- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analyzing (Re)Capping of mRNA Using Transcript Specific 5' End Sequencing

Published: Vol 10, Iss 20, Oct 20, 2020 DOI: 10.21769/BioProtoc.3791 Views: 4593
Reviewed by: Imre GáspárIndranil MalikRakesh Chatrikhi
Original research article
The authors used this protocol in:

Apr 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The 5′ cap is a ubiquitous feature of eukaryotic mRNAs. It is added in the nucleus onto newly synthesized pre-mRNA, and in the cytoplasm onto mRNAs after decapping or endonuclease cleavage. Cytoplasmic recapping can occur after loss of the cap at the native 5′ end, or downstream within the body of the mRNA. The identification and location of recapping events is key to understanding the functional consequences of this process. Here we present an approach that addresses this problem, using the Lexogen TeloPrime® cDNA synthesis kit to tag recapped 5′ ends. TeloPrime uses a proprietary DNA ligase to add a double stranded DNA oligonucleotide onto the 3′ end of cDNA while it is base paired with mRNA. Specificity for capped ends is obtained by the oligonucleotide having an unpaired C residue that base pairs weakly with m7G on the mRNA 5′ end. This is followed by PCR amplification of double-stranded cDNA using primers to the appended oligonucleotide and the mRNA of interest. The resulting products are gel purified and sequenced directly (if a single band) or cloned and sequenced. The sequence at the junction between the ligated oligonucleotide and the target mRNA provides the location of the cap on the corresponding transcript. This assay is applicable to all capped transcripts. It can be used with Sanger sequencing for small numbers of transcripts or adapted for use with Illumina library sequencing.
Keywords: 5′ CapBackground
The N7-methylguanosine cap is a distinguishing feature of all eukaryotic mRNAs. Proteins binding to the cap function all stages of the mRNA lifecycle, including nuclear processing, export, translation and mRNA decay. The 5′ cap is added co-transcriptionally to all mRNAs, and a number of genome wide techniques (e.g., Capped Analysis of Gene Expression, or CAGE) (Morioka et al., 2020) have been developed that use the identification of capped ends as a way to mark transcription initiation sites. In addition to marking transcription start sites approximately 25% of CAGE tags map downstream within spliced introns (Djebali et al., 2012). Evidence of the biochemical basis for this came from our lab’s identification in 2009 of a cytoplasmic complex capable of restoring the N7-methylguanosine cap onto transcripts with a 5′-monophosphate end, but not onto transcripts with a 5′-hydroxyl end (Otsuka et al., 2009). Cytoplasmic capping is catalyzed by a complex of enzymes including capping enzyme (RNGTT), cap methyltransferase (RNMT) and its activating subunit (RAM), and a kinase that converts the 5′-monophosphate ends of decapped transcripts to a 5′-diphosphate substrate for GMP addition (Trotman and Schoenberg, 2019). Our early work was based on use of a dominant negative form of capping enzyme that blocks cytoplasmic capping at the GMP addition step. Expression of this protein resulted in the appearance of a number of uncapped transcripts whose ends were mapped using 5’-RACE to the vicinity of downstream CAGE tags (Kiss et al., 2015; Berger et al., 2019). While encouraging, this approach has three major drawbacks; a) it requires separation of capped and uncapped RNAs, b) it assumes uncapped transcripts remain sufficiently stable to be detected, and c) it assumes uncapped ends detected in this manner have undergone limited additional exonucleolytic trimming.
These issues were resolved by the development of a new approach to inhibiting cytoplasmic capping that instead targets cap methylation (Del Valle Morales et al., 2020). Inhibition of cytoplasmic cap methylation results in the creation of recapped mRNAs with unmethylated G caps which are degraded by cap surveillance enzymes (Grudzien-Nogalska and Kiledjian, 2017). This makes it possible to identify cytoplasmic capping targets by quantitative changes in RNA-Seq data, thus eliminating assumptions about the stability and detectability of uncapped RNAs, the need to separate uncapped from capped RNAs, and concerns for exonucleolytic trimming. By simplifying the process of identifying cytoplasmic capping targets this approach makes it possible to study this process in any cells or tissues expressing the cytoplasmic cap methylation inhibitor. Finally, because these transcripts retain their caps, we paired this approach with a facile method for identifying capped ends that can be scaled from a few mRNAs to larger sequencing libraries.
