- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dual sgRNA-based Targeted Deletion of Large Genomic Regions and Isolation of Heritable Cas9-free Mutants in Arabidopsis

Published: Vol 10, Iss 20, Oct 20, 2020 DOI: 10.21769/BioProtoc.3796 Views: 6999
Reviewed by: Caiji GaoMatthew A. EscobarLifeng Liu
Original research article
The authors used this protocol in:

May 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
CRISPR/Cas9 system directed by a gene-specific single guide RNA (sgRNA) is an effective tool for genome editing such as deletions of few bases in coding genes. However, targeted deletion of larger regions generate loss-of-function alleles that offer a straightforward starting point for functional dissections of genomic loci. We present an easy-to-use strategy including a fast cloning dual-sgRNA vector linked to efficient isolation of heritable Cas9-free genomic deletions to rapidly and cost-effectively generate a targeted heritable genome deletion. This step-by-step protocol includes gRNA design, cloning strategy and mutation detection for Arabidopsis and may be adapted for other plant species.
Keywords: CRISPR/Cas9Background
Dual sgRNA-directed gene knockout by CRISPR/Cas9 has been successfully used for genome editing in a variety of organisms (Wang et al., 2013; Chen et al., 2014; Char et al., 2017; Cai et al., 2018; Durr et al., 2018; Cui et al., 2019; Do et al., 2019; Liu et al., 2020). Targeted deletions of genomic DNA regions offer a valuable starting point for functional genomics studies (Hilton and Gersbach, 2015; Ford et al., 2019; Gowthaman et al., 2020). CRISPR/Cas9-based methods to delete genomic regions benefit from two gRNAs flanking the target DNA regions (Xiao et al., 2013; Canver et al., 2014; Kistler et al., 2015; Song et al., 2016). In plants, one key bottleneck to perform multiplex gene targeting from a single transformation event is to include multiple gBlocks into one binary vector. A gBlock is composed of a RNA Polymerase III (RNAPIII) promoter, a gene specific sgRNA protospacer, a sgRNA scaffold and a RNAPIII terminator. However, gBlock DNA sequences are usually long and repetitive, rendering design expensive for synthetic DNA and laborious by traditional assembly methods (Gao et al., 2016; Peterson et al., 2016; Zhang et al., 2016; Char et al., 2017; Durr et al., 2018; Pauwels et al., 2018; Schuster, 2018; Wu et al., 2018; Hui et al., 2019; Fonseca et al., 2020). For example, Durr et al. (2018) developed a dual-sgRNA vector by first modifying a pEN-Chimera entry vector to generate two gBlocks, then inserting two gRNAs into the modified entry vector by restriction enzymes, respectively, and finally cloning two gBlocks into the binary vectors. The multiple steps necessary are laborious and time-consuming. Although multiplex CRISPR/Cas9 platforms by introducing repetitive gBlocks for targeted genome editing were reported (Ordon et al., 2016; Schuster, 2018), several studies have noted that transformation of highly repetitive DNA sequences can trigger recombination and silencing of the RNA expression cassettes in a variety of species (Ma and Mitra, 2002; Lovett et al., 2004; Brake et al., 2008). To simplify targeted genomic regions and minimize potential recombination in Arabidopsis, we combined and modified existing cloning-based assembly steps. First, we amplified the middle border of our target specific two-gBlocks from a previously developed vector pHEE2E-TRI (Wang et al., 2015) in a single step. Second, we cloned the middle border into a known CRISPR/Cas9 binary vector pKIR1.1 (Maruyama et al., 2013; Tsutsui and Higashiyama, 2017), allowing two-gBlocks with different Poll III-dependent promoters to reduce the repetitiveness. This vector harbors an RPS5A-Cas9 cassette driving high constitutive expression of Cas9 protein at all developmental stages including egg cells, thus achieves highly efficient mutation in the T1 generation of Arabidopsis. In addition, the expression cassette OLE1-tagRFP in this system shows red fluorescence in seeds, allowing rapid screening for heritable Cas9-free Arabidopsis mutants in the seed of primary transformants. We combined the advantages of the two vectors by a single PCR and a single cloning step, thus providing a simple and reliable protocol to generate stable inherited deletion mutants. Our strategy promises to save cost and time to delete any chromosomal region in Arabidopsis, and can be likely adapted for genome editing of multiple genes simultaneously. It also has the potential to simplify genomic deletion in other plant species.
