- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Understanding Docking Complexes of Macromolecules Using HADDOCK: The Synergy between Experimental Data and Computations

Published: Vol 10, Iss 20, Oct 20, 2020 DOI: 10.21769/BioProtoc.3793 Views: 7465
Reviewed by: Vivien Jane Coulson-ThomasReynier SuardiazPrashanth N Suravajhala
Original research article
The authors used this protocol in:

Jun 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
This protocol illustrates the modelling of a protein-peptide complex using the synergic combination of in silico analysis and experimental results. To this end, we use the integrative modelling software HADDOCK, which possesses the powerful ability to incorporate experimental data, such as NMR Chemical Shift Perturbations and biochemical protein-peptide interaction data, as restraints to guide the docking process. Based on the modelling results, a rational mutagenesis approach is used to validate the generated models. The experimental results allow to select a final structural model best representing the bona fide protein-peptide complex. The described protocol can also be applied to model protein-protein complexes. There is no size limit for the macromolecular complexes that can be characterized by HADDOCK as long as the 3D structures of the individual components are available.
Keywords: HADDOCKBackground
Proteins function in most cases not as single molecules, but rather as part of networks by interacting with a variety of other biomolecules. Therefore, the description at atomic level of the macromolecular complexes they form is an essential milestone in the understanding of molecular mechanisms at the basis of physiological and/or pathophysiological processes, and, most importantly, for the rational design of pharmacologically relevant drugs.
Here, we present a computational protocol to predict the three dimensional (3D) structure of a protein-peptide complex, whose quality is comparable to that of structures derived from conventional experimental methods such as X-ray, NMR and recently cryo-EM.
The described protocol may overcome several limitations of the experimental methods, such as, for instance, low binding affinities of the protein partners. Indeed, many macromolecular interactions are often weak at physiological conditions, as they mediate transient/highly dynamic processes. There may be also intrinsic limits of the protein sample, which preclude the use of one or more experimental methods.
In this context, computational approaches offer valuable alternatives, opening the possibility to describe, at atomic level, physiologically relevant macromolecular complexes (Saponaro et al., 2018) and concomitant conformational rearrangements induced by the interaction (Weißgraeber et al., 2017, Groß et al., 2018; Porro et al., 2019), otherwise elusive to the conventional structural biology methods.
Among the docking software used for modelling macromolecular complexes, HADDOCK, an integrative modelling platform, possesses the powerful advantage of being able to implement experimental data directly into the docking process to guide the computations (Dominguez et al., 2003). Indeed, HADDOCK is specifically designed as a data-driven docking program, although it has two ab-initio docking modes for cases when no information is available.
Docking calculations in HADDOCK are driven by ambiguous interaction restraints (AIRs). Those are derived from experimental and/or bioinformatics data and are integrated into the docking calculations in order to restrain the conformational search space (Dominguez et al., 2003) and satisfy the a priori information about the protein-protein interaction. AIRs are defined through a list of residues that fall under two categories: Active and passive. Active residues are defined as the solvent exposed residues directly involved in the interaction between the two proteins (typically defined based on the available experimental data). Passive residues correspond to the solvent exposed ones, which are close to the active residues. Passive residues are introduced in order to account for the fact that often the experimental data are scarce and not fully covering the true binding interface. The main difference between those two types of residues is that active residues are “forced” to be at the interface, while “passive” residues can be at the interface, but are not penalized if not.
A flowchart highlighting the main steps of HADDOCK docking program is shown in Figure 1. The process consists of three successive stages, each of them possessing a specific aim:
1) Initial docking by rigid body energy minimization (it0)
In this initial docking stage, the interacting molecules are treated as rigid bodies. They are separated in space and randomly rotated for each docking trial. The docking step is a rigid body energy minimization during which AIRs, based on the experimental data, are included in the energy function in order to restrict the sampling of the conformational space and guide the docking.
2) Semi-flexible simulated annealing in torsion angle space (it1)