Materials and Reagents
- 0.2 ml PCR tubes (e.g., BioExpress, GeneMate, catalog number: T-3225-1 )
- 1.7 ml plastic, sterile, RNase-free microcentrifuge tubes (e.g., BioExpress, GeneMate, catalog number: C-3262-1 )
- 10 cm or 15 cm culture dishes (e.g., Alkali Scientific, catalog numbers: TD0100 , TD0150)
- Sterile nitrile gloves
- 18 G needle
- Cell lifters (e.g., BioExpress, GeneMate, catalog number: T-2443-4 )
- Sterile, RNase-free tips for P10, P20, P100, and P1000 pipets
- CleanCap® mCherry mRNA (Trilink, catalog number: L-7203 )
- mCherry forward primer: ATGGTGAGCAAGGGCGAGGAG
- mCherry reverse primer: GCCACCCTTGGTCACCTTCAGC
- Gene specific primer
- U2OS cells (ATCC, catalog number: HTB-96 ) or HEK293 cells (ATCC, catalog number: CRL-1573 )
- RNase-free water (e.g., from a Millipore Synergy water purification system, 18.2 MΩ cm)
- TeloPrime Full-Length cDNA Amplification Kit V2 (Lexogen, catalog number: 013.24)
- RNA buffer (part of TeloPrime kit)
- DNA buffer (part of TeloPrime kit)
- Reverse transcription mix (RT) (part of TeloPrime kit)
- Reverse transcription primer (RTP) (part of TeloPrime kit)
- Enzyme mix 1 (E1) (part of TeloPrime kit)
- Ligation mix (LM) (part of TeloPrime kit)
- Enzyme mix 2 (E2) (part of TeloPrime kit)
- Second strand synthesis mix (SS) (part of TeloPrime kit)
- Enzyme mix 3 (E3) (part of TeloPrime kit)
- PCR forward primer (FP) (part of TeloPrime kit, 5’-TGGATTGATATGTAATACGACTCACTATAG)
- PCR reverse primer (RP) (part of TeloPrime kit)
- TeloPrime PCR mix (Telo PCR) (part of TeloPrime kit)
- Column binding buffer 1 (CB1) (part of TeloPrime kit)
- Column binding buffer 2 (CB2) (part of TeloPrime kit)
- Column wash buffer (CW) (part of TeloPrime kit)
- Purifications columns (part of TeloPrime kit)
- Collection tubes (part of TeloPrime kit)
- SensiFASTTM SYBR® No-ROX (Bioline, catalog number: BIO-98050 )
- SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S11494 )
- DNA size marker (e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: AM7778 )
- 2x DNA loading dye (e.g., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0641 )
- 40 U/μl RNaseOUT Recombinant Ribonuclease Inhibitor (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10777019 )
- RNA Clean and Concentrator-25 (Zymo Research, catalog number: R1017 )
- DNA Clean and Concentrator-5 (Zymo Research, catalog number: D4013 )
- pGEM®-T Easy Vector Systems (Promega, catalog number: A1360 )
- pGEM®-T Easy Vector (part of pGEM®-T Easy Vector Systems kit)
- T4 DNA Ligase (part of pGEM®-T Easy Vector Systems kit)
- 2X Rapid Ligation Buffer ( part of pGEM®-T Easy Vector Systems kit)
- StellarTM Competent Cells (Clontech, catalog number: 636766 )
- Bacterial culture tube (e.g., VWR, catalog number: 20170-579 )
- LB Agar Ampicillin-100 (Sigma-Aldrich, catalog number: L5667 )
- NucleoSpin® Plasmid DNA Purification Kit (Clontech, catalog number: 740588.50)
- McCoy’s 5A medium (for U2OS cells; Thermo Fisher Scientific, GibcoTM, catalog number: 16600082 )
- Dulbecco’s modified Eagle medium (DMEM; for HEK293 cells; Thermo Fisher Scientific, GibcoTM, catalog number: 21013024 )
- Fetal bovine serum (FBS; Atlanta Biologicals, catalog number: S10350 )
- Phosphate-buffered saline (PBS; e.g., Thermo Fisher Scientific, GibcoTM, catalog number: 10010049 )
- RNase-free 10x Tris/borate/EDTA (TBE) buffer (e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: 15581 )
- Ethanol
- Isopropanol
- Sodium chloride (NaCl; e.g., Fisher Scientific, catalog number: BP358 )
- 5 M NaCl (e.g., Thermo Fisher Scientific, catalog number: AM9759 )
- 3 M sodium acetate, pH 5.2 (e.g., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R1181 )
- Ammonium persulfate (e.g., Sigma-Aldrich, catalog number: A3678 )
- 1 M Tris-HCl, pH 7.5 (e.g., AMRESCO, catalog number: E691 )
- 1 M magnesium chloride (MgCl2, e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9530G )
- 2 M potassium chloride (KCl; e.g., Alfa Aesar, catalog number: J75896 )
- 0.1 M DTT (Thermo Fisher Scientific, catalog number: 707265ML )
- MyTaqTM Mix (Bioline catalog number: BIO-25041 )
- Tetramethylethylenediamine (TEMED) (Thermo Fisher Scientific, catalog number: 17919 )
- 40% Acrylamide/Bis Solution (Bio-Rad, catalog number: 1610144 )
- GlycoBlueTM Coprecipitant (Thermo Fisher Scientific, catalog number: AM9515 )
- 1x TBE (running buffer for urea-PAGE) (see Recipes)
- 10% (w/v) ammonium persulfate (see Recipes)
- Cytoplasmic lysis buffer (see Recipes)
- 8% native PAGE gel (see Recipes)
Equipment
- Eye protection
- NanoDrop spectrophotometer (Thermo Fisher Scientific, model: NanoDropTM 1000 , catalog number: ND-1000 ) or other instrument suitable for quantifying nucleic acids
- P10, P20, P100, and P1000 pipets
- Thermal cycler (e.g., MJ Research, model: PTC-200 )
- Quantitative PCR instrument (e.g., Bio-Rad CFX Connect Real-Time System)
- Heating block (e.g., Bioer, model: MB 101 )
- -80 °C freezer
- Water-jacketed humidified incubator (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: Forma® Series II ) set to 37 °C with 5% CO2
- Refrigerated centrifuge (Eppendorf, model: 5415 R )
- Programmable rotator-mixer (Grant Instruments, model: PTR-30 ) at 4 °C and set to 10 rpm orbital rotation
- Electrophoresis system for PAGE (e.g., Bio-Rad Laboratories, catalog number: 1660828EDU )
- Azure Sapphire Biomolecular Imager (or similar imaging system)
- UV transilluminator (BT Lab Systems, model: BT503 )
Software
- Microsoft Excel software
- ImageJ software
- Bio-Rad CFX Maestro software
- Adobe Photoshop
- Azure Sapphire Capture Software
Procedure
A schematic overview of TeloPrime mapping of capped ends is presented in Figure 1.