Materials and Reagents
- Consumables
- Sterile pipette tips (Axygen, catalog numbers: TF-300-R-S [10 µl], T-350-C-L-R-S [300 µl], TF-1000-R-S [1,000 µl])
- PCR microtubes (BioExpress, catalog number: T-3135-2 )
- 60 mm x 15 mm round Petri dishes (VWR, catalog number: 100488-404 )
- Competent cells
- Escherichia coli HST08 competent cell (homemade, protocol 1), store at -80 °C
- Agrobacterium tumefaciens GV3101 competent cells (homemade, protocol 2), store at -80 °C
- Vectors
- pHEE2E-TRI (Addgene, catalog number: 71288 ), store at -20 °C
- pKIR1.1 (Addgene, catalog number: 85758 ), store at -20 °C
pHEE2E-TRI sequence: https://www.addgene.org/71288/sequences/
pKIR1.1 sequence: https://www.addgene.org/85758/sequences/ - Oligonucleotides 10 µM
Dual-sgRNA1_F: 5′-CACCTGCATACATTGN20(protospacer 1)GTTTTAGAGCTAGAAATAGC-3′
Dual-sgRNA2_R: 5′-CACCTGCATACAAACN20(protospacer 2 reverse complement)CAATCTCTTAGTCGA CTCTAC-3′
Mlo 1938: 5′-TCCCAGGATTAGAATGATTAGG-3′
Primer_F: 5′-TTCTCTCTTCGCTCTCTCGTAG-3′
Primer_R: 5′-GGCCCAAATACTCTTTTCCAAGAC-3′
Cas9_F: 5′-CAGCCGACAAGAAGTACAGC-3′
Cas9_R: 5′-ATGGTGGGGTACTTCTCGTG-3′ - Enzymes and buffers
- AarI (Thermo Fisher Scientific, catalog number: ER1581 ), store at -20 °C
- T4 DNA Ligase (NEB, catalog number: M0202L ), store at -20 °C
- T4 Polynucleotide Kinase (NEB, catalog number: M0201L ), store at -20 °C
- T4 DNA Ligase Reaction Buffer (10x) (NEB, catalog number: B0202S ), store at -20 °C
- Alkaline Phosphatase, Calf Intestinal (CIP) (NEB, catalog number: M0290 ), store at -20 °C
- Phusion High-Fidelity DNA Polymerase Kits (New England Biolabs, catalog number: M0530S ), store at -20 °C
- HotMaster Taq DNA Polymerase (VWR, catalog number: QUNT2200330 ), store at -20 °C
- Wizard® SV Gel and PCR Clean-Up System (Promega, catalog number: A9282 ), store at room temperature
- DNA plasmid kit (VWR, catalog number: D6943-02 ), store at room temperature
- Reagents
- MES (Sigma-Aldrich, catalog number: 4432-31-9 ), store at room temperature
- KOH (Sigma-Aldrich, catalog number: 1310-58-3 ), store at room temperature
- Sucrose (Sigma-Aldrich, catalog number: 57-50-1 ), store at room temperature
- Plant agar (Sigma-Aldrich, catalog number: 9002-18-0 ), store at room temperature
- Bacto agar (BD Biosciences, catalog number: 214030 ), store at room temperature
- Bacto tryptone (BD Biosciences, catalog number: 211699 ), store at room temperature
- Bacto yeast extract (BD Biosciences, catalog number: 212730 ), store at room temperature
- Sodium chloride (Fisher Scientific, catalog number: 7647-14-5 ), store at room temperature
- Murashige & Skoog medium (Duchefa Biochemie, catalog number: M524 ), store at 4 °C
- Antibiotics
- Spectinomycin (VWR, catalog number: 101454-196 ), store at -20 °C
- Rifampicin (VWR, catalog number: 13292-46-1 ), store in a dry and well-ventilated place
- Gentamycin (VWR, catalog number: 97062-974 ), store at 4 °C
- Kanamycin (VWR, catalog number: 25389-94-09 ), store at 4 °C
- Media (see Recipes)
- LB liquid medium + 100 mg/L spectinomycin (store at 4 °C for one month)
- LB agar plates + 100 mg/L spectinomycin (store at 4 °C for one month)
- LB liquid medium + 100 mg/L spectinomycin + 20 mg/L rifampicin + 25 mg/L gentamycin + 25 mg/L kanamycin (store at 4 °C for one month)
- LB agar plates + 100 mg/L spectinomycin + 20 mg/L rifampicin + 25 mg/L gentamycin + 25 mg/L
- ½ MS-medium plates
Equipment
- Pipettes (Thermo Scientific, FinnpipetteTM F2, catalog numbers: 4642010 [0.2-2 µl], 4642030 [1-10 µl], 4642060 [2-20 µl], 4642080 [20-200 µl], 4642090 [100-1,000 µl])
- Incubator (Thermo Scientific, catalog number: 51028132 )
- Shakers (Eppendorf, model: New BrunswickTM Innova® 44 , catalog number: M1282-0000)
- Stereo Fluorescence Microscope (Leica, model: M205FA )
- Gel Doc EQ System (Bio-Rad, Universal Hood II, model: BGDII )
- Heating blocks (Eppendorf, catalog number: T1317-1EA )
- ThermoMixer® C (Eppendorf, catalog number: 5382000015 )
- PCR Thermo Cycler (Bio-Rad, model: T100 , catalog number: 1861096 )
- Tabletop centrifuge (Thermo Fisher Scientific, catalog number: 75008801 )
- NanoDrop (Thermo Scientific, model: NanoDropTM 2000C , catalog number: ND2000)
- Agarose gel electrophoresis equipment (Bio-Rad, catalog number: 1704489EDU )
- Plant growth chamber (photoperiod: 16 h light/ 8 h dark, temperature: 22 °C in the day/20 °C in the darkness, humidity: 65%, light intensity: 100 μE m−2 s−1)
Note: No equipment from specific manufacturers is required. Any equivalent device can be used.
Software
- SnapGene® (SnapGene, https://www.snapgene.com/)
- Image Lab Software (Bio-Rad, https://www.bio-rad.com/)
Procedure
- Overview
The process that generates dual sgRNA-directed large deletion by CRISPR/Cas9 is shown in Figure 1. The vector pKIR1.1 can be ordered from Addgene. A gBlock is composed of a U6 promoter, a gene specific sgRNA protospacer, and a sgRNA scaffold and a terminator.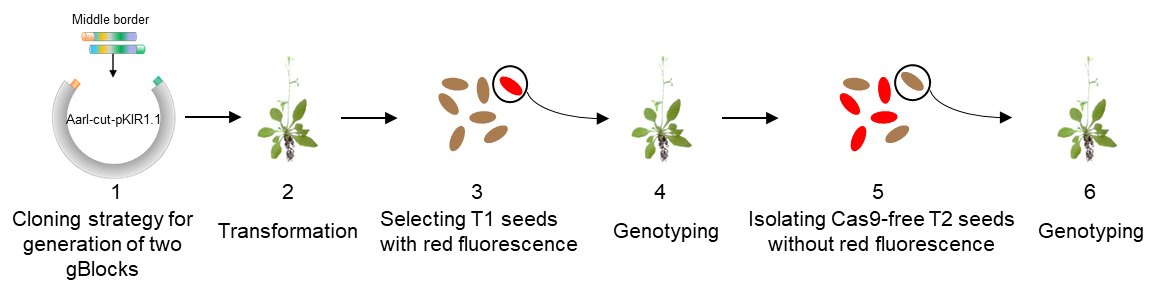
Figure 1. Flowchart for isolation of Cas9-free target mutants. 1. Cloning strategy for generation of two gBlocks. 2. Transformation by floral dipping. 3. Screening T1 seeds by red fluorescence. 4. Genotyping and sequencing T1 plants with red fluorescence to identify candidates and harvest seeds from each individual plant. 5. Isolating Cas9-free T2 seeds without red fluorescence. 6. Genotyping T2 plants to obtain stable and heritable null mutants. - Generate the middle border of two-gBlocks
- Design two gRNAs to target the same gene of interest (GOI) with CRISPRdirect (Naito et al., 2015) (Figure 2).