This is a refinement stage during which flexibility is introduced stepwise, first along the side chains and then along both side chains and backbone of the interfacial residues. Those are automatically defined for each model based on their proximity to the partner molecule. The flexible refinement protocol is based on simulated annealing using torsion angles molecular dynamics.
3) Final refinement in explicit solvent (water)
In this final refinement stage the complexes are typically solvated in a layer of water and subjected to a short restrained molecular dynamic in Cartesian space. The final solutions are clustered and the resulting clusters are ranked based on the average score of their top 4 members (4 is the minimum number of models required to define a cluster).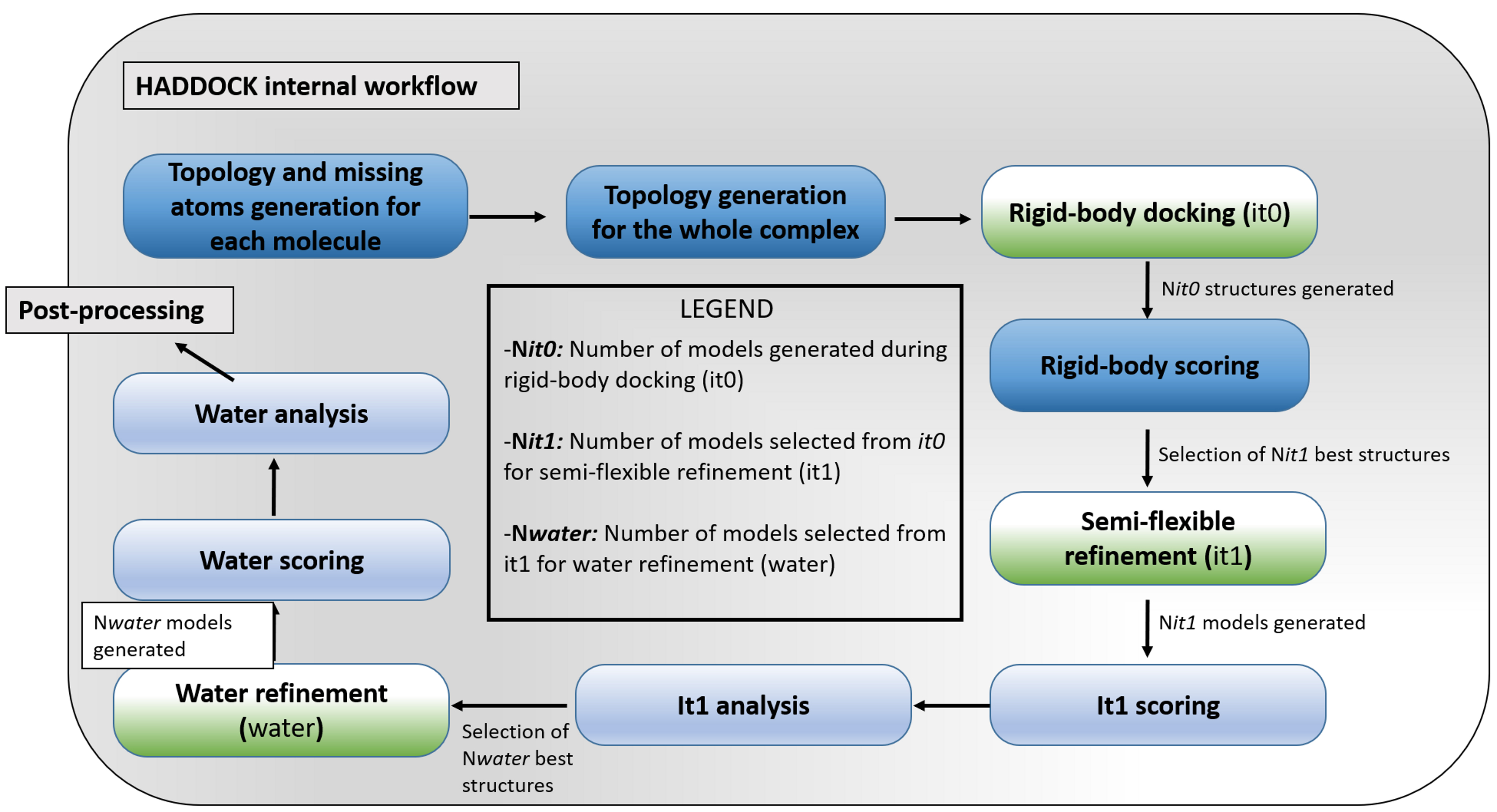
Figure 1. Flowchart of the HADDOCK protocol
HADDOCK, by default, randomly discards a percentage (50%) of the AIRs for each docking trial to account for possible false positives in the data. The percentage of discarded AIRs can be turned off, or even increased. The latter option is typically performed in the case AIRs derived from bioinformatics interface prediction data.
This protocol aims at providing a detailed description of the procedure for generating a 3D model structure of a protein-peptide complex using HADDOCK guided by experimental data. We illustrate the protocol by modelling the structure of the complex formed by the Cyclic Nucleotide Binding Domain (CNBD) of the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels bound to the TRIP8bnano peptide. This peptide represents the minimal portion of the brain protein TRIP8b, a regulative subunit of HCN channels, that binds to HCN CNBD (Saponaro et al., 2018). Solution NMR data are used to guide the modelling and the resulting models are then validated following rational mutagenesis/biochemical assays.
Equipment
- A computer with internet access and a web browser (preferably Chrome, Safari or FireFox)
- A visualization program such as UCSF Chimera, or PyMOL
Software
- HADDOCK 2.2
HADDOCK 2.2 (High Ambiguity Driven protein-protein DOCKing) is freely available to non-profit users via its web interface here (van Zundert et al., 2016).
Access does however require registration, which is free for non-profit users. - Program for protein solvent accessibility calculation
The NACCESS program is freely available here for researchers at academic and non-profit-making institutions. NACCESS is used to identify solvent accessible residues. It is worth noting that Naccess program requires Linux/Unix systems. A free alternative to NACCESS is freeSASA, freely available here.
Data requirements
- 3D coordinates of the two components of the protein complex, preferably in the bound conformation, provided in PDB or mmCIF format.
Note: The 3D coordinates in PDB format of HCN2 CNBD and TRIP8bnano are available from the following link: (LINK TO THE PDB). Those structures were solved by solution NMR as described in Saponaro et al., 2018. For docking purposes, the unfolded regions of both CNBD and TRIP8bnano structures, which are not involved in the binding, were removed. This allows to simplify the docking calculation and avoids spurious interactions between the disordered regions of CNBD and TRIP8bnano. In the case of TRIP8bnano the unfolded C-terminal tail was removed (I272-N275), whereas in the case of CNBD the unfolded N-terminal region was removed (E521-F533). - Definition of Active and Passive residues
Active residues can be identified by using a large variety of experimental data, including solution state NMR and biochemical protein-protein interaction data, as described in this protocol. Passive residues can be manually assigned by the operator, as performed in this protocol, or automatically defined by HADDOCK. Indeed, the program uses a 6.5 Å distance cutoff from the heavy atoms of the active residues to define the passive ones. Active and Passive residues are incorporated in the input page as comma-separated list of residue numbers (see Procedure).
Note: In the representative case described in this protocol, we define the following active and passive residues for CNBD and TRIP8bnano respectively:
CNBD:
Active residues: A546, N547, R650, E653, T654, I657, D658, R659, R662, I663, K665.
Passive residues: P543, D549, P646, R649, G664 and K666. Manually defined by the operator.
The above residues are defined based on the NMR Chemical Shift Perturbations (CSP) analysis. CSP highlights the residues of the interacting protein whose HN NMR signal features a significant chemical shift perturbation upon interaction with the peptide. A necessary prerequisite to conduct CSP is the availability of the assignment of the NMR backbone signals of the protein of interest.
The CSP (ΔHN) is given by the following equation:ΔHN={((HNfree-HNbound)2+(((Nfree-Nbound))⁄5)2)⁄2)}1⁄2where HNfree, Nfree and HNbound, Nbound, are the chemical shift of amide protons (HN) and amide nitrogen (N) in the peptide-free and bound state respectively. In order to select a meaningful set of residues involved in the binding to the peptide, a cutoff which corresponds to the average CSP value plus one standard deviation should be selected.
