to 6.0 using HCl. Store at room temperature.
TE buffer pH 8.0 contains 100 mM Tris pH 8.0 and 10 mM EDTA pH 8.0
Stock solution Volume required 1 M Tris pH 8.0 10 ml 0.5 M EDTA pH 8.0 2 ml Nuclease-free water Make up volume to 100 ml Mix well and store at room temperature.
Hybridization buffer
Contains 1 M Sodium Trichloroacetate, 5 mM EDTA, 50 mM Tris pH 7.4, 0.2 mg/ml Heparin and 0.1% LDS
Stock solution Volume required 2 M Sodium trichloroacetate 500 ml 0.5 M EDTA 10 ml 1 M Tris pH 7.4 50 ml Nuclease-free water Make up volume to 1,000 ml Mix well and store at room temperature. When you start a PLISH stain, take the required volume of buffer, add Heparin (final concentration–0.2 mg/ml) and LDS (final concentration 0.1%). Mix well
Note: LDS does not fully dissolve in this buffer at room temperature. Warm buffer in a water bath or oven at 37 °C to ensure LDS dissolves completely. Keep this buffer at 37 °C to avoid precipitation.
High Salt Buffer
Contains 0.25 M NaCl, 50 mM Tris pH 7.4, 2 mM EDTA, 0.1% LDS in nuclease-free water
Stock solution Volume required 3 M NaCl 83.33 ml 1 M Tris pH 7.4 50 ml 0.5 M EDTA 10 ml 10% LDS 10 ml Nuclease-free water Make up volume to 1,000 ml Mix well and store at room temperature
Circle hybridization buffer
Contains 2x SSC, 20% Formamide, 0.1% LDS and 0.2 mg/ml Heparin in nuclease-free water
Stock solution Volume required 20x SSC 1 ml Formamide 2 ml 10% LDS 100 µl 20 mg/ml Heparin 100 µl Nuclease-free water Make up volume to 10 ml Prepare fresh on the day of PLISH, mix well and pre-warm at 37 °C before use
1x Ligase Buffer
10x T4 Ligase buffer is supplied along with the T4 DNA ligase enzyme. Dilute the 10x stock buffer in nuclease-free water to 1x. Prepare fresh on the day of PLISH, mix well and pre-warm at 37 °C before use
1x phi29 DNA polymerase buffer
10x phi29 DNA polymerase buffer is supplied along with the phi29 DNA polymerase enzyme. Dilute the 10x stock buffer in nuclease-free water to 1x. Prepare fresh on the day of PLISH, mix well and pre-warm at 37 °C before use
Label probe buffer
Contains 2x SSC, 20% Formamide and 0.2 mg/ml Heparin in nuclease-free water
Stock solution Volume required 20x SSC 1 ml Formamide 2 ml 20 mg/ml Heparin 100 µl Nuclease-free water Make up volume to 10 ml Prepare fresh on the day of PLISH, mix well and pre-warm at 37 °C before use
Acknowledgments
We thank Andres Andalon and Yana Kazadaeva for technical assistance. This work was supported by the NIH HubMap Consortium 1UG3HL14562301 (PH, TD), NHLBI 1R01HL14254901 (TD), Chan Zuckerberg Initiative (TD), and a Stanford Discovery Innovation Fund award (PH). TD is the Woods Family Endowed Faculty Scholar in Pediatric Translational Medicine of the Stanford Child Health Research Institute.
Competing interests
Tushar J Desai, Pehr B Harbury, and Monica Nagendran have filed a patent “Molecular Profiling Using Proximity Ligation in situ Hybridization” (Application #62/475,090).
Ethics
Human subjects: Adult human lung was obtained from Stanford Healthcare with patient informed consent and consent to publish in strict accordance with protocol 18891, approved by the Institutional Review Board Administrative Panel on Human Subjects in Medical Research of Stanford University, in compliance with requirements for protection of human subjects.
References
- Beliveau, B. B., Kishi, J. Y., Nir, G., Sasaki, H. M., Saka, S. K., Nguyen, S. C., Wu, C. and Yin, P. (2018). OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115(10): E2183-E2192.
- Eichman, B. F., Vargason, J. M., Mooers, B. H. and Ho, P. S. (2000). The Holliday junction in an inverted repeat DNA sequence: sequence effects on the structure of four-way junctions. Proc Natl Acad Sci U S A 97(8): 3971-3976.
- Hays, F. A., Schirf, V., Ho, P. S. and Demeler, B. (2006). Solution formation of Holliday junctions in inverted-repeat DNA sequences. Biochemistry 45(8): 2467-2471.
- Khuu, P. A., Voth, A. R., Hays, F. A. and Ho, P. S. (2006). The stacked-X DNA Holliday junction and protein recognition. J Mol Recognit 19(3): 234-242.
- Liu, J., Declais, A. C., McKinney, S. A., Ha, T., Norman, D. G. and Lilley, D. M. (2005). Stereospecific effects determine the structure of a four-way DNA junction. Chem Biol 12(2): 217-228.
- Miick, S. M., Fee, R. S., Millar, D. P. and Chazin, W. J. (1997). Crossover isomer bias is the primary sequence-dependent property of immobilized Holliday junctions. Proc Natl Acad Sci U S A 94(17): 9080-9084.
- Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. and Desai, T. J. (2018). Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359(6380): 1118-1123.
- Nagendran, M., Riordan, D. P., Harbury, P. B. and Desai, T. J. (2018). Automated cell-type classification in intact tissues by single-cell molecular profiling. Elife 7: e30510.
- Wilbrey-Clark, A., Roberts, K. and Teichmann, S. A. (2020). Cell atlas technologies and insights into tissue architecture. Biochem J 477(8): 1427-1442.
Article Information
Copyright
![]() Nagendran et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Nagendran et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Nagendran, M., Andruska, A. M., Harbury, P. B. and Desai, T. J. (2020). Advances in Proximity Ligation in situ Hybridization (PLISH). Bio-protocol 10(21): e3808. DOI: 10.21769/BioProtoc.3808.
- Nagendran, M., Riordan, D. P., Harbury, P. B. and Desai, T. J. (2018). Automated cell-type classification in intact tissues by single-cell molecular profiling. Elife 7: e30510.
Category
Developmental Biology > Morphogenesis > Organogenesis
Cancer Biology > General technique > Molecular biology technique > Antisense oligonucleotide
Molecular Biology > RNA > RNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.