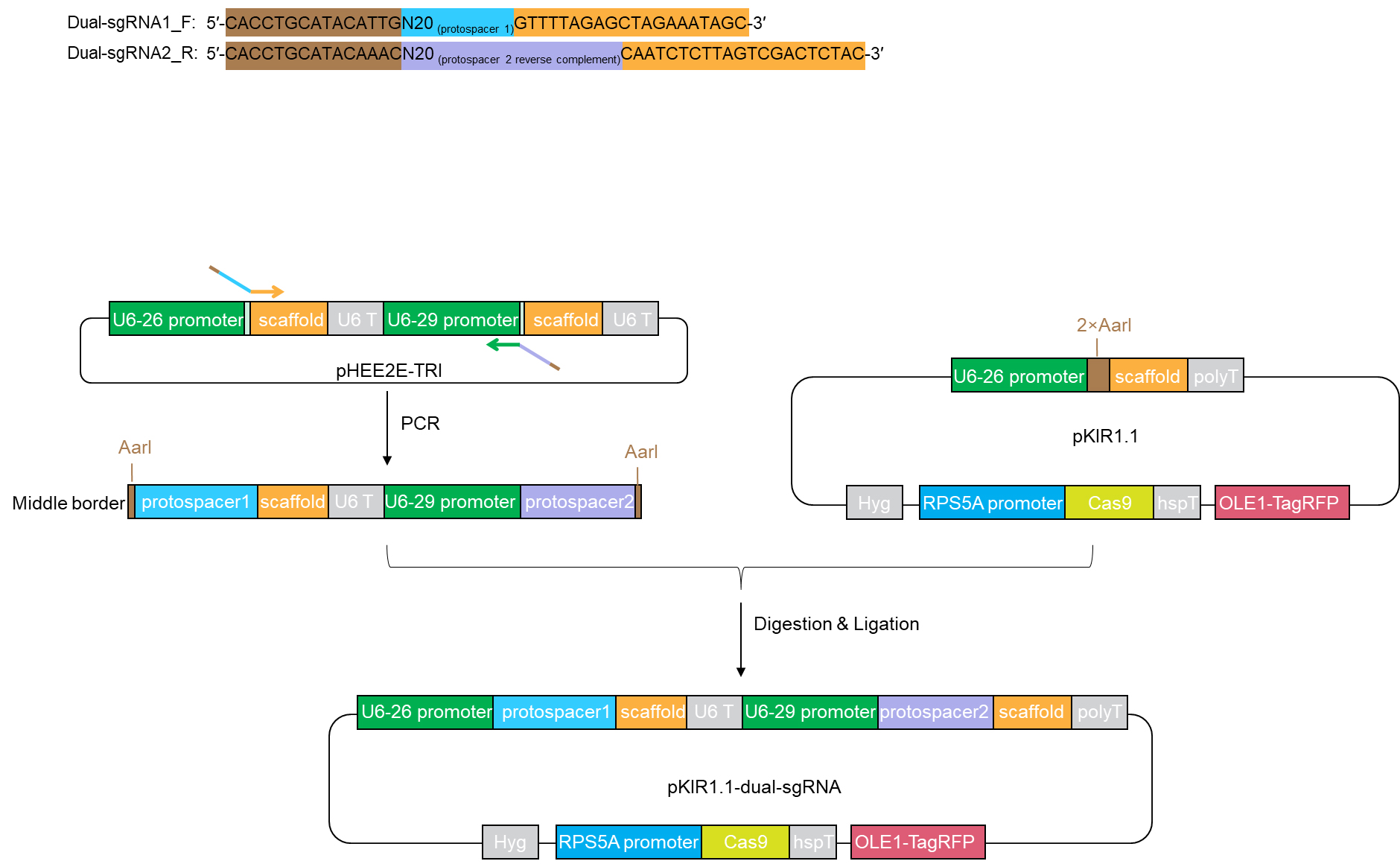
Figure 2. Overview of dual-sgRNAs cloning strategy. Schematic representation of the cloning strategy for the introduction of the middle border of two-gBlocks into the pKIR1.1 backbone. pHEE2E-TRI harbors two sets of gBlocks including gBlock1 (a U6-26 promoter, a 19 bp target sequence 1, a sgRNA scaffold, a terminator) and gBlock2 (a U6-29 promoter, a 19 bp target sequence 2, a sgRNA scaffold, a terminator). Therefore, pHEE2E-TRI can serve as a template to amplify the middle border (AarI-overhang1-protospacer1-scaffold-terminator-U6-29 promoter protospacer2-overhang2-AarI) using a pair of dual-sgRNA primers. After digestion of pKIR1.1 plasmid and the middle border both by AarI, the middle border can be integrated into the linearized pKIR1.1 backbone to generate pKIR1.1-dual-sgRNA for plant transformation.- Open the CRISPRdirect webpage (http://crispr.dbcls.jp/) and paste a target genomic sequence into text field in a FASTA format or a plain nucleotide sequence up to 10 kb.
Note: You can also enter an accession number (e.g., NM_001187) or genome location (e.g., hg19: chr7: 900000-901000) to retrieve sequence, or upload a sequence plain text file in a FASTA format or a plain nucleotide sequence up to 10 kb. - NGG is selected on the ‘PAM sequence requirement’ panel.
- Select Thale cress (Arabidopsis thaliana) genome, TAIR10 as the organism.
- Click on ‘design’.