While more residues might be affected by the binding, we assume that only solvent-expose residues will form contacts. Accordingly, the residues identified by CSP need to be filtered based on their solvent accessibility (either main chain or side chain relative accessibility should be typically > 40-50%). Solvent accessibility can be calculated with the program NACCESS. Passive residues are defined as the residues close in space to active residues and with at least 50% of solvent accessibility. The same procedure here reported is also valid for protein-protein docking calculations and can be applied for both interacting proteins.
TRIP8bnano:
Active residues: we initially define as active residues a solvent exposed (≥ 50% of solvent accessibility) stretch of four residues (E239, E240, E241, F242, E243) previously identified as crucial for the binding to the CNBD (Santoro et al., 2011).
Passive residues: Since TRIP8bnano is a small peptide (40 amino acids), all other solvent accessible (≥ 50% of relative solvent accessibility) residues of the peptide are defined as passive. - Experimental data (rational mutagenesis and protein-protein interaction assay). Experimental data can be used to validate the HADDOCK output.
Procedure
Submitting a HADDOCK calculation:
- HADDOCK calculation can be launched from the web-sever available at: https://haddock.science.uu.nl/services/HADDOCK2.2.
Note: In the representative case described in this protocol, the “Guru interface” is used to run HADDOCK since it allows to specify/modify more parameters to fine tune the docking settings. Access to this interface does require “guru” level, which can be requested by users in their own registration page. - Fill in the HADDOCK input page (Figure 2):
- Provide a name for the job.
- Provide the input data, i.e., the 3D coordinates and the lists of the active and passive residues as a comma-separated list of residue numbers for each components of the complex.
- It is possible also to personalize the docking run by modifying several default parameters, if needed:
- Histidine protonation state for each component.
- Semi-Flexible and Fully Flexible segments.
- Additional restraints such as, for example, distance restraints, dihedral and hydrogen bonds restraints, noncrystallographic symmetry restraints, residual dipolar coupling.
- Sampling parameters.
- Parameters for clustering.
- Energy and interaction parameters.
- Scoring parameters.
- Advanced sampling parameters.
- Solvated docking parameters.
- Analysis parameters.
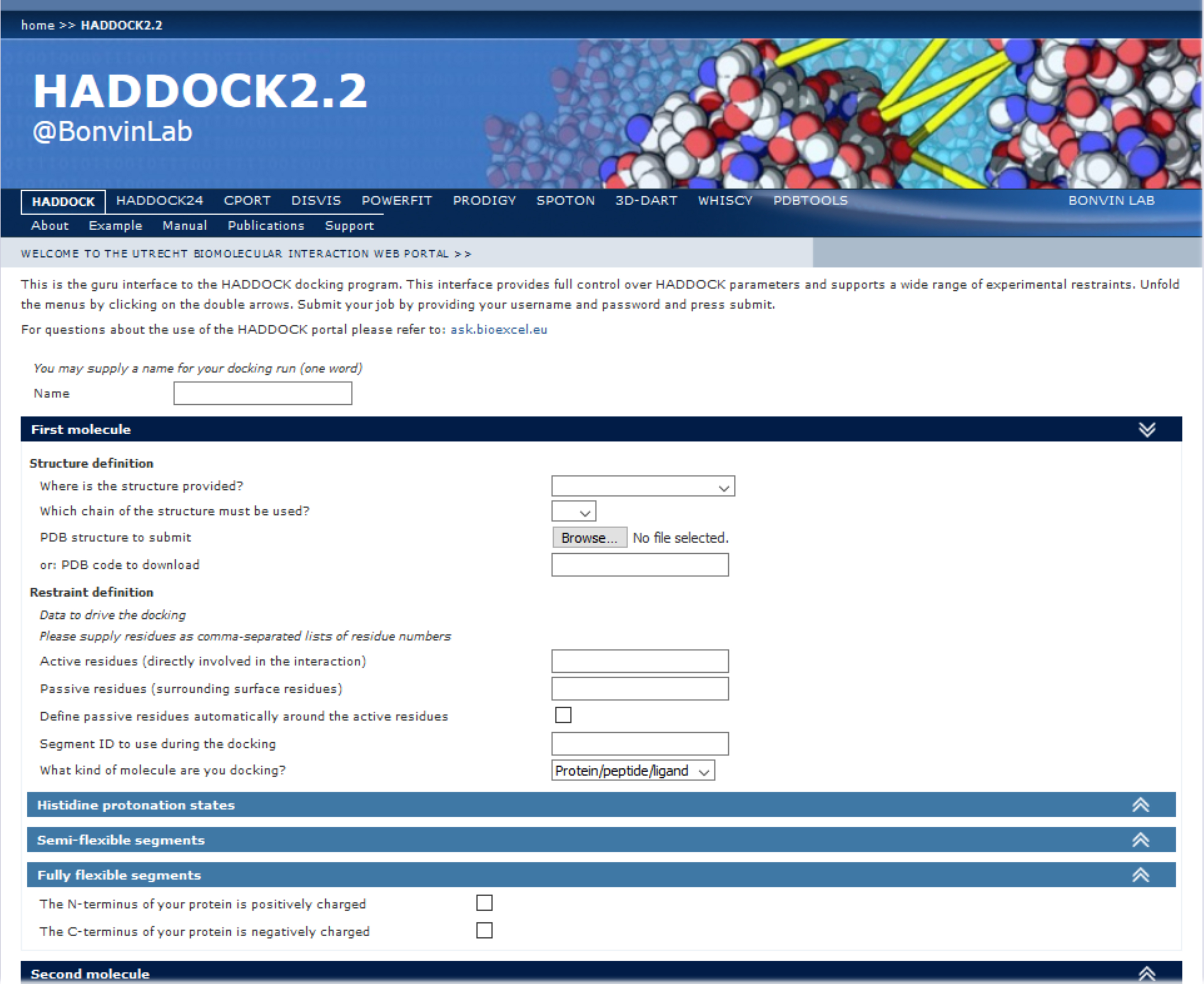
Figure 2. Example view of an input page of the HADDOCK2.2 web-server
Note: A detailed description of the various parameters of the HADDOCK scoring function and of the details about the HADDOCK molecular docking process can be found in the online manual here. - Histidine protonation state for each component.
