- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Simple Time-lapse Imaging for Quantifying the Hydrostatic Production of Oxygenic Photogranules



Published: Vol 10, Iss 19, Oct 5, 2020 DOI: 10.21769/BioProtoc.3784 Views: 3539
Reviewed by: Dennis J NürnbergJoseph Gitau GikonyoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Oxygenic photogranules (OPGs) are dense, three-dimensional aggregates containing a syntrophic, light-driven microbial community. Their temporal and spatial development interests microbial ecologists working at the bioprocess engineering interface, as this knowledge can be used to optimize biotechnological applications, such as wastewater treatment and biomass valorization. The method presented here enables the high-throughput quantification of photogranulation. OPGs are produced from a loose sludge-like microbial matrix in hydrostatic batch cultures exposed to light. This matrix transforms into a consolidated, roughly spherical aggregate over time. Photogranulation is quantified by time-lapse imaging coupled to automated image analysis. This allows studying the development of many OPGs simultaneously and in a fully automated way to systematically test what factors drive photogranulation. The protocol can also be used to quantify other types of (a)biotic aggregation.
Keywords: Oxygenic photogranulesBackground
OPGs are dense, roughly spherical aggregates with diameters of several millimeters containing a syntrophic community of heterotrophic and phototrophic microorganisms (Milferstedt et al., 2017). Microbial ecologists study photogranulation to understand what factors drive the formation of the three-dimensional (3D) structure. This knowledge can be applied to steer ecosystem function towards a desired function for biotechnological processes, such as wastewater treatment and the production of value-added products (Abouhend et al., 2018; Quijano et al., 2017). OPGs can be produced from a sludge-like microbial matrix, i.e., activated sludge from the aeration basin of a wastewater treatment plant (Milferstedt et al., 2017; Park and Dolan, 2015). The transformation of this sludge takes place in closed, unagitated vials exposed to light. Over the course of several weeks, the sludge bed compacts (i.e., reduces in height) and contracts (i.e., reduces in diameter) and transforms into one consolidated, 3D aggregate per vial (Figure 1A). Experimental images are automatically acquired through the bottom of vials at a pre-set interval (Figure 1B) of multiple replicates simultaneously. Images are treated in ImageJ (Schneider et al., 2012), extending a macro developed for the quantification of naturally occurring OPGs called cryoconite granules (Irvine-Fynn et al., 2010). Dynamics of biomass contraction is calculated and plotted in the software environment R (R Core Team, 2019). This protocol enables testing photogranulation in a large number of repetitions, e.g., using different sludge sources or environmental conditions to advance the understanding of photogranulation. The protocol can also be used to quantify other types of (a)biotic aggregation.

Figure 1. Typical course of photogranulation, including compaction and contraction. Note the displayed images are for illustration purposes and were not obtained with the protocol presented here. A. Temporal progression of the transformation of loose activated sludge into a consolidated OPG in a 10 ml serum bottle with an outer diameter of 24 mm (adapted from Milferstedt et al., 2017). Arrows illustrate the terms compaction and contraction; B. Temporal progression of biomass contraction seen through the bottom of one well of a 24-well microplate with an outer diameter of 16 mm.
Materials and Reagents
- Fresh activated sludge from the aeration basin of a wastewater treatment plant
- 0.5-5 ml pipette with tips (Thermo Fisher Scientific, FinnpipetteTM F1, catalog number: 4641110N ; 5 ml FinntipTM, catalog number: 9402030 )
- Grid for aligning and spacing vials on scanner surface. Custom-made dark gray metal grid measuring 30.4 × 22.2 × 0.2 cm (L × W × thickness) (Figures 2A-2B)
The grid should be sufficiently low so that it does not shadow the biomass from the side, e.g., it should not be much higher than the thickness of the vial bottom (Figure 3A-3B). The required cut-out where to place the vials can be produced using computer numerical control (CNC) metal milling or 3D printing. You can also make the grid out of other materials, such as paper. - Light-impermeable box for covering distance between vials and light source to prevent loss of light and uncontrolled illumination. Home-made cardboard box of the approximate dimensions of 65 × 35 × 65 cm (L × W × H), enclosing the scanner and the lighting device.

Figure 2. Grid. A. Grid design; B. Custom-made dark gray metal grid positioned on top of the scanner.
Equipment
- 1 L break resistant bottle with a wide neck for sampling (Fisher Scientific, Gosselin, catalog number: 11728643 )
- 2 L polypropylene beaker for mixing (Thermo Fisher Scientific, Nalgene, catalog number: 1201-2000 )
- 4 ml clear glass vials measuring 15 mm × 45 mm × 8 mm (outer diameter × height × inner diameter) with a screw top for cultivations (Sigma-Aldrich, Supelco, catalog number: 27111) (Figure 3A)
A clear, smooth and flat bottom is critical for the success of this experiment (Figure 3C). We used glass vials here, and successfully used polystyrene vials previously. - Screw caps with a contrasting color to the biomass for image treatment (Agilent Technologies, catalog number: 5183-4305 )
- Magnetic stirrer and stir bar (Bioblock Scientific, AM AMC BBS 3000 ) to homogenize activated sludge so that differences in sample composition between vials are minimized
- Holding device for mounting a light source, e.g., copy stand with camera arm (Kaiser, RS1, RA1, catalog number: 205510 )
- Flat light source, e.g., light emitting diode (LED) panel measuring 59.5 × 59.5 (cropped to 30) × 1.06 cm (L × W × H) (Rexel, LEDVANCE, PANEL LED 600, 40W, 6500K, 4000 lm, catalog number: 4058075000582 )
We used cool white light (6,500 K) for this experiment, but other light temperatures were also successfully tested (e.g., 5,600 and 6,000 K) - Photoactive synthetically radiation (PAR) quantum light sensor and display meter system (Skye Instruments Ltd, SKP 215/S 39520 , SKP 200 39521 )
- Desktop scanner (Epson, Perfection V500 Photo, model: J251A)

Figure 3. Vials. A. Glass vial with screw cap; B. Vial bottom thickness; C. Clear, smooth and flat vial bottom.
Software
- Scanner driver allowing time-lapse acquisition of images using a desktop scanner, e.g., VueScan version 9.5.51 (Hamrick Software, https://www.hamrick.com/)
- Tool to read, write and edit meta information in batches of images, e.g., ExifTool version 11.78 (Phil Harvey, https://exiftool.org/)
- ImageJ version 1.52a (image processing program) (National Insitutes of Health (NIH), https://imagej.nih.gov/ij/) (Schneider et al., 2012)
MorphoLibJ plugin to ImageJ version 1.4.0 (collection of mathematical morphology methods and plugins) (INRA-IJPB Modeling and Digital Imaging lab, https://imagej.net/MorphoLibJ) (Legland et al., 2016). - R version 3.6.0 (software environment for statistical computing and graphics) (R Core Team, https://www.r-project.org/) (R Core Team, 2019)
exiftoolr package version 0.1.3 (O’Brien, 2020)
ggplot2 package version 3.2.0 (Wickham, 2016)
plyr package version 1.8.4 (Wickham, 2011)
readr package version 1.3.1 (Wickham et al., 2018)
Procedure
The steps below describe the production of OPGs in hydrostatic batch cultivations and the automated time-lapse acquisition of experimental images. The image analysis follows at “Data analysis”.