- Select two highlighted protospacer sequence by clicking on ‘show highly specific target only’ as shown in the screen shot below (Figure 3).
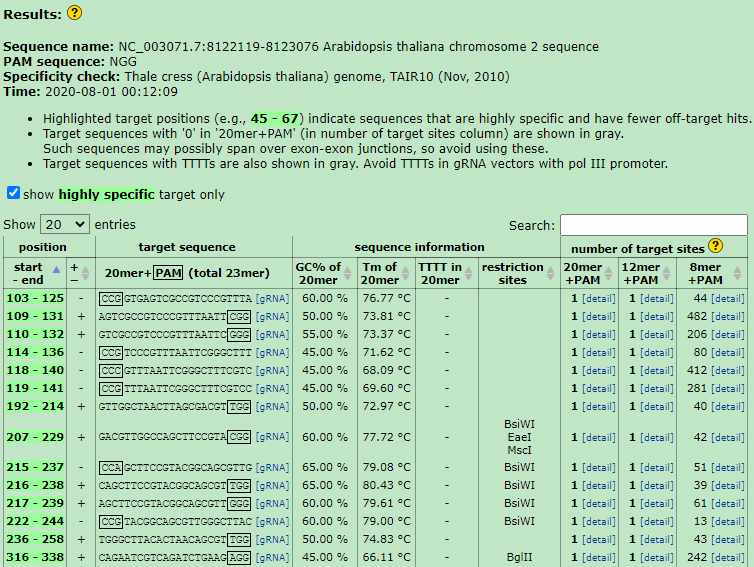
Figure 3. gRNA selection. Example screenshot for gRNA selection using the CRISPRdirect webpage.
Note: One limitation is to avoid presence of an AarI restriction site on your protospacer sequence. In addition, target sequence can be selected from both DNA strands and should be devoid of TTTTs. The distance between the two sgRNAs depends on your expected deletion regions. Small deletions (<100 bp) can be induced with relatively high frequencies and large one (up to 120 kb) with low frequencies (Ordon et al., 2016) in Arabidopsis. The deletion area at 5′ end or 3’ end of the non-coding region doesn’t show a frequency difference in this sequence from screen shot (NC_003071.7:8122119-8123076 Arabidopsis thaliana chromosome 2 sequence). The 20 nt-protospacer sequence does not have to start with a G, because our linearized plasmids retain a G overhang at the 3′ end of the U6 promoter that serves as the first G preferred for initiating transcription at the U6 promoter. - Open the CRISPRdirect webpage (http://crispr.dbcls.jp/) and paste a target genomic sequence into text field in a FASTA format or a plain nucleotide sequence up to 10 kb.
- Amplify the middle border from the templet pHEE2E-TRI (Figure 2)
- Order forward and reverse primers from idtDNA (https://www.idtdna.com/) and dilute them to a final concentration of 10 μM.
Dual-sgRNA1¬_F: 5′-CACCTGCATACATTGN20GTTTTAGAGCTAGAAATAGC-3′
Dual-sgRNA2¬_R: 5′-CACCTGCATACAAACN20CAATCTCTTAGTCGACTCTAC-3′
AarI: CACCTGC
20 nt-protospacer 1 sequence: N20
20 nt-protospacer 2 reverse complement sequence: N20
Overhang1 (ATTG) and overhang2 (AAAC) in yellow
Template specific forward sequence in orange
Template specific reverse sequence in green
Note: Here we take the first and the last gRNA designs in the screen shot as an example:
protospacer 1: TAAACGGGACGGCGACTCAC
protospacer 2: CAGAATCGTCAGATCTGAAG
protospacer 2 reverse complement sequence: CTTCAGATCTGACGATTCTG - PCR amplification with the following setup:
PCR program:5x HF buffer 5 µl dNTP (10 mM) 1 µl Dual-sgRNA1_F (10 µM) 1.25 µl Dual-sgRNA2_R (10 µM) 1.25 µl pHEE2E-TRI (100 ng/µl) 1 µl Phusion 0.25 µl Water 15.25 µl total 25 µl 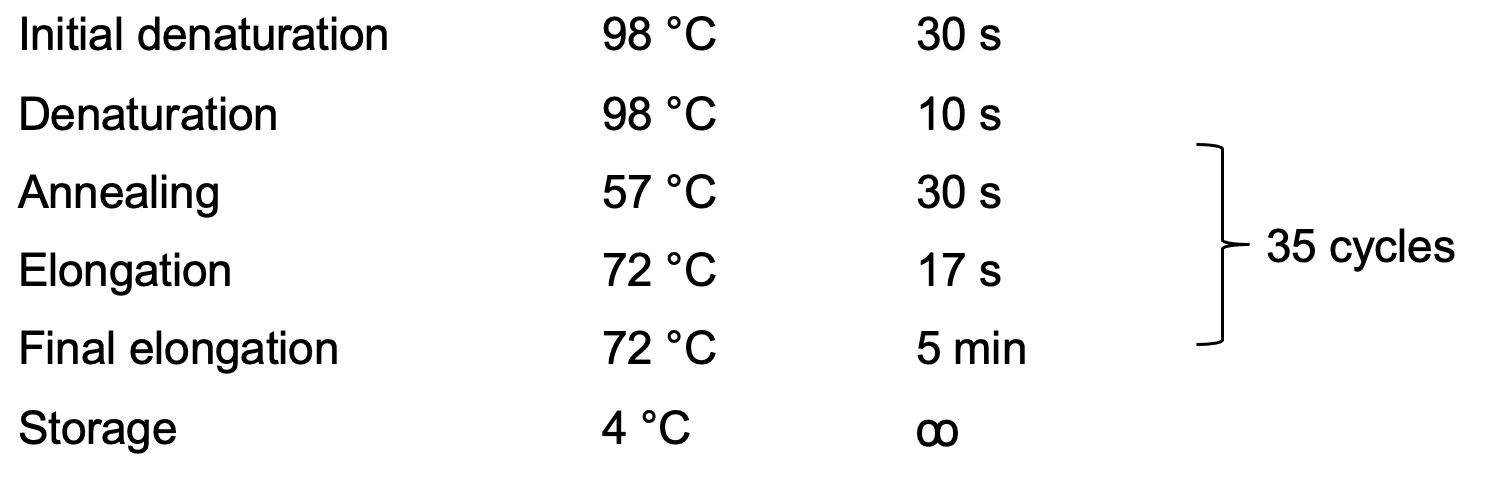
- Run 5 µl PCR product on an agarose gel to check a band with the expected size 620 bp and clean up the rest PCR product using Wizard® SV Gel and PCR Clean-Up System.