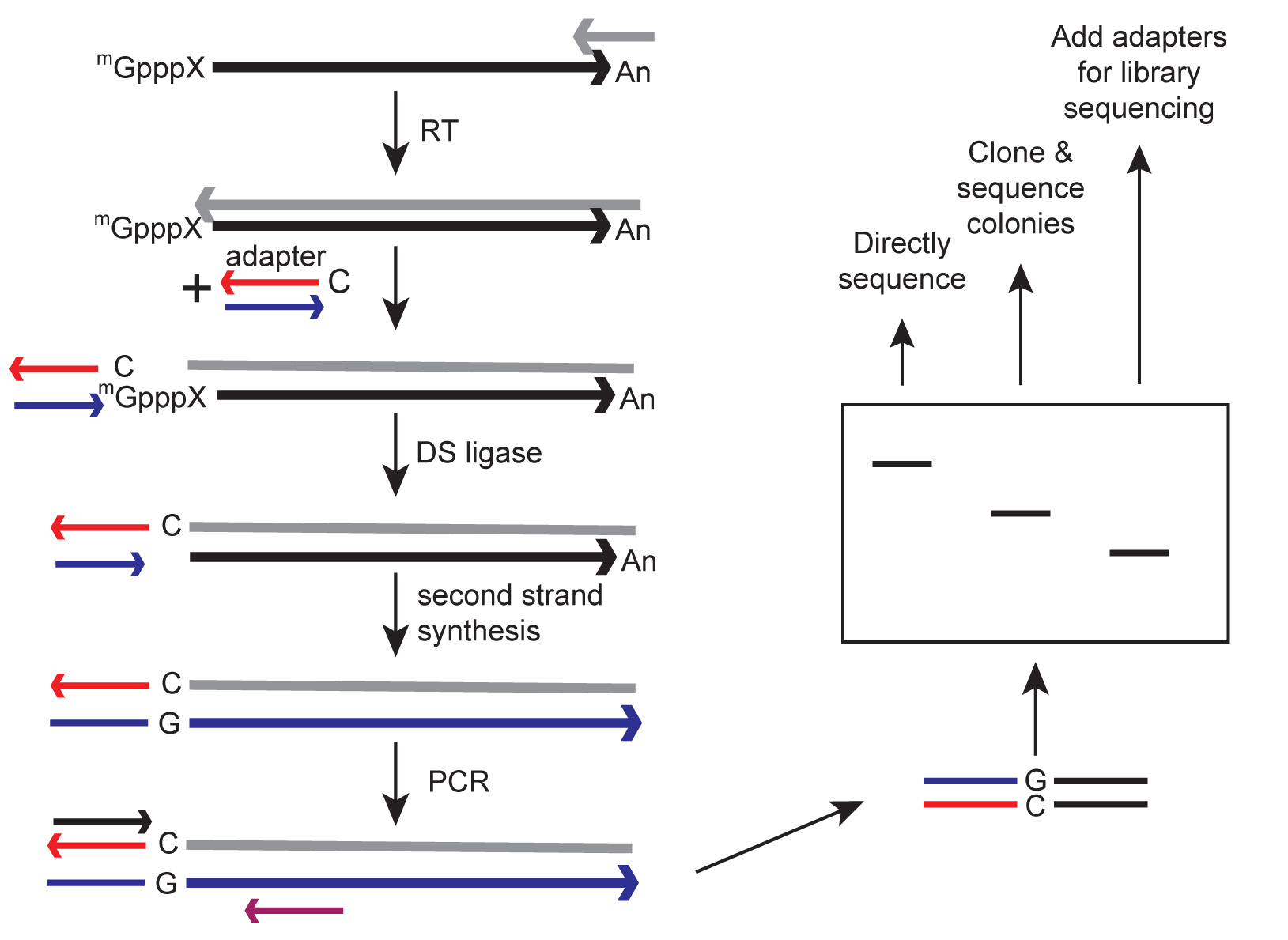
Figure 1. Schematic overview of cap methyltransferase activity assay. The population of cytoplasmic RNA (large black arrow) is reverse transcribed using the primer (small grey arrow) and reverse transcriptase (large grey arrow) included in the TeloPrime kit. The resulting mRNA:cDNA hybrid is ligated to a double-stranded DNA adapter that has a 5′ overhanging C residue on the antisense strand (red arrow) that weakly base pairs with m7G of the cap. Note that the G of the cap is not reverse transcribed. Ligation is performed using a proprietary double stranded DNA ligase included in the TeloPrime kit. RNA is then removed by second strand synthesis (large blue arrow) and specific transcripts are PCR amplified, using primers to the gene of interest (small purple arrow) and to the ligated adapter (small black arrow). Products are separated on a non-denaturing gel that is subsequently stained with SYBR Gold. DNA recovered from individual bands can be sequenced directly, cloned and sequenced, or secondarily processed to add Illumina sequencing adapters.