- Scanner setup (Figures 4A-4B)
- Connect a scanner to a computer and install scanner driver, e.g., VueScan.
- Set scanner settings and save program.
- 24-bit RGB image.
- 800 dots per inch (dpi) resolution yielding a pixel size of 32 µm. This resolution is sufficient to resolve the biological process and prevents the generation of excessively large files.
- Make sure there is no automatic color correction because this will complicate subsequent data analysis.
- Select the uncompressed Tagged Image File Format (TIFF) for saving images.
- Assign TIFF file names, e.g., YY-MM-DD_01+.tif and directory to save acquired scans.
- Set acquisition interval. Eight hours were sufficient to capture the dynamics of photogranulation. The interval can be increased as the experiment progresses.
- Take scanner image with a plain white sheet on top of the grid. This will be the background image for data analysis (Figure 5A).

Figure 4. Experimental setup. A. Light source and scanner setup (Note the cardboard box is not shown); B. Close-up of scanner setup with vials positioned on the grid.
- Light source setup (Figures 4A-4B)
- Install a flat light source above a scanner, preferably using a device that allows adjusting the distance between the light source and vials, for example by connecting it to the camera arm of a copy stand. A flexible distance makes it possible to adjust the light intensity that vials receive and allows easy placement and removal of vials.
- Light intensity is typically not homogeneously distributed over a LED panel. To ensure that the local light conditions are known, you can map local PAR at different points on the scanner surface, draw a contour map and decide where to place the vials, e.g., along contour lines of similar light intensities. You can design a grid with circles that indicate where to place the vials (Figure 2A). You can print the grid on paper, cut out the circles and place it on top of the desktop scanner for positioning the vials. If you want to use the grid regularly, you can make it out of a more solid material, e.g., metal (Figure 2B) using CNC metal milling or 3D printing.
- Adjust the distance between the vials and light source such that the light intensity at the vial positions corresponds to approximately 60 µmol·m2·s-1 PAR. This can be measured using a PAR quantum light sensor.
- Activated sludge sampling and characterization
- Sample 200 ml of activated sludge from the aeration basin of a wastewater treatment plant. This volume is largely sufficient for an experiment using 72 vials inoculated with 1.5 ml of activated sludge each, as described in the following. The sample volume needs to be adapted if you are interested in characterizing the activated sludge, e.g., total and volatile solids, chemical oxygen demand, nitrogen, phosphorus, pH, chlorophyll, microbial community.
- It is useful to have available the measurement of total solids. This can be used to adjust the sludge concentration to a value comparable between experiments. We typically run an experiment with a concentration between 4 and 5 g/L of activated sludge.
- Vial preparation and placement
- Keep activated sludge in suspension upon arrival at the lab, e.g., using a magnetic stirrer.
Note: Sludge can be stored at 4 °C when it cannot be used directly, but we recommend to use fresh activated sludge. - Pipette 1.5 ml well-mixed activated sludge into 4 ml vials. Close vials with a screw cap. Our typical incubation set contains 72 vials.
- Place vials unagitated at the assigned areas on top of desktop scanner under constant light illumination at ambient room temperature (22-26 °C) (Figures 4A-4B).
- Place a light-impermeable box around the scanner and light source to limit loss of light and uncontrolled illumination of your samples.
- Keep activated sludge in suspension upon arrival at the lab, e.g., using a magnetic stirrer.
- Vial incubation and time-lapse imaging
- Start scanner software to take an image of the bottom of the vials at the desired interval. Make sure the scanner takes an image at time 0 and that images are taken at indicated intervals.
- The unconsolidated sludge transforms into one OPG per vial that is typically situated at the bottom of the vial, unless it starts floating due to attached gas bubbles. Run the experiment until mature OPGs have been formed. A mature OPG is roughly spherical and remains its shape after vigorous shaking. Under the given experimental conditions, photogranulation can be expected to occur between three to six weeks. The experimental conditions, e.g., light intensity and sludge characteristics, may influence the time needed for photogranulation.
- Take a final scan before removing the vials from the scanner.
- Photogranulation success
- Note down whether OPGs are sitting on the bottom or floating for image analysis.
- Take camera images of OPGs to remember later what they looked like.
- Shake vials vigorously and note down in which vials a successful OPG has been formed. Photogranulation success is the percentage of successful OPGs among the total incubated vials.
- Characterize OPGs depending on the factors you are interested in, e.g., physicochemical parameters, microbial community.
Data analysis
For data analysis, we show the results of an experiment performed with 72 vials of a volume of 4 ml using two sludge sources, therefore having 36 replicates per condition. Some files are created as preparation for automatic data treatment (Procedures A-B). Experimental time-lapse images are treated in ImageJ (Schneider et al., 2012) using particle size analysis to measure particle characteristics including surface area (Procedures C-D). The surface area is subsequently transformed to equivalent diameters in the software environment R (R Core Team, 2019) and plotted over time. The decrease in equivalent diameter per sample is a measure for proceeding photogranulation (Procedure E). Text in green are comments from the authors, text in red require user input, text in blue show files that will be imported into R (Procedure E).
- Experimental conditions
- Create a text file (experimental_conditions.txt) containing the vial positions and their respective experimental conditions in three columns, i.e., Location, ExpCondition, ExpID. You can find an example of this file at https://doi.org/10.5281/zenodo.3938457.
- Create a text file (experimental_conditions.txt) containing the vial positions and their respective experimental conditions in three columns, i.e., Location, ExpCondition, ExpID. You can find an example of this file at https://doi.org/10.5281/zenodo.3938457.
- Extract acquisition date/ time and remove scan resolution
- The Exchangeable Image File (EXIF) data of the scanner images contains the filename and the creation date of the image. Both need to be available in a separate file for subsequent plotting of the data. You can automatically extract filename (e.g., 2018-03-14-01.tif) and creation date/ time (e.g., 2018:03:14 16:47:36) from EXIF data using the R script below.
- The EXIF data may also contain the scan resolution. This resolution in dpi interferes with the particle size quantification using ImageJ. Remove scan resolution from EXIF data running ExifTool from within R so that ImageJ does not use dpi as a scaling factor. You can later assign a measurement unit to the images in ImageJ.
- Install ExifTool.
- Open a new R script in an R editor, for example RStudio, by clicking on ‘file’, ‘new file’, ‘R script’.
- Copy the following script into the source window. Text in red require user input and adaptations.