- Order forward and reverse primers from idtDNA (https://www.idtdna.com/) and dilute them to a final concentration of 10 μM.
- Design two gRNAs to target the same gene of interest (GOI) with CRISPRdirect (Naito et al., 2015) (Figure 2).
- Ligate the middle border into pKIR1.1 (Figure 2)
- Digest and phosphorylate PCR product (the middle border)
- Digest the purified PCR product with AarI for 6 hrs at 37 °C.
Note: 50x oligo contains the AarI recognition sequence for stimulating AarI activity (Grigaitė et al., 2002). Both 10x Aarl buffer and 50x oligo are supplied with AarI enzyme from Thermo Fisher.10x Aarl buffer 5.0 µl Middle border X µl (=1.5 µg) AarI 1.5 µl 50x oligo 1.0 µl Water 42.4 – X µl Total 50 µl - Phosphorylate the digestion mixture for 30 min at 37 °C in the following step, then clean up the mixture using Wizard® SV Gel and PCR Clean-Up System.
Digestion mixture 25 µl 10x T4 ligase buffer 2.83 µl T4 Polynucleotide Kinase 0.5 µl
- Digest the purified PCR product with AarI for 6 hrs at 37 °C.
- Digest and dephosphorylate pKIR1.1
- Digest pKIR1.1 with AarI for 6 h at 37 °C.
10x Aarl buffer 5.0 µl pKIR1.1 X µl (= 1.5 µg) AarI 1.5 µl 50x oligo 1.0 µl Water 42.4 – X µl Total 50 µl - Dephosphorylate the pKIR1.1 digestion mixture by 1 μl of phosphatase (CIP) for 30 min at 37 °C and for 10 min at 80 °C.
Digestion mixture 50 µl CutSmart® Buffer (10x) 5.67 µl CIP 1 µl - Perform agarose gel electrophoresis with 5 µl of digestion mixture to test the digest. The expected size of digested pKIR1.1 is around 18.5 kb. Clean up the rest digestion mixture using Wizard® SV Gel and PCR Clean-Up System.
- Digest pKIR1.1 with AarI for 6 h at 37 °C.
- Ligate the digested middle border with the linearized pKIR1.1 at 16 °C for 30 min to generate the final vector pKIR1.1-dual-sgRNA:
Note: Here we used vector: insert molar ratio 1:3.AarI-digested pKIR1.1 vector X µl (50 ng) AarI-digested middle border X µl (5 ng) 10x T4 ligase buffer 1.0 µl Water X µl T4 Ligase 1.0 µl Total 10.0 µl - Transform E. coli competent cells using 10 µl of ligation product and spread the transformed cells on LB agar plates with 100 mg/L Spectinomycin, then incubate the selection plates overnight at 37 °C (transformation protocol is provided by Clontech: http://www.takara.co.kr/file/manual/pdf/tr_PT5055-2.pdf).
- Pick 8 colonies to verify the correct insertion (Figure 4) by colony PCR using forward primer Mlo 1938 and your own protospacer reverse primer with the following setup:
Pick half of a single colony with a sterile pipette tip and swirl in the PCR reaction.10x HotMasterTM Taq Buffer with Mg2+ 2 µl dNTP (10 mM) 0.4 µl Mlo 1938 (10 µM) 1 µl Dual-sgRNA2_R (10 µM) 1 µl HotMasterTM Taq DNA Polymerase 0.1 µl Water 15.5 µl
Use the other half for inoculation.
PCR program:Total 20 µl 
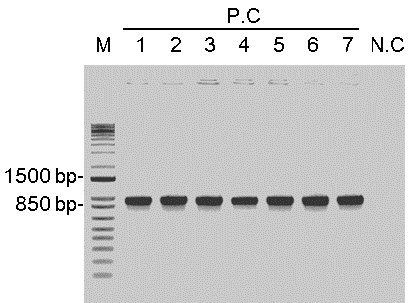
Figure 4. Electrophoresis of the colony-PCR products. The positive colonies show the correct insertion size with 868 bp. P.C: positive colony, N.C: negative colony. - Inoculate the positive colony in LB medium with 100 mg/L Spectinomycin and grow overnight.
- Extract the plasmid using the DNA plasmid kit.
- Sequence the plasmid and verify the sequence of the middle border insertion using primer Mlo 1938.
- Digest and phosphorylate PCR product (the middle border)
- Arabidopsis transformation with pKIR1.1-dual-sgRNA
- Transform competent A. tumefaciens GV3101cells with 1 μg of pKIR1.1-dual-sgRNA and spread the transformed cells on LB agar plates supplemented with 100 mg/L spectinomycin, 20 mg/L rifampicin, 25 mg/L gentamycin and 25 mg/L kanamycin (Höfgen and Willmitzer, 1988).
- After two days of growth at 30 °C, verify plasmid presence in at least three colonies by colony PCR as described before.
- Transform Arabidopsis plants using Agrobacterium-mediated T-DNA transfer with the floral dipping method (Clough and Bent, 1998).
- Detect dual sgRNA-directed deletion by Cas9
- Place the T1 seeds in a glass plate and pick up red fluorescent seeds with a slightly wet toothpick under a Stereo Fluorescence Microscope using an EL6000 external light source with magnification 10× and DsRED filter set (excitation, 546/10 nm; emission, 600/40 nm) (Figure 5).