- Isolation of cytoplasmic RNA
- Nuclear RNA must be removed to assay cytoplasmic recapping. The following steps are used to isolate cytoplasmic RNA.
- Two to three days in advance, seed 10-cm or 15-cm dishes with U2OS, HEK293, or other adherent mammalian cells, at a density that will reach ~80-90% confluence at the time of harvesting. U2OS cells should be grown in McCoy’s 5A medium containing 10% (v/v) FBS; HEK293 cells should be grown in DMEM containing 10% (v/v) FBS. Cells should be grown in a humidified incubator at 37 °C with 5% CO2.
- On the day of harvesting the cells, chill Cytoplasmic lysis buffer (see Recipes) on ice. Leave out dithiothreitol (DTT) and RNAseOUT until immediately before use.
- Remove culture medium from adherent cells and rinse with phosphate buffered saline (PBS). Harvest the cells by scraping into 1 ml of PBS using a cell lifter and transferring to 1.7 ml centrifuge tubes. Gently pellet the cells (70 x g for 10 min at 4°C), and then carefully remove the supernatant. Make note of the approximate volume of the cell pellets and place the tubes on ice.
- Add a volume of Cytoplasmic lysis buffer equivalent to 4-5 times the volume of each cell pellet. Gently resuspend the cells with 10 up-and-down strokes of a P1000 pipet. Incubate the cells on ice for 10 min, and then mix with 5 additional up-and-down strokes of the P1000 pipet.
- Remove nuclei by centrifuging at 16,100 x g for 10 min at 4 °C, then transfer the supernatants to new, pre-chilled 1.7 ml tubes on ice.
- Recover cytoplasmic RNA from the supernatant using Zymo RNA Clean and Concentrator-25 according to manufacturer’s instructions. Measure the concentration of RNA with a NanoDrop spectrophotometer.
- TeloPrime protocol
- TeloPrime is a reverse transcription protocol that will specifically enrich for capped mRNAs. The steps follows the protocol provided in the TeloPrime kit.
- Thaw Reverse transcription mix (RT), Reverse transcription primer (RTP), Ligation mix (LM), and RNA buffer from the TeloPrime kit at room temperature. Thaw Enzyme mix 1 and 2 (E1 and E2 from the TeloPrime Kit), CleanCap® mCherry mRNA, and RNA sample on ice.
- Prepare a 12 μl mixture in a 0.2 ml PCR tube containing the following: 1 ng of CleanCap® mCherry mRNA, 2 μg of RNA sample, and fill the remaining volume with RNA buffer. Keep the mixture at room temperature. Add 2 μl of RTP to the mixture.
- Denature the mixture for 30 s at 70 °C then cool to 37 °C in a thermocycler and maintain this at 37 °C while preparing a mixture of 4 μl of Reverse transcription mix (RT) and 2 μl of Enzyme mix 1 (E1). Add this to the sample tube while it is in the thermocycler. Mix the reaction by pipetting slowly, then incubate the reaction for 2 min at 37 °C and 50 min at 46 °C.
- Equilibrate Column binding buffer 1 (CB1) and Column wash buffer (CW) to room temperature. Place the purification column on a collection tube. Add 20 ml of 100% ethanol to the CW bottle.
- Add 160 μl of CB1 to the reaction and mix well by vortexing. Transfer the reaction to the purification column and centrifuge for 1 min at 12,000 x g at room temperature. Discard the flowthrough. Add 200 μl of CW to the column and centrifuge for 1 min at 12,000 x g at room temperature. Repeat for a total of two washes.
- Discard the flowthrough and centrifuge the column for 2 min at 12,000 x g at room temperature to remove excess liquid. Transfer the column to a new 1.5 ml tube and add 19 μl of RNA buffer. Centrifuge the tube for 2 min at 12,000 x g at room temperature.
- Transfer 18 μl of the eluate to a new 0.2 ml PCR tube. Prepare a mixture containing 20 μl of Ligation mix (LM) and 2 μl of Enzyme mix 2 (E2) for double stranded adapter ligation. Add this to the eluted cDNA and incubate overnight at 25°C (see Note 1).