- By default, HADDOCK generates 1,000 models in the initial rigid body docking. This parameter can be changed by unfolding the tab named “Sampling parameters” (Figure 3).
Note: In the specific case of CNBD-TRIP8bnano complex the number of structures is increased by a factor of five. - The default number of structures further subjected to the semi-flexible simulated annealing (it1) and final refinement in water (itw) is 200.
Note: In the specific case of CNBD-TRIP8bnano complex this parameter is increased by a factor of two (Figure 3, “Sampling parameters” tab). - The final water-refined models are clustered and ranked based on HADDOCK score value. Clustering is performed based on pairwise Root Mean Square Deviations (RMSD, the default value is set to a 7.5Å cutoff) and a minimum number of members to define a cluster (default value is set to four) (Figure 3, “Parameter for clustering” tab). The clusters are then ranked based on the average HADDOCK score of their top four members. The RMSD cut-off value is crucial for a proper clustering of the structures and different clustering cutoffs might be tried.
Note: In the specific case of the CNBD-TRIP8bnano complex the RMSD cut-off was kept to the default value.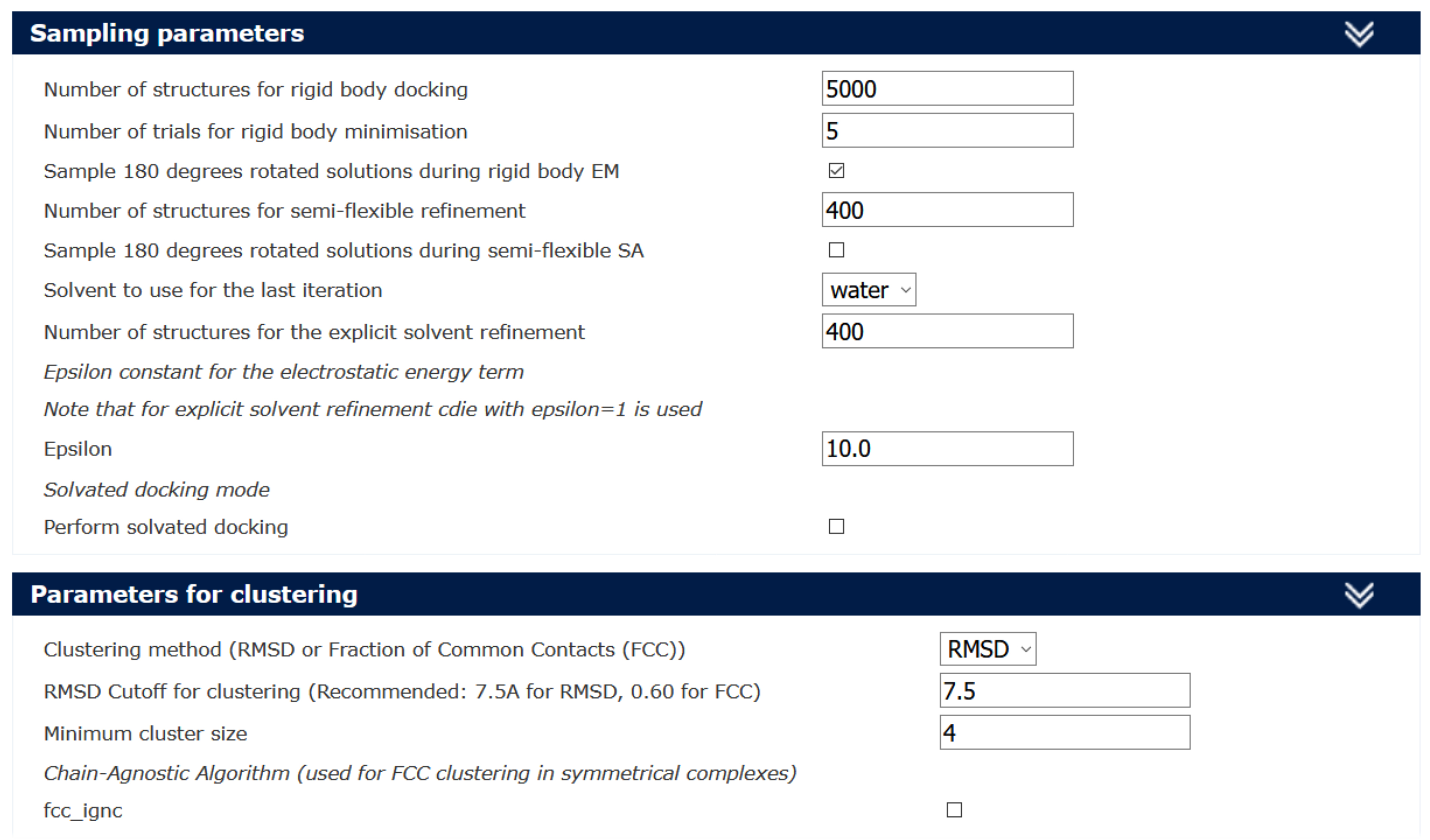
Figure 3. The sampling parameters and the parameters for clustering can be changed by the user in the Guru Interface level - It is recommended to set the “Advanced sampling parameters” (Figure 4) according to the specific protein-peptide docking calculation following the settings reported in Trellet et al., 2013.
Note: In the representative case described in this protocol, we have quadrupled the default number of molecular dynamics integrations steps for the rigid body dynamics at high temperature and the subsequent cooling stages with flexibility at the interface (from 500/500/1,000/1,000 to 2,000/2,000/4,000/4,000) (Figure 4, “Advanced sampling parameters” tab). The decision to increase the values is based on the fact that for one of the two component of the complex, TRIP8bnano, the number of the active residues was significantly limited (4 residues out of 40 forming TRIP8bnano). This can affect the sampling of the conformational space during the rigid body docking calculation phase. Furthermore, because CNBD contains a disordered C-terminal tail, which is necessary for the binding (Saponaro et al., 2014), the number of MD steps is also increased to allow the protein to adapt its conformation during the flexible refinement stage.