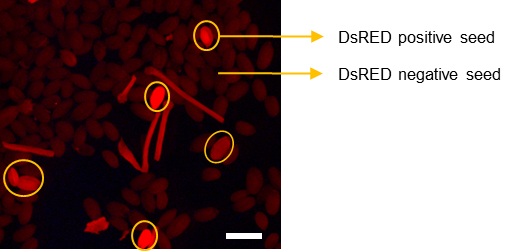
Figure 5. Visual screen for T1 seeds that harbor the CRISPR/Cas9 construct. pKIR1.1 vector can express the s.p. Cas9 protein driven by the RPS5A promoter, that allows high constitutive expression at all developmental stages including the germ cells. pKIR1.1 also contains an expression cassette of OLE1–TagRFP (red fluorescent protein) that exhibits red fluorescence in seeds. Therefore, the transformants containing CRISPR/Cas9 construct can be observed with red fluorescence in T1 seeds as indicated by circles. Scale bar = 1 mm.
Note: The transformation efficiency in this system is around 2.5%, which allows picking up 25 red T1 seeds within 10 min. - Genotype the targeted deletion of T1 plants observed with red fluorescence in seeds by isolating leaf genomic DNA (gDNA) and subsequent PCR (Edwards et al., 1991) using oligonucleotides flanking the deletion site (± 300-500 bp upstream and downstream) (Figure 6):
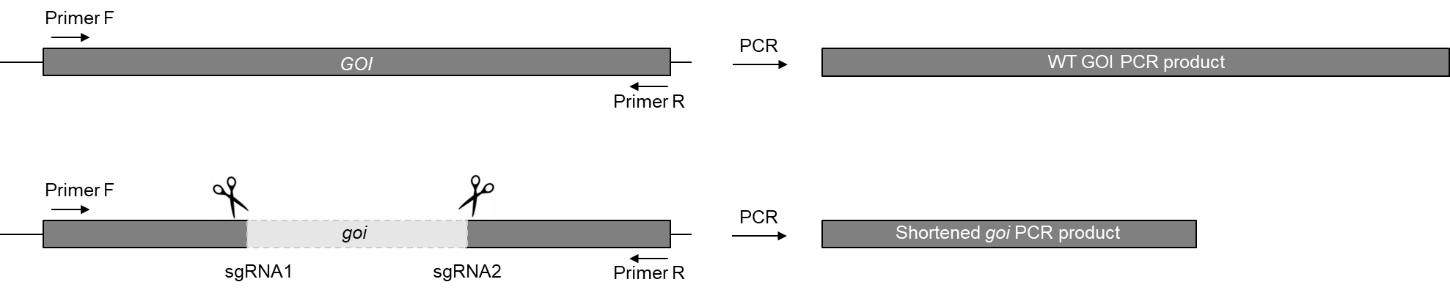
Figure 6. Genotyping the dual-sgRNA induced mutation on GOI. The large deletion of the GOI can be detected by PCR with a pair of primers flanking the deletion site (Primer F and Primer R). A shorter amplicon size can be detected in goi mutant compared to WT.10x HotMasterTM Taq Buffer with Mg2+ 2 µl dNTP (10 mM) 0.4 µl Primer F (10 µM) 1 µl Primer R (10 µM) 1 µl Leaf gDNA 1 µl (10 ng) HotMasterTM Taq DNA Polymerase 0.1 µl Water 14.5 µl Total 20 µl
PCR program:
- Place the T1 seeds in a glass plate and pick up red fluorescent seeds with a slightly wet toothpick under a Stereo Fluorescence Microscope using an EL6000 external light source with magnification 10× and DsRED filter set (excitation, 546/10 nm; emission, 600/40 nm) (Figure 5).
- Isolate Cas9-Free heritable mutation
- Harvest T2 seeds from individual genotyped T1 plants for the second round of observation under the Stereo Fluorescence Microscope to select the desired progeny. T2 seeds that do not contain the CRISPR/Cas9 construct can be identified since they lack red fluorescence and isolated.
- Genotype 10 Cas9-free T2 plants by PCR using oligonucleotides flanking the deletion site using phusion polymerase (Figure 7).
5x HF buffer 5 µl dNTP (10 mM 1 µl Primer F (10 µM) 1.25 µl Primer R (10 µM) 1.25 µl Leaf gDNA 1 µl (10 ng) Phusion 0.25 µl Water 15.25 µl Total 25 µl
PCR program:
- Separate the PCR products on an agarose gel and extract the shortened goi band with the correct size from the gel using the Wizard® SV Gel and PCR Clean-Up System.
- Sequence PCR product with Sanger sequencing and verify the sequence of the goi mutation. Usually heritable deletion events can be identified in plants lacking Cas9 activity after selection of seeds lacking the fluorescent reporter.