- The next day add 320 μl of CB1 to the reaction and mix well by vortexing. Transfer the reaction to a new purification column and collection tube, and centrifuge for 1 min at 12,000 x g at room temperature. Discard the flowthrough. Add 400 μl of CW to the column and centrifuge for 1 min at 12,000 x g at room temperature. Repeat for a total of two washes.
- Discard the flowthrough and centrifuge the column for 2 min at 12,000 x g at room temperature. Transfer the column to a new 1.5 ml tube and add 12 μl of RNA buffer. Centrifuge the tube for 2 min at 12,000 x g at room temperature.
- Thaw second strand mix (SS), forward primer (FP), reverse primer (RP), TeloPrime PCR mix (Telo PCR) and DNA buffer at room temperature. Thaw Enzyme mix 3 (E3) on ice. Equilibrate Column binding buffer 2 (CB2) at room temperature.
- Transfer 11 μl of the eluted cDNA to a new 0.2 ml PCR tube. Mix 8 μl of second strand mix (SS) with 1 μl of E3, add this to the eluted cDNA and mix well. Place the tube in a thermocycler and process for 90 s at 95 °C, 60 s at 62 °C, 5 min at 72 °C.
- Add 160 μl of CB2 to the reaction and mix well by vortexing. Transfer the reaction to a new purification column and collection tube, and centrifuge for 1 min at 12,000 x g at room temperature. Discard the flowthrough. Add 200 μl of CW to the column and centrifuge for 1 min at 12,000 x g at room temperature. Repeat the wash for a total of two washes
- Discard the flowthrough and centrifuge the column for 2 min at 12,000 x g at room temperature to remove excess liquid. Transfer the column to a new 1.5 ml tube and add 20 μl of DNA buffer. Centrifuge the tube for 2 min at 12,000 x g at room temperature to recover the double stranded cDNA.
- Optimizing cDNA amplification
- qPCR amplification of the double stranded cDNA is performed to determine the ideal number of cycles to use for each sample. This also serves as a quality control step.
- Prepare a qPCR reaction containing the following: 1 μl of cDNA, 0.5 μl of forward primer (FP), 0.5 μl of reverse primer (RP), 5 μl of SensiFASTTM SYBR® No-ROX, and 3 μl of RNase free water. Perform the following 40 cycle program on a qPCR thermocycler: 1 cycle of 95 °C for 30 s, 50 °C for 45 s and 72 °C for 5 min; 39 cycles of 95 °C for 30 s, 62 °C for 30 s and 72 °C for 5 min.
- Determine the maximum value in which fluorescence reaches a plateau on the qPCR amplification curve. Calculate where the fluorescence is at 50% (y-axis) and extrapolate the CT value (x-axis). See Figure 2A for an example. The CT value is the number of cycles used for cDNA amplification (see Note 2).
- Endpoint PCR amplification
- The samples are PCR amplified according to the cycles determined in Procedure C to generate enough cDNA for downstream analysis.
- Prepare a PCR reaction in a 0.2 ml PCR tube containing the following: 8 μl of TeloPrime PCR mix, 1 μl of reverse primer (RP), 1 μl of forward primer (FP), 9 μl of cDNA and 1 μl E3. Perform the following PCR reaction: 95 °C for 30 s, 50 °C for 45 s, 72 °C for 5 min; x the number of cycles determined in Step C2.
- Add 160 μl of CB2 to the reaction and mix well by vortexing. Transfer the reaction to the purification column and centrifuge for 1 min at 12,000 x g at room temperature. Discard the flowthrough. Add 200 μl of CW to the column and centrifuge for 1 min at 12,000 x g at room temperature. Repeat the wash for a total of two washes.
- Discard the flowthrough and centrifuge the column for 2 min at 12,000 x g at room temperature. Transfer the column to a new 1.5 ml tube and add 20 μl of DNA buffer. Centrifuge the tube for 2 min at 12,000 x g at room temperature. qPCR amplification of the double stranded cDNA is performed to determine the ideal number of cycles to use for each sample. This also serves as a quality control step.
- mCherry normalization for gene specific PCR
- Capped mCherry RNA is included in each sample as a normalization control. Prior to analyzing individual transcripts PCR is performed on the recovered cDNA using mCherry primers. The cDNA preparations are then adjusted prior to gene-specific amplification.
- Prepare a PCR reaction in a 0.2 ml PCR tube for each sample containing the following: 10 μl of myTAQ x2 mix, 1 μl of 10 μM mCherry forward primer, 1 μl of 10 μM mCherry reverse primer, 1 μl of cDNA sample and 7 μl of RNase-free water. Perform the following PCR reaction: 95 °C for 1 min, 20 cycles of 95 °C for 15 s, 60 °C for 15 s, 72 °C for 10 s.
- Transfer the PCR reaction to a 1.5 ml tube. Add 1 μl of GlycoBlueTM, 2 μl of 3 M sodium acetate, pH 5.2, and 46 μl of ice cold 100% ethanol. Vortex and place at -80 °C for 30 min. The product is recovered by centrifuging at 12,000 x g for 15 min, at 4 °C. Carefully remove the supernatant without disturbing the pellet. Add 200 μl of ice cold 70% ethanol to the pellet and centrifuge for 15 min at 12,000 x g at 4 °C. Carefully remove the supernatant without disturbing the pellet. Air dry the pellet at room temperature for 10-15 min. Resuspend the pellet with 10 μl of RNase-free water.