Figure 4. Advanced sampling parameters used for CNBD-TRIP8bnano complex
- Provide a name for the job.
- Provide both username and password to submit a job.
Data analysis
- Submission data
Once all data have been uploaded and the run submitted, the web-server offers the option to download a parameter file and provides a link to the results page. This link is also e-mailed to the user. - Analysis of the docking results
- The result page of a successful docking run presents the statistics for the top10 clusters ranked by HADDOCK. Those are ranked based on the average HADDOCK score of the top4 member of each cluster, which is a weighted sum of electrostatics, van der Waals, restraints energy, buried surface area and empirical desolvation energy terms (Fernandez-Recio et al., 2004). The page indicates how many water-refined models could be clustered and displays at the bottom a graphical overview of the results.
Note: For the specific case of CNBD-TRIP8bnano complex calculation, seventeen clusters are obtained and ranked according to their HADDOCK score. - For each cluster, detailed statistics are displayed, representing the average values calculated over the top four best-scoring structures within each cluster. The clusters require to be also manually screened for the relative orientation of the two components of the complex on the basis of available experimental data.
Note: In the specific case of CNBD-TRIP8bnano, Double Electron-Electron Resonance (DEER) data are available (DeBerg et al., 2015). They provide nanometer-scale distances between unpaired electrons in paramagnetic systems or tagged with paramagnetic probes. DEER data were used to screen the relative orientation of TRIP8bnano and CNBD and select two out of the seventeen clusters obtained (Figure 5). The two selected clusters lay within the 30% top ranked clusters. Table 1 summarizes the HADDOCK statistics of the two selected clusters.
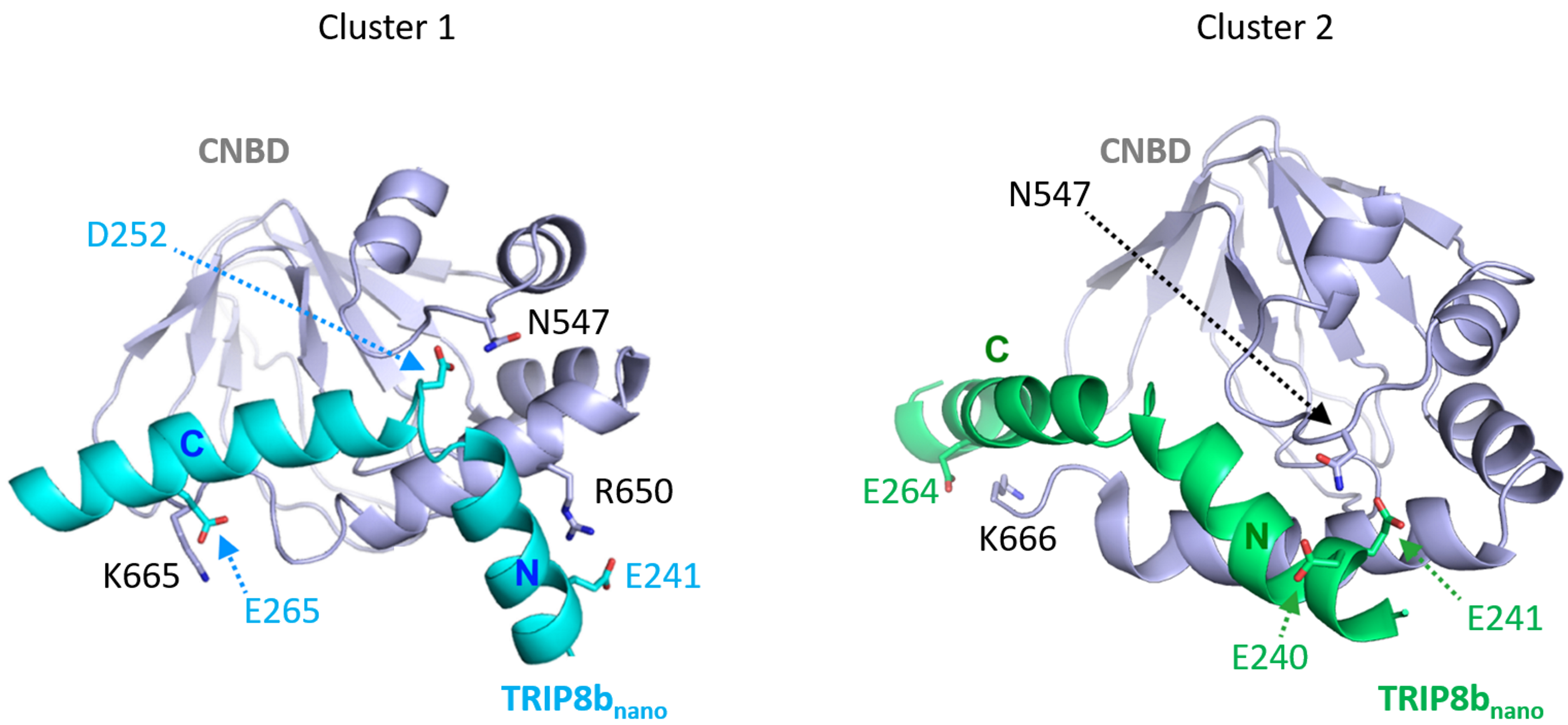
Figure 5. Representative poses of cluster families obtained from the first docking calculation. CNBD is colored in gray; TRIP8bnano is colored in light blue in cluster 1 and in green in cluster 2. Residues forming cluster-specific contacts are shown and labeled (modified from Saponaro et al., 2018 with permission).