Data analysis
The sequencing data can be analyzed by alignment against the wild-type sequence using software SnapGene. Large deletion is generated in goi mutant of T2 (Figure 7).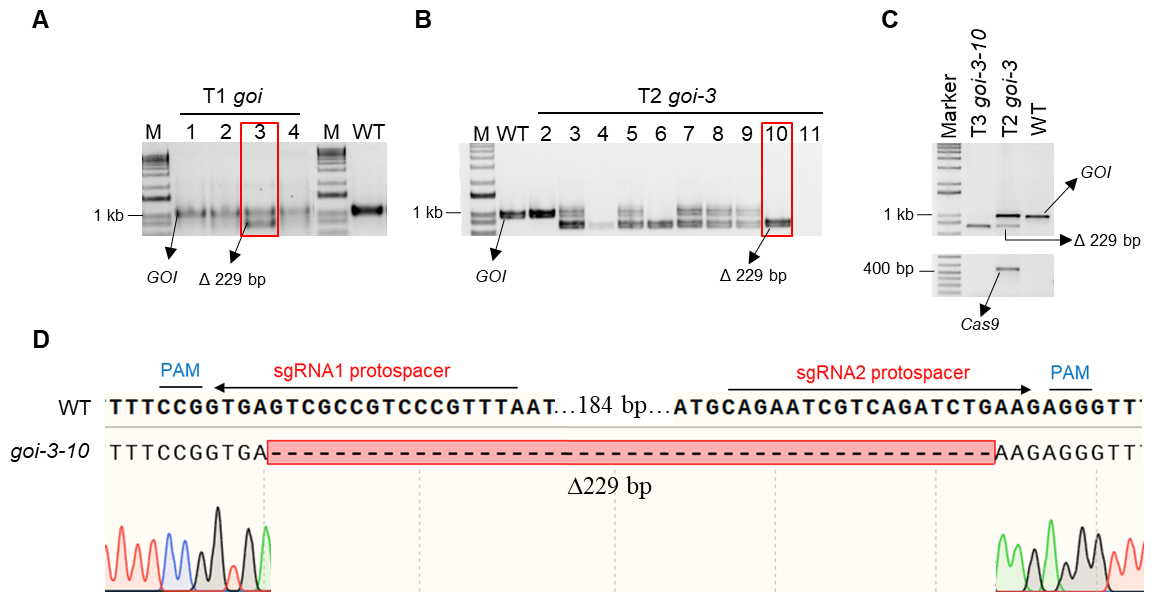
Figure 7. Representative chromatogram of PCR product from goi mutant. A. Genotyping of individual T1 plants that display red fluorescence. goi-3 in red box shows two bands, WT GOI amplicon and goi amplicon. This representative goi mutant with an expected 229 bp deletion is generated by dual-sgRNA targeting at the genomic region Chr2: 8122228-8122453. PCR product with 1 kb in WT represents no deletion of the GOI. B. In the T2 generation, seeds from T1 goi-3 are harvested and grown for screening Cas9-free plants. goi3-10 in red box (without red fluorescence in seeds) shows only the expected size of the goi amplicon. C. Bulked seedlings of goi-3-10 in the T3 generation are genotyped. Top, only the expected goi amplicon is amplified, suggesting a heritable homozygous deletion. Bottom, no amplification of the Cas9-specific band (406 bp) validates fluorescence-based counter selection of the Cas9 transgene. goi-3 in the T2 generation (no fluorescence selection) and WT are used as a positive control and negative control, respectively. D. Sequences of the representative deletion fragment of GOI (∆229 bp). The alignment is generated with SnapGene. PAM are in blue, sgRNA protospacers are in red, and deleted bases are replaced by a dash in goi3-10.
Note: It is mandatory to inspect the sequencing chromatograms carefully and check for the presence of overlapping peaks. Overlapping peaks are a sign of genetic heterogeneity in the sequenced sample.
Recipes
- LB-liquid medium
10 g/L Bacto-tryptone
5 g/L yeast extract
5 g/L NaCl - LB-agar medium
10 g/L Bacto tryptone
5 g/L Bacto yeast extract
5 g/L Sodium chloride (NaCl)
15 g/L Bacto agar for plates - ½ MS-medium plate
4 g/L Murashige & Skoog medium
0.5 g/L MES
10 g/L Sucrose
Adjust pH to 5.7 with KOH
10 g/L plant agar for plates
Acknowledgments
This work was funded by the Novo Nordisk Foundation NNF15OC0014202, Copenhagen Plant Science Centre Young Investigator Starting grant and the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme StG2017-757411 (S.M.).
Competing interests
No competing interests.
References
- Brake, T. O., Liu, Y. P., Centlivre, M., Von Eije, K. J., and Berkhout, B. (2008). Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol Therapy 16(3): 557-564.
- Cai, Y., Chen, L., Sun, S., Wu, C., Yao, W., Jiang, B., Han, T. and Hou, W. (2018). CRISPR/Cas9-mediated deletion of large genomic fragments in soybean. Int J Mol Sci 19(12).
- Canver, M. C., Bauer, D. E., Dass, A., Yien, Y. Y., Chung, J., Masuda, T., Maeda, T., Paw, B. H. and Orkin, S. H. (2014). Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem 289(31): 21312-21324.
- Char, S. N., Neelakandan, A. K., Nahampun, H., Frame, B., Main, M., Spalding, M. H., Becraft, P. W., Meyers, B. C., Walbot, V., Wang, K. and Yang, B. (2017). An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol J 15(2): 257-268.
- Chen, X., Xu, F., Zhu, C., Ji, J., Zhou, X., Feng, X. and Guang, S. (2014). Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci Rep 4: 7581.
- Clough, S. J. and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16(6): 735-743.
- Cui, X., Balcerzak, M., Schernthaner, J., Babic, V., Datla, R., Brauer, E. K., Labbe, N., Subramaniam, R. and Ouellet, T. (2019). An optimised CRISPR/Cas9 protocol to create targeted mutations in homoeologous genes and an efficient genotyping protocol to identify edited events in wheat. Plant Methods 15: 119.
- Do, P. T., Nguyen, C. X., Bui, H. T., Tran, L. T. N., Stacey, G., Gillman, J. D., Zhang, Z. J. and Stacey, M. G. (2019). Demonstration of highly efficient dual gRNA CRISPR/Cas9 editing of the homeologous GmFAD2-1A and GmFAD2-1B genes to yield a high oleic, low linoleic and alpha-linolenic acid phenotype in soybean. BMC Plant Biol 19(1): 311.
- Durr, J., Papareddy, R., Nakajima, K. and Gutierrez-Marcos, J. (2018). Highly efficient heritable targeted deletions of gene clusters and non-coding regulatory regions in Arabidopsis using CRISPR/Cas9. Sci Rep 8(1): 4443.
- Edwards, K., Johnstone, C., and Thompson, C. (1991). A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res 19(6): 1349.
- Fonseca, J. P., Bonny, A. R., Town, J. and El-Samad, H. (2020). Assembly of Genetic Circuits with the Mammalian ToolKit. Bio-Protocol 10(5): e3547.
- Ford, K., McDonald, D. and Mali, P. (2019). Functional Genomics via CRISPR-Cas. J Mol Biol 431(1): 48-65.