- Add 10 μl of 2x DNA loading dye to the reaction. Load 2 μl of DNA size marker on the first well and each sample to the remaining wells of an 8% native page gel. Electrophorese at 120 V for 30 min in 1x Tris-Borate-EDTA (TBE) buffer. Place the gel in a plastic container slightly larger than itself and stain with a mixture of 20 ml of 1x TBE containing 2 μl of SYBRTM Gold nucleic acid gel stain. Incubate the gel at room temperature for 15 min. Image the stained gel on an Azure Sapphire or other suitable imaging instrument. Normalize samples using the intensity of the mCherry bands as determined with ImageJ.
- Gene specific PCR
- Gene specific PCR is performed on the normalized samples to determine the 5’ cap site of the gene of interest.
- Prepare a PCR reaction in a 0.2 ml PCR tube for each sample containing the following: 10 μl of myTAQ 2x mix, 1 μl of forward primer (FP), 1 μl of 10 μM gene specific reverse primer (see Notes 2 and 3), 1 μl of cDNA sample adjusted to mCherry in Procdure E and 7 μl of RNase-free water. Perform the following PCR reaction: 95 °C for 1 min, 20 cycles of 95 °C for 15 s, 50 °C (see Note 3) for 15 s, 72 °C for 10 s.
- Transfer the PCR reaction to a 1.5 ml tube. Add 1 μl of GlycoBlueTM, 2 μl of 3 M sodium acetate, pH 5.2, and 46 μl of ice cold 100% ethanol, vortex and place at -80 °C for 30 min. Recover the product by centrifuging at 12,000 x g for 15 min at 4 °C. Carefully remove the supernatant without disturbing the pellet. Add 200 μl of ice cold 70 % ethanol to the pellet and centrifuge for 15 min at 12,000 x g at 4 °C. Carefully remove the supernatant without disturbing the pellet. Air dry the pellet at room temperature for 10-15 min. Resuspend the pellet with 10 μl of RNase-free water.
- Add 10 μl of 2x DNA loading dye to the reaction. Load DNA size markers in the first 1 or 2 wells, mCherry in the next well, and experimental samples in the remaining wells of an 8% native page gel. Electrophorese in 1x TBE at 120 V for 30 min and stain with SYBRTM Gold nucleic acid gel stain as in Step E4, and visualize this on a gel imager (see Note 5). An example of this is shown in Figure 2. This figure shows results for the mCherry control, one mRNA with a single capped end (RPS3) and two mRNAs that have multiple capped end species (EIF3K and EIF3D). The solid dots indicate canonical RefSeq cap sites as determined by sequencing. Save the gel for band purification.
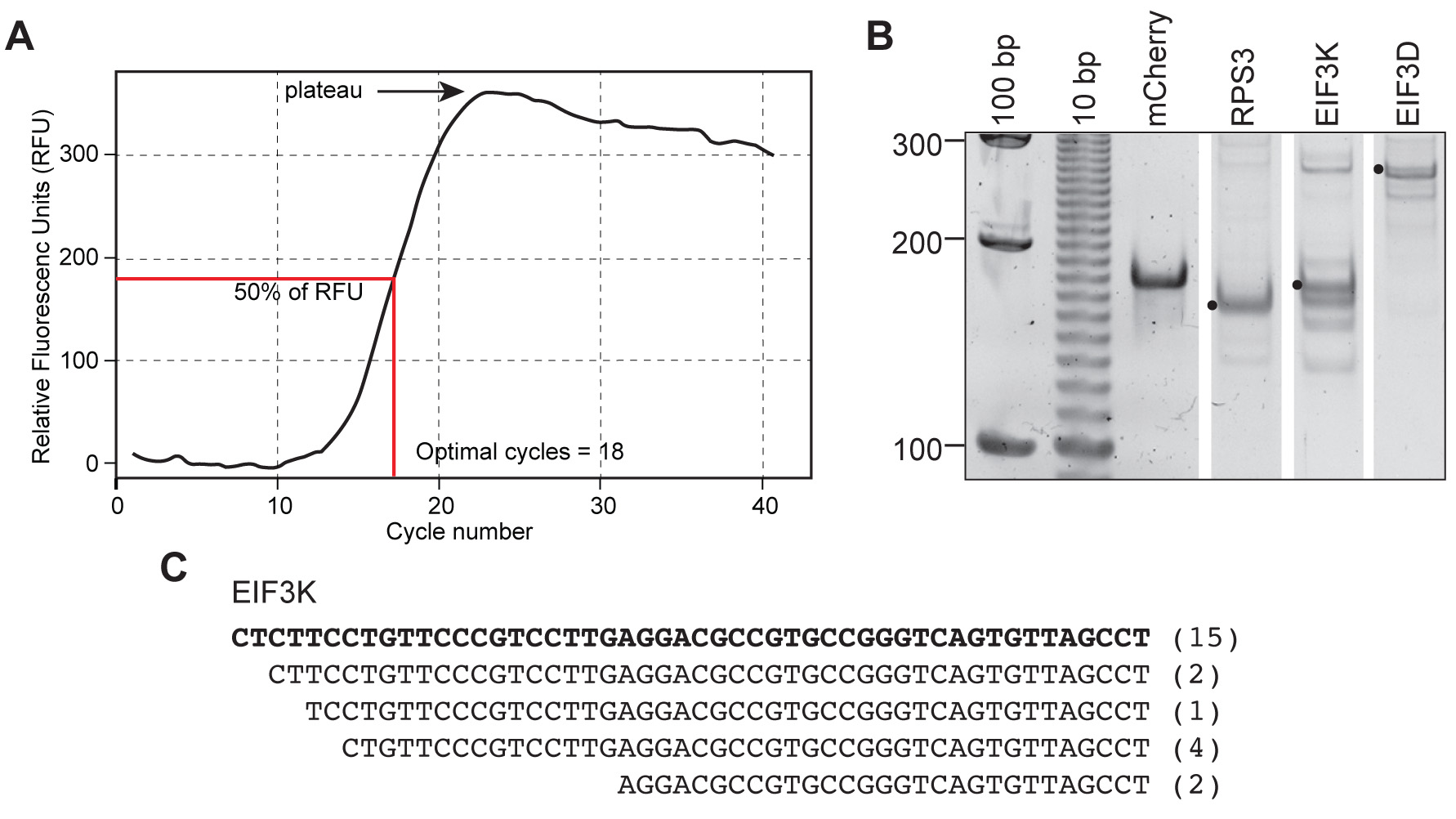
Figure 2. Examples of an amplification curve, a gel of TeloPrime generated 5′ end PCR products, and sequencing results for EIF3K. A. An example of a gene-specific amplification curve is shown with the plateau, 50% point and optimal number of cycle for this sample indicated. B. Shown is a gel for cap end PCR products for mCherry, one mRNA that has only a single capped species (RPS3), and two mRNAs that have capped ends located both at the canonical RefSeq cap site (indicated with a black dot) and further downstream. C. PCR amplification of EIF3D and EIF3K generated closely spaced bands. These were excised in a single gel piece and cloned, and multiple colonies were sequence to identify capped ends. The sequences of 24 colonies from EIF3K are shown beneath the figure, with the RefSeq sequence in bold and the number of colonies of a particular sequence indicated in parentheses (data are from Del Valle Morales et al., 2020).