##Packages --------------------------------------
require(exiftoolr) #(O’Brien, 2020)
##Working directory --------------------------------------
rm(list=ls())
directory <- ("/path-to-directory/")
setwd(directory)
##Extract acquisition date/ time and remove scan resolution --------------------------------------
image.vec <- list.files(directory, pattern = ".tif") #Generates a list of all *.tif files in the directory
exif.images <- exif_read(path = image.vec) #Reads the EXIF data from the specified *.tif images
image.data <- cbind(exif.images$FileName, exif.images$CreateDate)
colnames(image.data) <- c("Name", "DateTime")
write.csv(image.data, file = "image_acquisition.csv", quote = F, row.names = F) #Writes the tags file name and creation date to a separate file to be used later. You can find an example of this file at https://doi.org/10.5281/zenodo.3938457.
exiftool_cmd <- paste("exiftool -ResolutionUnit= -XResolution= -YResolution= ", "*.tif",sep='') #Defines the ExifTool command to erase the Resolution data in the EXIF data of all images with the *.tif extension
system(exiftool_cmd) #Executes ExifTool from within R. You can also run ExifTool outside R. Original scanner images are automatically saved as *.tif_original by ExifTool. Created images without scan resolution (*.tif) will be used for subsequent data analysis in this protocol.
The method presented below shows an example of how to define and measure regions of interest. ImageJ macros are adapted from Irvine-Fynn et al. (2010) who quantified granule geometry of cryoconite, microbial aggregates that share similarities with OPGs, under laboratory conditions. Extracting information from experimental time-lapse images is a means to an end and not the main interest of this protocol. Other methods may be used equally well to obtain particle characteristics, including area and X-, Y-coordinates.
- Marker image
- Here we convert the background image (Figure 5A) into a marker image (Figure 5B). The marker image will be used to determine the X-, Y-coordinates of areas that could possibly contain particles, i.e., the circles that vials occupy. These data are used to assign measured particle characteristics to unique samples and their respective experimental conditions (Procedure E).
- Open an empty macro in ImageJ by clicking on ‘plugins’, ‘new’, ‘macro’.
- Copy the following script into the macro.txt window.
jobdirectory = getDirectory("Please choose a directory for saving result files and the marker image.");
waitForUser("Please open the background image. Then click ok.");
backgroundimage = getTitle();
run("Split Channels");
selectImage(backgroundimage + " (red)");
close();
selectImage(backgroundimage + " (green)");
close();
selectImage(backgroundimage + " (blue)");
//Converts RGB to grayscale image. Retains only the channel which presents most contrast between the areas on which vials are placed (white) and the grid (black).
run("Invert");
//Vial areas now appear black and the grid white.
setAutoThreshold("Default");
run("Threshold...");
waitForUser("Please set the threshold manually. Then press OK.");
setOption("BlackBackground", false);
run("Convert to Mask");
run("Close");
//Creates binary image from grayscale image: choose and apply cut-off value to divide image into foreground, i.e., vial areas, and background, i.e., grid. This works well when the grid is darker than the biomass. Otherwise you will have to setOption (“Black background”, true).
run("Fill Holes (Binary/Gray)");
//Fills holes in areas occupied by vials to achieve a solid area (Legland et al., 2016).
run("Morphological Filters", "operation=Opening element=Disk radius=10");
//Removes isolated pixels and breaks connections between areas occupied by vials and the grid so that it becomes less likely that the grid is accidentally considered part of the vial area.
run("Morphological Filters", "operation=Erosion element=Disk radius=35");
//Decreases the size of detected particles to exclude areas that are not part of the biomass, e.g., a ring corresponding to a vial that connects the biomass to the grid, resulting in a larger biomass particle than it actually is.
//Note: You may adjust the parameter disk radius depending on your images and experiment.
saveAs("Tiff", jobdirectory + "marker.tif");
run("Set Measurements...", "area centroid fit shape display redirect=None decimal=3");
//Specifies which measurements are recorded.
run("Analyze Particles...", " show=Overlay display exclude");
//Generates a result table containing information about each particle in the image, including a running number to label detected particles (i.e., the area that a vial occupies), the particle area and the centroid X-, Y-coordinates of the particles (Figure 6). It furthermore overlays the particle labels with the marker image to visually relate the particle characteristics in the table to the detected particles on the image.
saveAs("Results", jobdirectory + "results_marker.csv");
//You can find an example of this file at https://doi.org/10.5281/zenodo.3938457. - Run the macro by clicking on ‘macros’, ‘run macro’. During execution of the macro, manually select the output directory to save the created marker image and open the background image. You can use your own background image or run a demo with the image that can be downloaded from https://doi.org/10.5281/zenodo.3938457.
- Take a screenshot of the resulting marker image with particle labels overlaid that is not automatically saved running the macro (Figure 5B). We numbered vial locations 1 to 72 from top left to bottom right. The order in which ImageJ detected the areas occupied by vials on the marker image does not necessarily correspond to this numbering as the particle analyzer in ImageJ scans the image until it finds an edge of an object, which becomes particle 1 (see number 1 in Figure 5B), which in this case also corresponds to location 1 according to our numbering. In the R script (Procedure E1b), we use the screenshot to relate ImageJ particle numbers (lines in result table) to our vial locations.