- Gao, X., Chen, J., Dai, X., Zhang, D. and Zhao, Y. (2016). An effective strategy for reliably isolating heritable and Cas9-Free Arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol 171(3): 1794-1800.
- Gowthaman, U., García-Pichardo, D., Jin, Y., Schwarz, I., and Marquardt, S. (2020). DNA processing in the context of non-coding transcription. doi: 10.20944/preprints202007.0744.v1.
- Grigaitė, R., Manelienė, Z., and Janulaitis, A. (2002). Aar I, a restriction endonuclease from Arthrobacter aurescens SS2-322, which recognizes the novel non-palindromic sequence 5’-CACCTGC (N) 4/8-3. Nucleic Acids Res 30(21): e123-e123.
- Hilton, I. B. and Gersbach, C. A. (2015). Enabling functional genomics with genome engineering. Genome Res 25(10): 1442-1455.
- Höfgen, R., and Willmitzer, L. (1988). Storage of competent cells for Agrobacterium transformation. Nucleic Acids Res 16(20): 9877.
- Hui, L., Zhao, M., He, J., Hu, Y., Huo, Y., Hao, H., Hao, Y., Zhu, W., Wang, Y. and Xu, M. (2019). A simple and reliable method for creating PCR-detectable mutants in Arabidopsis with the polycistronic tRNA–gRNA CRISPR/Cas9 system. Acta Physiologiae Plantarum 41(10): 170.
- Kistler, K. E., Vosshall, L. B. and Matthews, B. J. (2015). Genome engineering with CRISPR-Cas9 in the mosquito Aedes aegypti. Cell Rep 11(1): 51-60.
- Liu, K., Sun, B., You, H., Tu, J. L., Yu, X., Zhao, P. and Xu, J. W. (2020). Dual sgRNA-directed gene deletion in basidiomycete Ganoderma lucidum using the CRISPR/Cas9 system. Microb Biotechnol 13(2): 386-396.
- Lovett, S. T. (2004). Encoded errors: mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol Microbio 52(5): 1243-1253.
- Ma, C. and Mitra, A. (2002). Intrinsic direct repeats generate consistent post-transcriptional gene silencing in tobacco. Plant J 31 (1): 37-49.
- Maruyama, D., Hamamura, Y., Takeuchi, H., Susaki, D., Nishimaki, M., Kurihara, D., Kasahara, R.D. and Higashiyama, T. (2013). Independent control by each female gamete prevents the attraction of multiple pollen tubes. Dev Cell 25(3): 317-23.
- Naito, Y., Hino, K., Bono, H. and Ui-Tei, K. (2015). CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31(7): 1120-1123.
- Ordon, J., Gantner, J., Kemna, J., Schwalgun, L., Reschke, M., Streubel, J., Boch, J. and Stuttmann, J. (2017). Generation of chromosomal deletions in dicotyledonous plants employing a user-friendly genome editing toolkit. Plant J 89:155-168.
- Pauwels, L., De Clercq, R., Goossens, J., Inigo, S., Williams, C., Ron, M., Britt, A. and Goossens, A. (2018). A Dual sgRNA approach for functional genomics in Arabidopsis thaliana. G3 (Bethesda) 8(8): 2603-2615.
- Peterson, B. A., Haak, D. C., Nishimura, M. T., Teixeira, P. J., James, S. R., Dangl, J. L. and Nimchuk, Z. L. (2016). Genome-wide assessment of efficiency and specificity in CRISPR/Cas9 mediated multiple site targeting in Arabidopsis. PLoS One 11(9): e0162169.
- Schuster, M., Trippel, C., Happel, P., Lanver, D., Reissmann, S. And Kahmann, R. (2018). Single and multiplexed gene editing in Ustilago maydis using CRISPR-Cas9. Bio-Protocol 8(14): e2928.
- Song, Y., Yuan, L., Wang, Y., Chen, M., Deng, J., Lv, Q., Sui, T., Li, Z. and Lai, L. (2016). Efficient dual sgRNA-directed large gene deletion in rabbit with CRISPR/Cas9 system. Cell Mol Life Sci 73(15): 2959-2968.
- Tsutsui, H. and Higashiyama, T. (2017). pKAMA-ITACHI vectors for highly efficient CRISPR/Cas9-mediated gene knockout in Arabidopsis thaliana. Plant Cell Physiol 58(1): 46-56.
- Wang, H., Yang, H., Shivalila, C. S., Dawlaty, M. M., Cheng, A. W., Zhang, F. and Jaenisch, R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4): 910-918.
- Wang, Z. P., Xing, H. L., Dong, L., Zhang, H. Y., Han, C. Y., Wang, X. C. and Chen, Q. J. (2015). Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol 16: 144.
- Wu, R., Lucke, M., Jang, Y. T., Zhu, W., Symeonidi, E., Wang, C., Fitz, J., Xi, W., Schwab, R. and Weigel, D. (2018). An efficient CRISPR vector toolbox for engineering large deletions in Arabidopsis thaliana. Plant Methods 14: 65.
- Xiao, A., Wang, Z., Hu, Y., Wu, Y., Luo, Z., Yang, Z., Zu, Y., Li, W., Huang, P., Tong, X., Zhu, Z., Lin, S. and Zhang, B. (2013). Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res 41(14): e141.
- Zhang, Z., Mao, Y., Ha, S., Liu, W., Botella, J. R. and Zhu, J. K. (2016). A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep 35(7): 1519-1533.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jin, Y. and Marquardt, S. (2020). Dual sgRNA-based Targeted Deletion of Large Genomic Regions and Isolation of Heritable Cas9-free Mutants in Arabidopsis. Bio-protocol 10(20): e3796. DOI: 10.21769/BioProtoc.3796.
Category
Plant Science > Plant molecular biology > DNA > Mutagenesis
Plant Science > Plant molecular biology > Genetic analysis
Molecular Biology > DNA > Chromosome engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.