Table 1. HADDOCK Score and energetic values for the selected clusters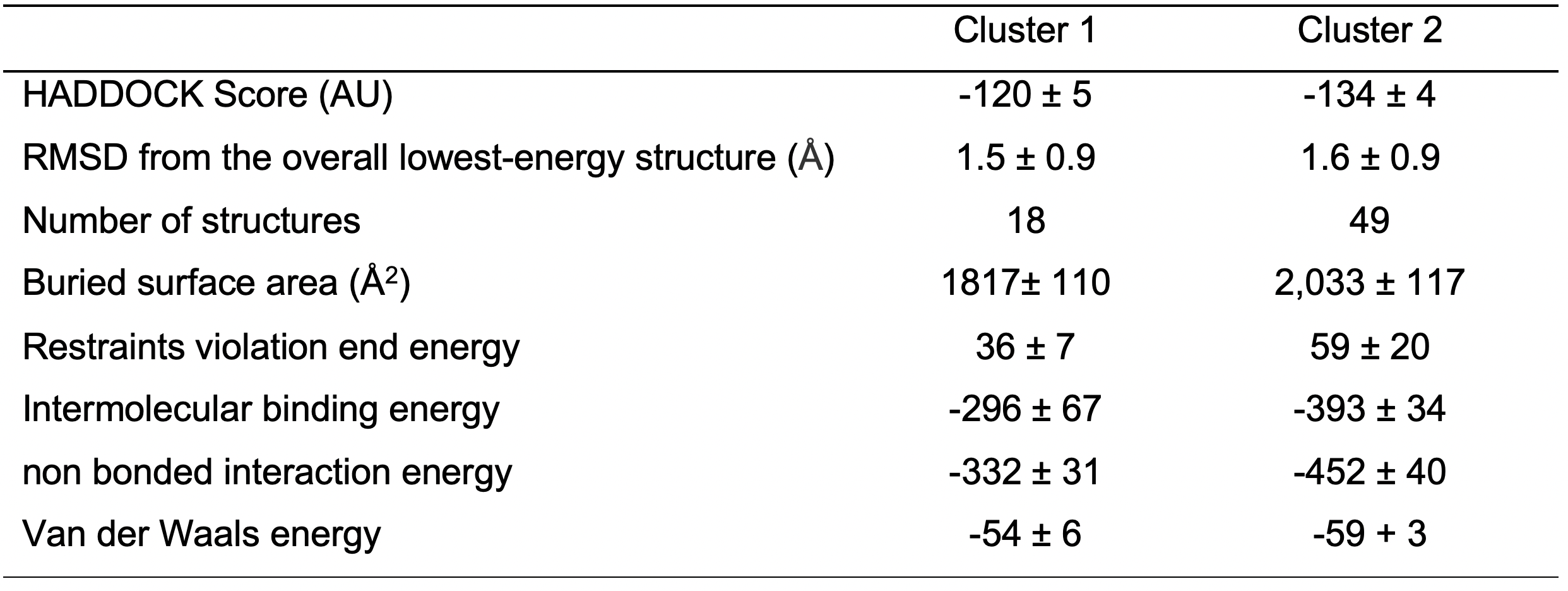
- The result page of a successful docking run presents the statistics for the top10 clusters ranked by HADDOCK. Those are ranked based on the average HADDOCK score of the top4 member of each cluster, which is a weighted sum of electrostatics, van der Waals, restraints energy, buried surface area and empirical desolvation energy terms (Fernandez-Recio et al., 2004). The page indicates how many water-refined models could be clustered and displays at the bottom a graphical overview of the results.
- Validation of the generated models by mutagenesis
Rational mutagenesis studies, coupled with an appropriate protein-peptide assay, can be used to validate a cluster-specific contact and eventually perform a second round of modelling introducing as active those residues whose mutations abolish the interaction.
Note: This approach was used for determining the structural model of CNBD-TRIP8bnano complex.
In order to select residues for mutagenesis we examine the intermolecular contacts formed in each of the two selected clusters. This reveals that:- Residues E264 or E265 of TRIP8bnano are found to interact with residues K665 or K666 of CNBD in both clusters (see Figure 5).
- In cluster 1, TRIP8bnano is oriented in such a way that allows E239-E243 residues to interact with R650 of CNBD; whereas, in cluster 2, TRIP8bnano is oriented, in almost all the conformers of the cluster, in such a way that CNBD R650 cannot developed contacts with TRIP8bnano.
- Residues E264 or E265 of TRIP8bnano are found to interact with residues K665 or K666 of CNBD in both clusters (see Figure 5).
- Refining the model by a second round of docking
On the basis of the experimental observations described above, we performed a second molecular docking calculation including E264 and E265 as additional active residues for TRIP8bnano. The docking setup was similar to the first docking calculation described above.