Figure 5. Marker image created from background image. Screenshots of A. the background image, that is converted into B. the marker image, here displayed with the particle label overlay. The images that are saved in the macro do not have the headers and overlay as displayed in this figure. Columns are indicated in blue and rows in green.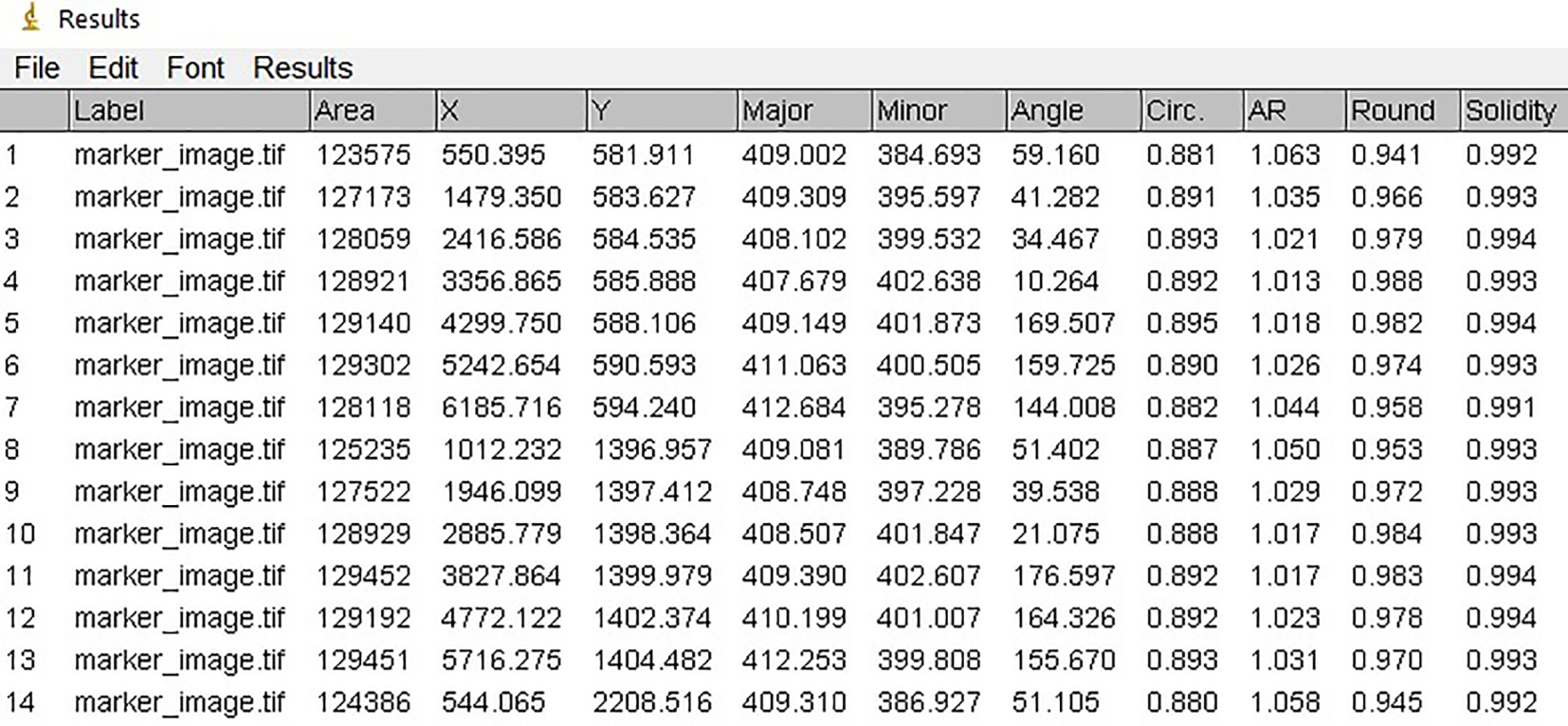
Figure 6. Result table of marker image. Screenshot of the result table showing characteristics of the first 15 out of 72 particles of the marker. This is not the result table that is saved in the macro, which is a comma-separated value (csv) file.
- Open an empty macro in ImageJ by clicking on ‘plugins’, ‘new’, ‘macro’.
- Here we convert the background image (Figure 5A) into a marker image (Figure 5B). The marker image will be used to determine the X-, Y-coordinates of areas that could possibly contain particles, i.e., the circles that vials occupy. These data are used to assign measured particle characteristics to unique samples and their respective experimental conditions (Procedure E).
- Experimental time-lapse images
- Here we convert scanner color images to black and white binary images, displaying biomass particles in black and the grid in white. Characteristics of each particle will be saved in a corresponding result table. The grid for positioning the vials will be automatically removed from the images. Possible erratic particles that are not part of the OPGs are equally removed during the process. The macro includes a manual thresholding step which is relatively time-consuming, but automatic thresholding may not always result in particles that reflect well the visual impression on the scanner images. In theory, we obtain 72 biomass particles per image for an incubation set of 72 vials. We acquired experimental images over a period of six weeks, resulting in 110 color images, each of a size of 182 MB. An image that is taken halfway through the experiment is shown as an example in Figure 7A.
- An image containing the background (i.e., areas that cannot possibly contain biomass, for example the grid) is required. This image needs to be a grayscale image in which the grid is white and the areas potentially containing particles during the experiment are black. In the simplest case, the image could be the marker image generated in Procedure C, or, for improved particle detection, a manually curated image based on either the background or an experimental image. During manual curation, the area available for photogranulation may be redrawn as disks with the inner diameter of the vials. A foreground limited to the potential areas where particles can be detected avoids artefacts interfering with image detection that can be caused by reflections of the vial walls or shadowing effects of the grid during scanning.
- Open an empty macro in ImageJ by clicking on ‘plugins’, ‘new’, ‘macro’.
- Copy the following script into the macro.txt window.
setBatchMode(false);
//Enters or remains in batch mode and hide active images during macro execution.
function action(input, output, filename){
open(input + filename);
jobname = getTitle();
jobnamemod = indexOf(jobname, ".");
jobname = substring(jobname, 0, jobnamemod);
//The file paths and filenames entered when starting the macro are converted to variables that become useable to the commands where they are used.
run("Split Channels");
selectImage(jobname + ".tif (blue)");
close();
selectImage(jobname + ".tif (green)");
close();
selectImage(jobname + ".tif (red)");
red_image = getTitle();
//Converts RGB to grayscale image. Retains only the channel which presents most contrast between the areas on which vials are placed (white) and the grid (black).
open(path);
//Opens the grid image (e.g., the marker image) selected in Procedure D1a using the file specified below.
//Vial areas now appear blackish and the grid whitish.
grid = getTitle();
imageCalculator("Add create", red_image, grid);
close("\\Others");
//Making grid appear as true white so that it will be detected as background in the subsequent binarization step.
run("Set Scale...", "distance=0 known=0 pixel=1 unit=pixel");
//Removes spatial scale of active image so that measurement results (e.g., area measurements) have the unit pixel.
run("Threshold...");
waitForUser("Please set the threshold manually. Then press OK, or cancel to exit macro");
setOption("BlackBackground", false);
run("Convert to Mask");
selectWindow("Threshold");
run("Close");
//Creates binary image from grayscale image: choose and apply cut-off value to divide image into foreground, i.e., vial areas, and background, i.e., grid. Define your own criteria and try to treat each image of your dataset in the same way.
//Note: This can be tricky because we normally do not find a threshold that works ideally for every particle on an image. We recommend testing a small subset (e.g., first, middle and last images) to get an idea of what works well.
run("Fill Holes (Binary/Gray)");
//Fills holes in the areas occupied by vials to achieve a solid area (Legland et al., 2016).
//Note: This may fill up entire vial area when a biomass particle is surrounded by a ring corresponding to a vial. If this happens, you can reduce the size of the respective particle on the marker image using for example image editor Gimp and run the macro again (see also Procedure D1a).
run("Morphological Filters", "operation=Opening element=Disk radius=10");
close("\\Others");
//Removes isolated pixels and breaks connections between areas occupied by vials and the grid so that it becomes less likely that the grid is accidentally considered part of the vial area.
mask_image = getTitle();
//Created image becomes mask image.
run("Morphological Filters", "operation=Erosion element=Disk radius=40");
marker_image = getTitle();
//Drastically decreases the size of detected particles on the mask image to create an image that will be used for morphological reconstruction.