- Purification of gel bands
- The bands of interest are excised and purified from the gel. The purified PCR products can be directly sequenced or in the case of multiple bands, cloned into plasmids for sequencing.
- Place the gel on a UV transilluminator set to 300 nm wavelength. While wearing gloves, lab coat, and a face shield, use a clean razor blade to excise the band of interest of the gel (see Note 6).
- Place the gel slice in a 0.6 ml microfuge tube having a hole at the bottom made with an 18 G needle. Note that if there are multiple closely spaced (as in Figure 2 with EIF3K and EIF3D) these should be recovered in a single gel slice and processed for cloning as described in Procedure H.
- Place the punctured microfuge tube with the gel slice in a 1.5 ml microfuge tube and centrifuge for 2 min at 12,000 x g at room temperature to shred the gel. Add 5x volume of RNase-free water to the shredded gel slice. Incubate overnight at room temperature with rotation.
- The next day, centrifuge the tube for 2 min at 12,000 x g at room temperature to collect the gel pieces at the bottom of the tube. Using a 200 μl pipette tip, carefully transfer the supernatant without disturbing the gel pieces to a new 1.5 ml tube.
- To the recovered supernatant add 1 μl of GlycoBlueTM, 0.1 volume of 3 M sodium acetate, pH 5.2, and 1 volume of ice-cold 100% isopropanol. Place this in a -80 °C freezer for 30 min, then centrifuge for 15 min at 12,000 x g at 4 °C. Carefully remove the supernatant without disturbing the pellet. Add 200 μl of ice cold 70% ethanol to the pellet and centrifuge for 15 min at 12,000 x g at 4 °C. Carefully remove the supernatant without disturbing the pellet. Air dry the pellet at room temperature for 10-15 min. Resuspend the pellet with 10 μl of RNase-free water. The recovered product of a single PCR band can be directly sequenced by Sanger sequencing. Recovered products from multiple bands need to be cloned and sequenced as described below.
- Cloning products from multiple closely spaced bands
- For multiple closely spaced bands, the recovered PCR product are cloned using TA cloning. The transformed bacterial colonies are then selected and sequenced to determine the capping site (see the legend to Figure 2).
- Prepare an A-tailing reaction in a 0.2 ml PCR tube containing 10 μl of recovered PCR products and 10 μl of myTAQ x2 mix. Incubate the reaction in a thermocycler for 20 min at 70 °C. Purify the reaction using Zymo DNA Clean and Concentrator-5 according to the manufacturer's instructions. Quantify the concentration of the recovered cDNA with a nanodrop spectrophotometer.