From this second HADDOCK calculation fourteen clusters are obtained. Four of these are consistent with the DEER and mutagenesis data (Figure 6). In order to validate these models, we performed a second round of mutagenesis based again on identified unique contacts. In particular, in cluster 1 D252 of TRIP8bnano, located at the junction between helix N and helix C, contacts residue N547 of the CNBD, while this contact is absent in the other clusters.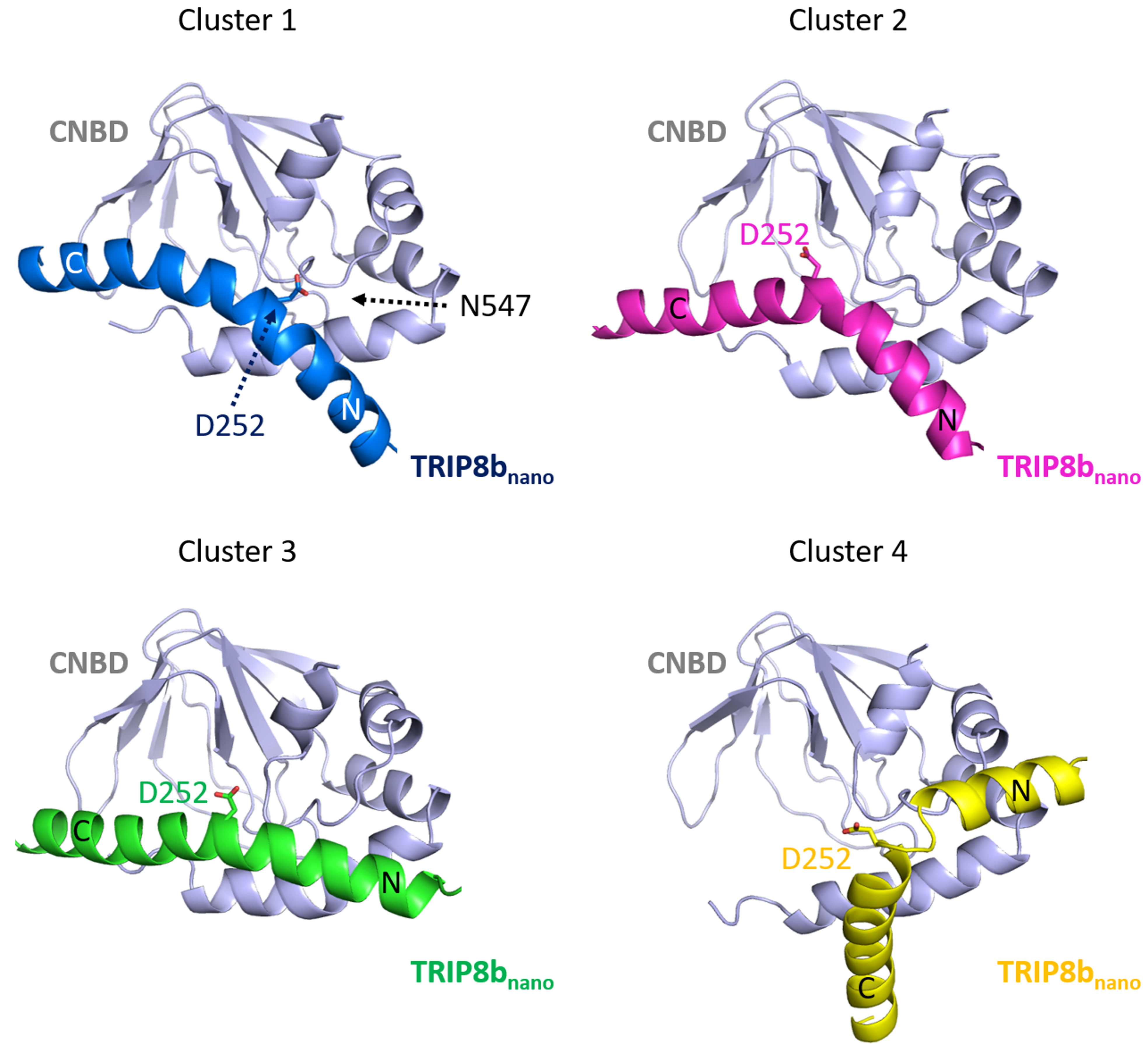
Figure 6. Representative poses of cluster families obtained from the second docking calculation. CNBD is colored in gray in all the four poses; TRIP8bnano is colored in blue in cluster 1, in magenta in cluster 2, in green in cluster 3 and in yellow in cluster 4. D252 of TRIP8bnano is shown in all the four poses; N547 of CNBD is shown in the pose representing cluster 1 as in this cluster N547 contacts D252. Helices N and C of TRIP8bnano are labeled.
N547 in CNBD was mutated into aspartate (N547D) to generate an electrostatic repulsion with the supposed partner residue D252, while the carboxyl group in D252 of TRIP8bnano was removed by mutating it into an asparagine (D252N). Both mutations reduced binding to TRIP8b (for details see Saponaro et al., 2018). These results suggest that Cluster 1 can be chosen as the bona fide model for the CNBD-TRIP8bnano complex.
It is worth noting that cluster 1 represents also the top-ranking cluster for energetic, scoring function and number of structures populating the cluster (Table 2). This is explained by the fact that the second docking calculation was further implemented with more ambiguous interaction restraints, which significantly improved HADDOCK results.
Table 2. HADDOCK Score and energetic values for the selected clusters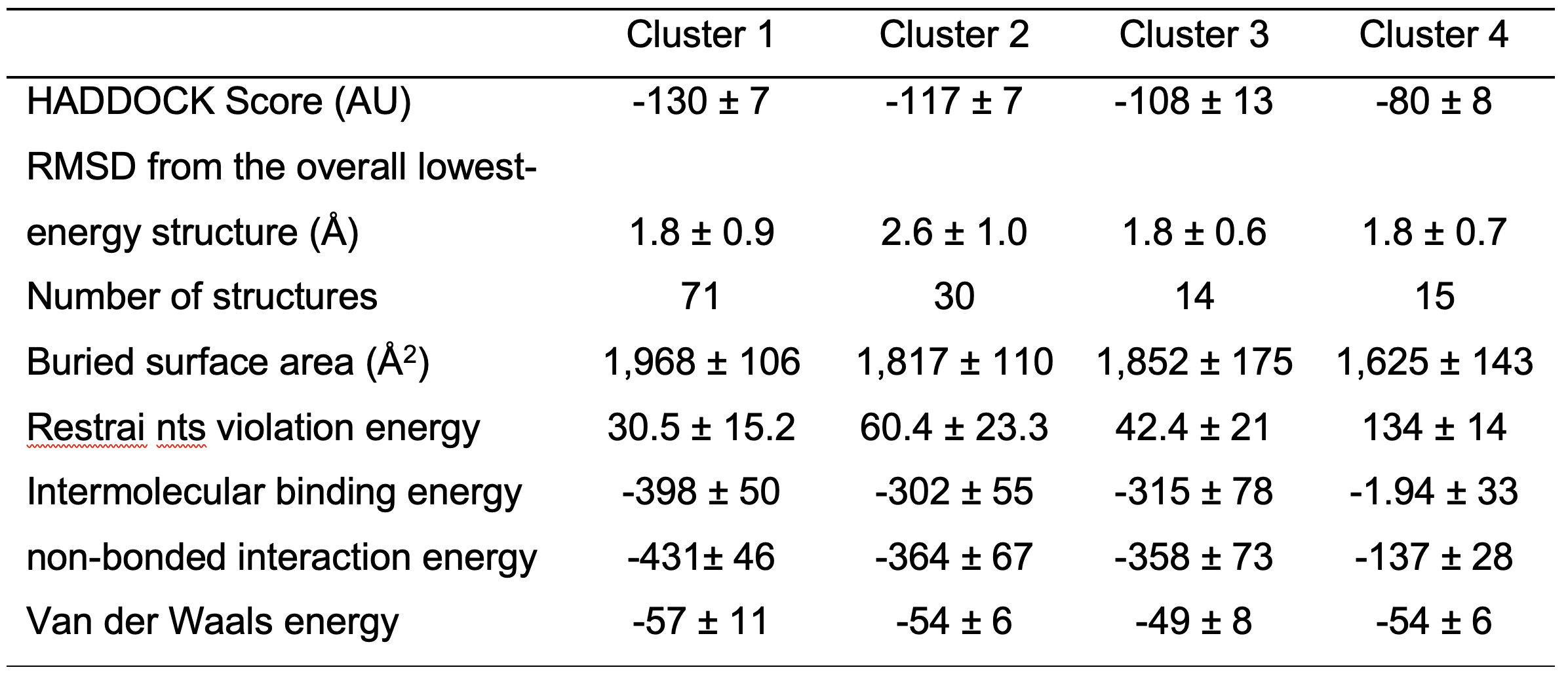
Notes
Tutorials and video describing HADDOCK program and its use for various scenarios can be found at http://www.bonvinlab.org/education.