//Note: Images of small granules may be removed when the disk radius is too large, you may adjust the parameter disk radius depending on your images and experiment.
print("The marker image name is " + marker_image);
print("The mask image name is " + mask_image);
//Prints active marker and mask images.
run("Morphological Reconstruction", "marker=["+marker_image+"] mask=["+mask_image+"] type=[By Dilation] connectivity=4");
//Keeps particles from the mask image that overlap with at least one pixel on the marker image and remove other pixels. This step assures that only particles are retained that overlap with vial areas and that possible erratic particles that are not part of OPGs are removed.
run("Set Measurements...", "area centroid fit shape display redirect=None decimal=3");
rename(jobname);
//Specifies which measurements are recorded.
run("Analyze Particles...", "size=0-Infinity circularity=0.00-1.00 show=[Overlay Outlines] display exclude");
//Generates result table (see explanation at Procedure C).
save(output + jobname + ".final.tif");
close();
saveAs("Results", output+ jobname + "_results.csv" );
run("Clear Results");
selectWindow("Log");
run("Close All");
}
//Erases any previous measurement results.
input = getDirectory("Choose an input directory with the raw images, please.");
output = getDirectory("Choose an output directory for result images and tables, please.");
waitForUser("Please open the grid image. Then click ok.");
//Selects the grid image (e.g., the marker image) selected in Procedure D1a. This image allows the removal of the grid from the scanner images.
grid = getTitle();
dir = getDirectory("image");
path = dir+grid;
close();
//Define input directory with experimental images and output directory to save created images. Open the grid image selected in Procedure D1a. In the chronology of the script, results from these commands will be displayed once when starting the macro.
list = getFileList(input);
for (i = 0; i < list.length; i++)
action(input, output, list[i]);
setBatchMode(false);
//Definition of a loop structure that will execute the macro function as many times as there are images found in the input directory.
- Run the macro by clicking on ‘macros’, ‘run macro’. During execution of the macro, manually select the input directory with experimental images, output directory to save created images and open the grid image selected in Procedure D1a. You can use your own experimental images and grid image or run a demo with exemplary images that can be downloaded from https://doi.org/10.5281/zenodo.3938457. Actions are automatically repeated for all images in the input directory.
Note: Make sure to not have unrelated files in the folder as otherwise ImageJ attempts to treat them as images. - Manually check whether the particles on the obtained images (Figure 7B) correspond to what you visually detect on the raw images.
Note: This sanity check is important because the automated analysis is supposed to mimic and automate the visual inspection but is not infallible. You can create a series of thumbnails by rescaling the original scanner images to a reduced size to browse more easily through the images. - Concatenate (combine) result tables into one csv file to simplify the analysis.
- Open a new R script in an R editor, for example RStudio (R Core Team, 2019), by clicking on ‘file’, ‘new file’, ‘R script’.
- Copy the following script into the source window. Text in red require user input and adaptations.
##Packages --------------------------------------
require(plyr) #(Wickham, 2011)
require(readr) #(Wickham et al., 2018)
##Working directory --------------------------------------
rm(list=ls())
directory <- ("/path-to-directory/")
setwd(directory)
##Concatenate result tables --------------------------------------
results.df <- list.files(path = directory, pattern = "*.csv", full.names = T) #Generates a list of all *.csv files in the directory
results.df <- ldply(results.df, read_csv) #Combines result tables of all images into a single data frame
colnames(results.df)[1] <- "ObjNum" #Assigns name ObjNum to first column
write.csv(results.df, file = "results_scanner_images.csv", quote = F, row.names = F) #Writes result tables of all images to a separate file to be used later. You can find an example of this file at https://doi.org/10.5281/zenodo.3938457.

Figure 7. Result image created from scanner image. Screenshots of A. a scanner image taken halfway through the experiment, that is converted into B. the result image for which the particle characteristics are determined. The images show particles contracted to varying degrees for the two sludge sources. The images that are saved in the macro do not have the headers as displayed in this figure.
- Here we convert scanner color images to black and white binary images, displaying biomass particles in black and the grid in white. Characteristics of each particle will be saved in a corresponding result table. The grid for positioning the vials will be automatically removed from the images. Possible erratic particles that are not part of the OPGs are equally removed during the process. The macro includes a manual thresholding step which is relatively time-consuming, but automatic thresholding may not always result in particles that reflect well the visual impression on the scanner images. In theory, we obtain 72 biomass particles per image for an incubation set of 72 vials. We acquired experimental images over a period of six weeks, resulting in 110 color images, each of a size of 182 MB. An image that is taken halfway through the experiment is shown as an example in Figure 7A.
- Quantification of photogranulation
- The experimental results are now imported into the software environment R (R Core Team, 2019). The challenge is to automatically convert the particle characterizations done per image into one time series per vial. The steps of the R script are as follows:
•Calculate average X-, Y-coordinates of vial areas identified on the marker image (Figure 5B);
•Assign experimental time to all experimentally measured particles per image by adding a column with experimental time to the particle data obtained with ImageJ. This facilitates the plotting to temporal dynamics;
•Identify particles from a vial at a specific physical location on each image using the X-, Y-coordinates of vial areas. Relate particles to their location by assigning a location number and match them to the appropriate experimental conditions;
•Transform the detected surface area of particles into equivalent diameters;
•Plot the average decrease in particle diameter per experimental condition over time.- Open a new R script in an R editor, for example RStudio, by clicking on ‘file, ‘new file’, ‘R script’.
- Copy the following script into the source window. Text in red require user input and adaptations.
##Packages --------------------------------------
require(plyr) #(Wickham, 2011)
require(ggplot2) #(Wickham, 2016)
##Working directory --------------------------------------
rm(list=ls())
directory <- ("/path-to-directory/")
setwd(directory)
##Start time --------------------------------------
start.time <- c("2018:03:14 17:51:19")
#Manually enter start date/ time of the first image of the experiment. Experimental time will be calculated based on this time point, using the acquisition time of the scanner images.
##Scaling factor --------------------------------------
scaling.factor <- 475.417/1.5 #pixels/cm
#Manually enter the scale of the images, i.e., number of pixels to cover the diameter of a vial. This value can be easily measured in ImageJ.
##Margin --------------------------------------
margin.vial <- 212
#Manually enter average number of pixels between the center and edges of a particle as measured in ImageJ, typically the inner radius of the vials in pixel.
##Files to be imported --------------------------------------
#You can run a demo with the files presented at https://doi.org/10.5281/zenodo.3938457.
coordinates.name <- "results_marker.csv" #Information about each particle on marker image (generated using ImageJ).
raw.data.name <- "results_scanner_images.csv" #Joined result tables of scanner images (generated using ImageJ).
acquisition.time.name <- "image_acquisition.csv" #Filename and creation date of scanner images (generated using ExifTool from within R).
experimental.condition.name <- "experimental_conditions.txt" #Experimental conditions per sample (manually generated).