- Prepare a TA-cloning ligation using the pGEM®-T Easy Vector Systems. Prepare the ligation reaction containing the following: 5 μl of 2x Rapid Ligation Buffer, 1 μl of T4 DNA ligase, an insert to vector (pGEM®-T Easy Vector) ratio of 1:3 and fill to 10 μl with RNAse-free water. Incubate the reaction at room temperature for 1 h.
- Transform 3 μl of the ligated vector to Stellar competent cells following the manufacturer's instructions. Plate the transformed cells on LB/Ampicillin plates and incubate overnight at 37 °C. The next day pick individual colonies from the LB/ampicillin plate with a sterile 10 μl pipette tip and transfer the colonies to a 5 ml bacterial culture tube containing liquid medium (LB/ampicillin). Incubate the bacterial culture tube in an incubated shaker overnight at 37 °C. To control for the recovery of plasmids without inserts at least 50 colonies should be sequenced.
- The following day purify the plasmid DNA from the bacterial culture tube using NucleoSpin Plasmid kit according to the manufacturer's instructions. The purified plasmid can be sequenced by Sanger sequencing to identify the insert of the vector.
Notes
- The double stranded ligation can be done on a thermocycler overnight.
- The ideal CT value for cDNA amplification ranges from 10-25 and will differ depending on the mRNA of interest and its relative representation in the sample.
- The number of cycles used for gene specific PCR will depend on the gene of interest. 20 cycles are used as a baseline.
- Ensure that the Tm of the gene specific primer is within 5 °C of the FP.
- The polyacrylamide gel concentration can be altered to improve resolution of closely spaced bands.
- When excising the band from the gel, try to cut out the smallest gel piece possible that contains the band. Excess gel can affect the recovery of the band.
- This protocol can be modified for use with Illumina sequencing to identify capped ends on multiple targets. This is done by performing PCR on the final cDNA product using gene specific primers fused to the Illumina PE1 adapter and TeloPrime Forward Primer (FP) fused to barcoded Illumina PE2 adapter.
Recipes
- 1x TBE (running buffer for urea-PAGE)
Bring 100 ml of RNase-free 10x TBE to 1 L with RNase-free water
Store at room temperature - 10% (w/v) ammonium sulfate
Dissolve 1 g of ammonium persulfate in RNase-free water to 10 ml
Store at 4 °C - Cytoplasmic lysis buffer
20 mM Tris-HCl, pH 7.5
150 mM NaCl
5 mM MgCl2
1 mM DTT*
0.2% NP-40
80 U/ml RNaseOUT*
*Note: Add immediately before using the buffer. - 8% Native PAGE gel
2.4 ml of 40% Acrylamide/Bis Solution
9.6 ml of 1x TBE
200 μl of 10% (w/v) ammonium persulfate*
10 μl of TEMED*
*Note: Add immediately before pouring the gel.
Acknowledgments
This work was supported by R01 grant GM084177 from the National Institutes of Health (to D.R.S). The protocol presented here was used for experiments in Del Valle Morales et al. (2020).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Berger, M. R., Alvarado, R. and Kiss, D. L. (2019). mRNA 5’ ends targeted by cytoplasmic recapping cluster at CAGE tags and select transcripts are alternatively spliced. FEBS Lett 593(7): 670-679.
- Del Valle Morales, D., Trotman, J. B., Bundschuh, R. and Schoenberg, D. R. (2020). Inhibition of cytoplasmic cap methylation identifies 5’ TOP mRNAs as recapping targets and reveals recapping sites downstream of native 5’ ends. Nucleic Acids Res 48(7): 3806-3815.
- Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., Tanzer, A., Lagarde, J.Lin, W. and Schlesinger, F., et al. (2012). Landscape of transcription in human cells. Nature 489(7414): 101-108.
- Grudzien-Nogalska, E. and Kiledjian, M. (2017). New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip Rev RNA 8(1): e1379.
- Kiss, D. L., Oman, K., Bundschuh, R. and Schoenberg, D. R. (2015). Uncapped 5’ ends of mRNAs targeted by cytoplasmic capping map to the vicinity of downstream CAGE tags. Febs Lett 589(3): 279-284.
- Morioka, M. S., Kawaji, H., Nishiyori-Sueki, H., Murata, M., Kojima-Ishiyama, M., Carninci, P. and Itoh, M. (2020). Cap Analysis of Gene Expression (CAGE): A Quantitative and Genome-Wide Assay of Transcription Start Sites. Methods Mol Biol 2120: 277-301.
- Otsuka, Y., Kedersha, N. L. and Schoenberg, D. R. (2009). Identification of a cytoplasmic complex that adds a cap onto 5’-monophosphate RNA. Mol. Cell. Biol. 29(8), 2155-2167.
- Trotman, J., and Schoenberg, D. R. (2019). A recap of RNA recapping. Wiley Interdiscip Rev RNA 10(1): e1504.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
del Valle Morales, D. and Schoenberg, D. R. (2020). Analyzing (Re)Capping of mRNA Using Transcript Specific 5' End Sequencing. Bio-protocol 10(20): e3791. DOI: 10.21769/BioProtoc.3791.
Category
Molecular Biology > RNA > RNA capping
Molecular Biology > RNA > Transcription
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.