Acknowledgments
This protocol was adapted from Saponaro et al. (2018). A.M.J.J.B. acknowledges financial support from the BioExcel CoE (www.bioexcel.eu), a project funded by the European Union Horizon 2020 program under grant agreements 675728 and 823820.
Competing interests
There are no conflicts of interest or competing interest.
References
- DeBerg, H. A., Bankston, J. R., Rosenbaum, J. C., Brzovic, P. S., Zagotta, W. N. and Stoll, S. (2015). Structural mechanism for the regulation of HCN ion channels by the accessory protein TRIP8b. Structure 23(4): 734-744.
- Dominguez, C., Boelens, R. and Bonvin, A. M. (2003). HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125(7): 1731-1737.
- Fernandez-Recio, J., Totrov, M. and Abagyan, R. (2004). Identification of protein-protein interaction sites from docking energy landscapes. J Mol Biol 335(3): 843-865.
- Groß, C., Saponaro, A., Santoro, B., Moroni, A., Thiel, G. and Hamacher, K. (2018). Mechanical transduction of cytoplasmic-to-transmembrane-domain movements in a hyperpolarization-activated cyclic nucleotide–gated cation channel. J Biol Chem 293(33): 12908-12918.
- Porro, A., Saponaro, A., Gasparri, F., Bauer, D., Gross, C., Pisoni, M., Abbandonato, G., Hamacher, K., Santoro, B., Thiel, G. and Moroni, A. (2019). The HCN domain couples voltage gating and cAMP response in hyperpolarization-activated cyclic nucleotide-gated channels. Elife 8: e49672.
- Santoro, B., Hu, L., Liu, H., Saponaro, A., Pian, P., Piskorowski, R. A., Moroni, A. and Siegelbaum, S. A. (2011). TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J Neurosci 31(11): 4074-4086.
- Saponaro, A. (2018). Isothermal titration calorimetry: a biophysical method to characterize the interaction between label-free biomolecules in solution. Bio-protocol 8(15): e2957.
- Saponaro, A., Cantini, F., Porro, A., Bucchi, A., DiFrancesco, D., Maione, V., Donadoni, C., Introini, B., Mesirca, P., Mangoni, M. E., Thiel, G., Banci, L., Santoro, B. and Moroni, A. (2018). A synthetic peptide that prevents cAMP regulation in mammalian hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Elife 7: 35753.
- Saponaro, A., Pauleta, S. R., Cantini, F., Matzapetakis, M., Hammann, C., Donadoni, C., Hu, L., Thiel, G., Banci, L., Santoro, B. and Moroni, A. (2014). Structural basis for the mutual antagonism of cAMP and TRIP8b in regulating HCN channel function. Proc Natl Acad Sci U S A 111(40): 14577-14582.
- Saponaro, A., Porro, A., Chaves-Sanjuan, A., Nardini, M., Rauh, O., Thiel, G. and Moroni, A. (2017). Fusicoccin activates KAT1 channels by stabilizing their interaction with 14-3-3 proteins. Plant Cell 29(10): 2570-2580.
- Trellet, M., Melquiond, A. S. and Bonvin, A. M. (2013). A unified conformational selection and induced fit approach to protein-peptide docking. PLoS One 8(3): e58769.
- van Zundert, G. C. P., Rodrigues, J. P. G. L. M., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., Melquiond, A. S. J., van Dijk, M., de Vries, S. J. and Bonvin, A. M. (2016). The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol 428(4):720-725.
- Weißgraeber, S, Saponaro, A., Thiel, G. and Hamacher, K. (2017). A reduced mechanical model for cAMP-modulated gating in HCN channels. Sci Rep 7:40168.
Article Information
Copyright
![]() Saponaro et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Saponaro et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Saponaro, A., Maione, V., Bonvin, A. M. J. J. and Cantini, F. (2020). Understanding Docking Complexes of Macromolecules Using HADDOCK: The Synergy between Experimental Data and Computations. Bio-protocol 10(20): e3793. DOI: 10.21769/BioProtoc.3793.
- Saponaro, A., Cantini, F., Porro, A., Bucchi, A., DiFrancesco, D., Maione, V., Donadoni, C., Introini, B., Mesirca, P., Mangoni, M. E., Thiel, G., Banci, L., Santoro, B. and Moroni, A. (2018). A synthetic peptide that prevents cAMP regulation in mammalian hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Elife 7: 35753.
Category
Molecular Biology > Protein > Protein-protein interaction
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.