##Loading files --------------------------------------
coordinates <- read.csv(coordinates.name, header=T, as.is =T)
raw.data <- read.csv(raw.data.name, header = T, as.is = T)
acquisition.time <- read.csv(acquisition.time.name, header = T, as.is = T)
experimental.condition <- read.delim(experimental.condition.name, header = T, as.is = T)
####Calculate average X-, Y-coordinates of vial areas identified on the marker image (Figure 5B).
##Average X-, Y-coordinates of vial areas --------------------------------------
column.1 <- c(1,14,27,40,53,66) #Manually enter which particle label on the marker screenshot corresponds to which column, as indicated in blue (Figure 5B). The grid consists of columns alternating in starting position and size, e.g., the first particle detected corresponds to the first particle in the first column of the scanner grid, the fourteenth particle detected is situated on the grid in column 1, row 3, etc.
column.2 <- c(8,21,34,47,60)
column.3 <- c(2,15,28,41,54,67)
column.4 <- c(9,22,35,48,61)
column.5 <- c(3,16,29,42,55,68)
column.6 <- c(10,23,36,49,62)
column.7 <- c(4,17,30,43,56,69)
column.8 <- c(11,24,37,50,63)
column.9 <- c(5,18,31,44,57,70)
column.10 <- c(12,25,38,51,64)
column.11 <- c(6,19,32,45,58,71)
column.12 <- c(13,26,39,52,65)
column.13 <- c(7,20,33,46,59,72)
row.1 <- c(1,2,3,4,5,6,7) #Manually enter which particle label on the marker screenshot corresponds to which row, as indicated in green (Figure 5B). The grid consists of rows alternating in starting position and size, e.g., the first particle detected corresponds to the most left particle in the first row of the grid, etc.
row.2 <- c(8,9,10,11,12,13)
row.3 <- c(14,15,16,17,18,19,20)
row.4 <- c(21,22,23,24,25,26)
row.5 <- c(27,28,29,30,31,32,33)
row.6 <- c(34,35,36,37,38,39)
row.7 <- c(40,41,42,43,44,45,46)
row.8 <- c(47,48,49,50,51,52)
row.9 <- c(53,54,55,56,57,58,59)
row.10 <- c(60,61,62,63,64,65)
row.11 <- c(66,67,68,69,70,71,72)
pos.names <- c(row.1, row.2, row.3, row.4, row.5, row.6, row.7, row.8, row.9, row.10, row.11)
#Order of particle numbers from top left to bottom right.
average.x.1 <- mean(coordinates$X[coordinates$X.1 %in% column.1])
average.x.2 <- mean(coordinates$X[coordinates$X.1 %in% column.2])
average.x.3 <- mean(coordinates$X[coordinates$X.1 %in% column.3])
average.x.4 <- mean(coordinates$X[coordinates$X.1 %in% column.4])
average.x.5 <- mean(coordinates$X[coordinates$X.1 %in% column.5])
average.x.6 <- mean(coordinates$X[coordinates$X.1 %in% column.6])
average.x.7 <- mean(coordinates$X[coordinates$X.1 %in% column.7])
average.x.8 <- mean(coordinates$X[coordinates$X.1 %in% column.8])
average.x.9 <- mean(coordinates$X[coordinates$X.1 %in% column.9])
average.x.10 <- mean(coordinates$X[coordinates$X.1 %in% column.10])
average.x.11 <- mean(coordinates$X[coordinates$X.1 %in% column.11])
average.x.12 <- mean(coordinates$X[coordinates$X.1 %in% column.12])
average.x.13 <- mean(coordinates$X[coordinates$X.1 %in% column.13])
#Calculates average X-coordinates.
average.y.1 <- mean(coordinates$Y[coordinates$X.1 %in% row.1])
average.y.2 <- mean(coordinates$Y[coordinates$X.1 %in% row.2])
average.y.3 <- mean(coordinates$Y[coordinates$X.1 %in% row.3])
average.y.4 <- mean(coordinates$Y[coordinates$X.1 %in% row.4])
average.y.5 <- mean(coordinates$Y[coordinates$X.1 %in% row.5])
average.y.6 <- mean(coordinates$Y[coordinates$X.1 %in% row.6])
average.y.7 <- mean(coordinates$Y[coordinates$X.1 %in% row.7])
average.y.8 <- mean(coordinates$Y[coordinates$X.1 %in% row.8])
average.y.9 <- mean(coordinates$Y[coordinates$X.1 %in% row.9])
average.y.10 <- mean(coordinates$Y[coordinates$X.1 %in% row.10])
average.y.11 <- mean(coordinates$Y[coordinates$X.1 %in% row.11])
#Calculates average Y-coordinates.
average.x.a <-c(average.x.1, average.x.3, average.x.5, average.x.7, average.x.9, average.x.11, average.x.13)
average.x.b <- c(average.x.2, average.x.4, average.x.6, average.x.8, average.x.10, average.x.12)
#Calculates average Y-coordinates of rows having the same X-coordinate.
average.y.a <- c(average.y.1, average.y.3, average.y.5, average.y.7, average.y.9, average.y.11)
average.y.b <- c(average.y.2, average.y.4, average.y.6, average.y.8, average.y.10)
#Calculates average X-coordinates of rows having the same Y-coordinate.
first.half <- expand.grid(average.x.a, average.y.a)
second.half <- expand.grid(average.x.b, average.y.b)
#Couples X-, Y-coordinates for both a and b.
average.coordinates <- rbind(first.half, second.half)
colnames(average.coordinates) <- c("X","Y")
#Combines first.half and second.half by rows.
average.coordinates <- average.coordinates[order(average.coordinates$Y, average.coordinates$X),]
#Arranges coordinates to be in the same order as the particle numbers that can be found on the marker screenshot.
average.coordinates <- cbind(average.coordinates, pos.names, 1:72)
colnames(average.coordinates) <- c("X","Y", "ParticleLabel", "Location")
#Adds columns with order of particles numbers that can be found on the marker screenshot and experimental vial locations (1 to 72 from top left to bottom right).
####Assign experimental time to all experimentally measured particles per image by adding a column with experimental time to the particle data obtained with ImageJ. This facilitates the plotting to temporal dynamics.
##Experimental time --------------------------------------
start.time <- strptime(start.time, "%Y:%m:%d %H:%M:%S")
#Converts start time to date/ time representation.
date.time <- strptime(acquisition.time$DateTime, "%Y:%m:%d %H:%M:%S")
#Acquires acquisition time per image and converts it to a date/ time representation.
exp.time <- round(-1*(as.numeric(start.time - date.time, units="hours")),3)
#Converts date/ time to experimental time.
acquisition.time <- cbind(acquisition.time, exp.time)
#Adds experimental time to acquisition time file.
colnames(acquisition.time) <- c("Name", "Date", "ExpTime")
acquisition.time$Name <- gsub(".tif", "", acquisition.time$Name)
#Removes *.tif from file names.
unique.images <- unique(raw.data$Label)
#Checks number of unique images.
ExpTime.vec <- rep(0, nrow(raw.data))
#Creates vector with as many zeros as there are rows in the raw data file (equals number of particles per image times number of images).
for(i in 1:length(unique.images))
{time.i <- acquisition.time$ExpTime[which(acquisition.time$Name == unique.images[i])]
indices.1 <- which(raw.data$Label == unique.images[i])
ExpTime.vec[indices.1] <- time.i}
raw.data <- cbind(raw.data, ExpTime.vec, rep(NA, nrow(raw.data)), rep(NA,
nrow(raw.data)), rep(NA, nrow(raw.data)))
colnames(raw.data)[(ncol(raw.data)-3):ncol(raw.data)] <- c("ExpTime", "Location",
"ExpCondition", "ExpID")
raw.data <- raw.data[order(raw.data$ExpTime),]
#Loops over number of scanner images (i.e., 110 images), gives experimental time of image in acquisition.time file that corresponds to image i. Gives rows that correspond to image raw.data file that corresponds to image i (each image typically has 72 particles). Replaces zeros in vector belonging to these rows by experimental time belonging to image i.
####Identify particles from a vial at a specific physical location on each image using the X-, Y-coordinates of vial areas. Relate particles to their location by assigning a location number and match them to the appropriate experimental conditions.
##Identify particles per location and add experimental conditions --------------------------------------
for(j in 1:nrow(average.coordinates) ){indices.2 <- which(
sqrt(((raw.data$X- average.coordinates$X[j])^2) + ((raw.data$Y-average.coordinates$Y[j])^2)) < margin.vial)
if(length(indices.2) == length(unique.images)){print(paste(c("All looks fine for Location ", j, "."), sep = "", collapse = ""))}else{print(paste(c("Warning: there is an issue for Location ", j, "."), sep = "", collapse = ""))}
#If there is an issue for Location j, go manually through the images and make sure the issue is corrected. It may happen that a particle was not detected on all images, e.g., because it started to float, or more than one particle has been detected in the same well on one image, e.g., because there was a small piece of loose biomass. In the latter case, you can manually remove the data from the erroneous particle that is not a granule from the dataset.
raw.data$Location[indices.2] <- average.coordinates$Location[j]
indices.1 <- which(experimental.condition$Location == average.coordinates$Location[j])
raw.data$Location[indices.2] <- experimental.condition$Location[indices.1]
raw.data$ExpCondition[indices.2] <- experimental.condition$ExpCondition[indices.1]
raw.data$ExpID[indices.2] <- experimental.condition$ExpID[indices.1]}
if (length(which(raw.data$Location == 0)) == 0) {print("No 0s left and all coordinates are assigned a location")} else {"Watch out! There are unidentified particles left in the data."}
#Loops over number of rows in average.coordinates file (i.e., 72 rows for 72 particles). Returns the particle that falls within the X- , Y-coordinates for a specific position, i.e., 110 particles for each location because there are 110 images. Note: small and not-centered particles may not be detected. Relates the location number (i.e., 1 to 72) to the X, Y position so that the particle has the same label on every image. Couples experimental conditions to locations.
####Transform the detected surface area of particles into equivalent diameters.
##Calculation of equivalent diameter from area --------------------------------------
raw.data <- cbind(raw.data, 2*sqrt(raw.data$Area/pi), rep(NA, nrow(raw.data)))
colnames(raw.data)[(ncol(raw.data)-1):ncol(raw.data)] <- c("EquivDiam_pix",
"EquivDiam_cm")
#Calculates diameter of particle area assuming that it represents a circle.
raw.data$EquivDiam_cm <- raw.data$EquivDiam_pix/scaling.factor
#Converts pixels to centimeters.
####Plot the average decrease in particle diameter per experimental condition over time.
##Plot diameter over experimental time per experimental condition --------------------------------------
treated.final.data <- ddply(raw.data, c("ExpTime", "ExpID"), summarise,
average.diameter = mean(EquivDiam_cm), sd.diameter = sd(EquivDiam_cm),sem.diameter= sd(EquivDiam_cm)/sqrt(length(EquivDiam_cm)))
treated.final.data$ExpID <- as.character(treated.final.data$ExpID)
average.diameter <- treated.final.data$ average.diameter
sd <- treated.final.data$sd.diameter
p <- ggplot(treated.final.data, mapping = aes(x=ExpTime, y= average.diameter, shape=ExpID, color=ExpID))
p <- p + geom_point()
p <- p + scale_shape_discrete(name = "ExpID", breaks=c("1", "2"), labels=c("sludge1", "sludge2"))
p <- p + scale_color_discrete(name = "ExpID", breaks=c("1", "2"), labels=c("sludge1", "sludge2"))
p <- p + geom_errorbar(aes(ymin= average.diameter-sd, ymax= average.diameter+sd))
p <- p + labs(x = "Experimental time [h]", y = "Biomass diameter [cm]")
p #This is Figure 8. - You can use your own generated files or you can run a demo with the files presented at https://doi.org/10.5281/zenodo.3938457.
- Run the script by clicking on ‘Run’.
- Verify whether the generated plot (Figure 8) matches with what you would expect based on the experimental time-lapse images.
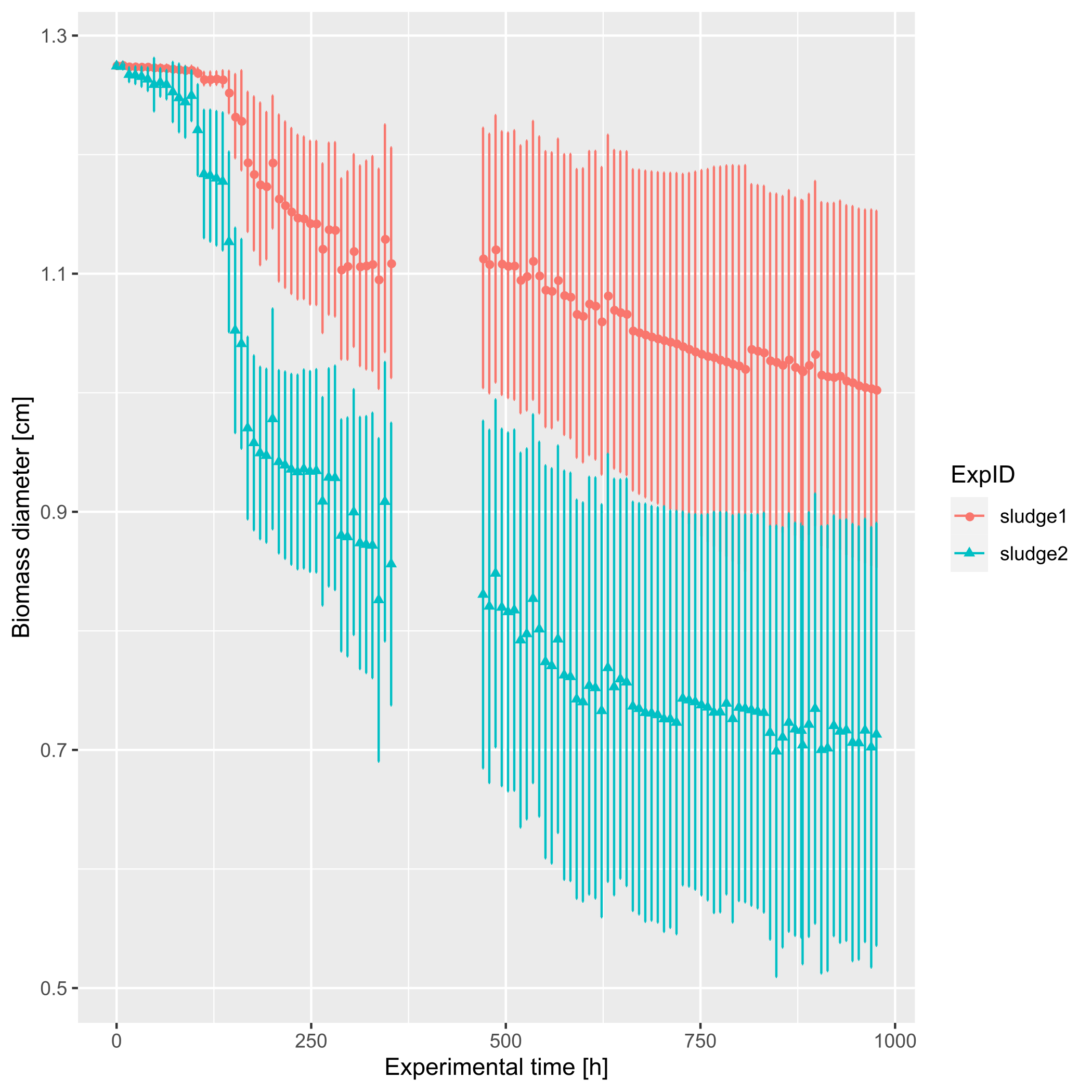
Figure 8. Progression of photogranulation shown as biomass contraction, i.e., decrease in diameter. Hydrotatic batch cultivations were started with two different sludge sources, each containing 36 replicates. Error bars present standard deviations.
Progression of photogranulation as quantified by biomass contraction, i.e., the decrease in diameter over time, is presented in Figure 8 for two different sludge sources. Initially, the sludge bed covers the entire bottom and corresponds to the inner diameter of the vial, i.e., 1.27 cm. During successful photogranulation, a final diameter of about half the initial value or less can be expected. Here the biomass did not granulate well in all 36 replicates per sludge source, and the formation of microbial mats was observed. This large variation in degree of contraction, i.e., the formation of two very different phenotypes, is reflected by the large standard deviations around the average diameters. Despite this heterogeneity, sludge source 2 resulted in more compact OPGs with a smaller average diameter than sludge source 1. Data was not obtained between 354 and 472 hours due to a technical issue. - The experimental results are now imported into the software environment R (R Core Team, 2019). The challenge is to automatically convert the particle characterizations done per image into one time series per vial. The steps of the R script are as follows:
- Data exclusion
- Failed experiments (e.g., overgrown vial bottoms, floating biomass, tipped vials) can be excluded manually from the analysis by removing the corresponding lines in the raw data or using a file specifying data points to be removed within the script (not described here).
- Failed experiments (e.g., overgrown vial bottoms, floating biomass, tipped vials) can be excluded manually from the analysis by removing the corresponding lines in the raw data or using a file specifying data points to be removed within the script (not described here).
- Statistical analysis
- Standard deviations around the average diameters of replicates are caculated at the end of the R script in Step E1b.
Notes
The biological phenomenon of photogranulation is not always reproducible: success rates greatly vary and may depend on the inoculum (i.e., the activated sludge) or experimental conditions, e.g., temperature, light intensity or light quality. This variability is subject of ongoing research using this protocol.
Acknowledgments
This work was funded by Graduate School GAIA of Montpellier University of Excellence (100% PhD fellowship for Esmee Joosten) and the ANR project PSST ANR- 16-CE04-0001.
Production of OPGs under hydrostatic batch conditions was earlier presented by Park & Dolan (2015) and Milferstedt et al. (2017).
Competing interests
The authors declare not to have any (non-)financial competing interests.
References
- Abouhend, A. S., McNair, A., Kuo-Dahab, W. C., Watt, C., Butler, C. S., Milferstedt, K., Hamelin, J., Seo, J., Gikonyo, G. J., El-Moselhy, K. M. and Park, C. (2018). The oxygenic photogranule process for aeration-free wastewater treatment. Environ Sci Technol 52(6): 3503-3511.
- Irvine-Fynn, T. D. L., Bridge, J. W. and Hodson, A. J. (2010). Rapid quantification of cryoconite: granule geometry and in situ supraglacial extents, using examples from Svalbard and Greenland. J Glaciol 56(196): 297-308.
- Legland, D., Arganda-Carreras, I. and Andrey, P. (2016). MorphoLibJ: Integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32(22): btw413.
- Milferstedt, K., Kuo-Dahab, W. C., Butler, C. S., Hamelin, J., Abouhend, A. S., Stauch-White, K., McNair, A., Watt, C., Carbajal-González, B. I., Dolan, S. and Park, C. (2017). The importance of filamentous cyanobacteria in the development of oxygenic photogranules. Sci Rep 7(1): 17944.
- O’Brien, J. (2020). exiftoolr: ExifTool Functionality from R. https://cran.r-project.org/package=exiftoolr
- Park, C. and Dolan, S. (2015). Algal-sludge granule for wastewater treatment and bioenergy feedstock generation. Pat Appl WO 2015112654 A2, Appl. number PCT/US2015/012332.
- Quijano, G., Arcila, J. S. and Buitrón, G. (2017). Microalgal-bacterial aggregates: Applications and perspectives for wastewater treatment. Biotechnol Adv 35(6): 772-781.
- R Core Team (2019). R: A language and environment for statistical computing. R Found. Stat. Comput. Vienna, Austria.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). ImageJ. Fundam Digit Imaging Med 9(7): 185-188.
- Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York.
- Wickham, H. (2011). The split-apply-combine strategy for data analysis. J Stat Software 40(1): 1-29.
- Wickham, H., Hester, J., Francois, R. (2018) readr: Read Rectangular Text Data. https://cran.r-project.org/package=readr
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Joosten, E. D., Hamelin, J. and Milferstedt, K. (2020). Simple Time-lapse Imaging for Quantifying the Hydrostatic Production of Oxygenic Photogranules. Bio-protocol 10(19): e3784. DOI: 10.21769/BioProtoc.3784.
Category
Microbiology > Microbial biofilm > Biofilm culture
Biological Sciences > Microbiology > Microbial communities
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.