- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Affinity Purification of GO-Matryoshka Biosensors from E. coli for Quantitative Ratiometric Fluorescence Analyses

Published: Vol 10, Iss 19, Oct 5, 2020 DOI: 10.21769/BioProtoc.3773 Views: 4889
Reviewed by: Wolf Dieter RötherJing LiYue (Yolanda) HuangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Genetically encoded biosensors are powerful tools for quantitative visualization of ions and metabolites in vivo. Design and optimization of such biosensors typically require analyses of large numbers of variants. Sensor properties determined in vitro such as substrate specificity, affinity, response range, dynamic range, and signal-to-noise ratio are important for evaluating in vivo data. This protocol provides a robust methodology for in vitro binding assays of newly designed sensors. Here we present a detailed protocol for purification and in vitro characterization of genetically encoded sensors, exemplified for the His affinity-tagged GO-(Green-Orange) MatryoshCaMP6s calcium sensor. GO-Matryoshka sensors are based on single-step insertion of a cassette containing two nested fluorescent proteins, circularly permutated fluorescent green FP (cpGFP) and Large Stoke Shift LSSmOrange, within the binding protein of interest, producing ratiometric sensors that exploit the analyte-triggered change in fluorescence of a cpGFP.
Keywords: BiosensorsBackground
The green fluorescent protein (GFP) was identified in 1962 in the jellyfish Aequorea Victoria (Shimomura et al., 1962). Thirty years later, its first use as a reporter gene was described (Chalfie et al., 1994). Since their discovery, GFP variants and other fluorescent proteins have contributed greatly to the principal advancements in the biological sciences, and are now common tools in biomedical research (Frommer et al., 2009).
A large number of fluorescent proteins (FP) and FP variants have been used as reporters or fused to proteins in organisms of all kingdoms of life (Chudakov et al., 2010; Valeur and Berberan-Santos, 2012). Continuously, new fluorescent proteins with enhanced properties have been identified or are being engineered, further improving and extending the toolkit for visualization of in vivo processes. These novel fluorophores allow us to monitor a wide range of real-time processes, from structural organization of cells to dynamic processes in living organisms. Photoactivatable FPs have been successfully used to track molecules and cells in space and time (Misteli and Spector, 1997; March et al., 2003). Recently, FPs enabled the design of biosensors. Different categories of genetically encoded biosensors are commonly used, including single fluorophore intensity-based sensors, and two-fluorophore sensors based on Förster resonance energy transfer (FRET) (Frommer et al., 2009). Various approaches are being used to carry out multiplex sensor analyses in the same cells (Mehta et al., 2018).
The mechanisms by which sensors operate can rely on modifications of the FPs themselves, in response to their interaction with a ligand (as in pHluorin or Clomeleon for protons and chloride, respectively) (Miesenbock et al., 1998; Kuner and Augustine, 2000). Alternatively, FPs can be grafted onto a ligand-binding domain. Ligand binding triggers conformational rearrangements that affect the fluorescence properties or relative positioning of the fluorophores (Deuschle et al., 2006; Kaper et al., 2008). Nowadays, genetically encoded biosensors are widely used in vivo to monitor levels and dynamics of ions and metabolites, the activity of transporters or tension (Frommer et al., 2009). While biosensors that rely on changes in the fluorescence ratio caused by changes in the relative positioning of a pair of donor and acceptor FPs were successfully used, many of the first generation sensors are limited by their comparatively low dynamic range and signal-to-noise ratio [SNR; for definitions of dynamic range and SNR, see Perez Koldenkova and Nagai (2013)]. Some of the advanced single fluorophore intensiometric biosensors, which involve the use of conformational sensitive FPs (csFPs), can have impressively high dynamic ranges and high SNR. However, changes in the expression level of the biosensor will affect the readout, raising concerns about reliability and artifacts for in vivo use. Actual suitability for in vivo measurements requires comparison of different sensor variants in the target tissues (Perez Koldenkova and Nagai, 2013). Nevertheless, the in vitro properties of such sensors are important pieces of information, including quantitative information on affinity and also on kinetics in the case of rapid processes such as action potentials. To avoid artifacts, intensiometric sensors can be converted into ratiometric sensors by coupling csFPs with a reference FP.
Recently, a new technology, termed Matryoshka, provided a universal platform to create dual-FP biosensors with a large dynamic range, good stability across a wide range of pH and buffer conditions, and single wavelength excitation thanks to the use of a Large Stokes Shift (LSS) FP (Ast et al., 2017). Nesting such a reference FP (e.g., LSSmOrange) within a reporter FP (e.g., circularly permuted green FP), permits excitation of both FPs at a single excitation wavelength. The technology was successfully applied to generate cytosolic calcium and ammonium transport sensors, GO-MatryoshCaMP and AmTryoshka variants, respectively (Ast et al., 2017).
Here, we present a detailed protocol for the purification and characterization by in vitro binding assays of genetically encoded His-tagged Matryoshka biosensors from Escherichia coli K12. The protocol presented here was specifically developed for purification and characterization of GO-MatryoshCaMP6s, but can be used more generally for other fluorescence biosensors with minor modifications.
Materials and Reagents
- Micropipette tips (GilsonTM PIPETMAN ClassicTM, catalog number: Gilson F12360x and, Starlab, TipOne®, catalog numbers: 1112-1840 , 1110-1840 , 1110-3800 )
Note: Pipetting accuracy is essential for the acquisition of reliable raw data. It may help to use of an electrical multichannel pipette (for 100 µl) (e.g., Sartorius AG) - 200 µl TipOne® pipette tips (sterile), yellow, conical, rack (STARLAB, catalog number: S1111-6701-C )
- 0.22-μm filter
- Single-use plastic serological pipettes (Sarstedt, catalog number: 86.1254.001 )
- 1 and 2 liter glass flasks for cell culture (SciLabware, catalog numbers: 1135/26D and 1135/30D )
- 50 ml tubes (Sarstedt, catalog number: 62.547.004 )
- 15 ml tubes (Sarstedt, catalog number: 62.554.002 )
- 1.5 ml microtubes (Sarstedt, catalog number: 72.690.001 )
- Stellar Scientific Centrifuge Tubes, High Speed (20,000 x g), 50 ml, Red Screw Cap (Stellar Scientific, catalog number: T15-701 )
- Stellar Scientific Centrifuge Tubes, High Speed (20,000 x g), 15 ml, Red Screw Cap (Stellar Scientific, catalog number: T15-701 )
- Round 92 x 16 mm Petri dishes (Sarstedt, catalog number: 82.1473 )
- Disposable plastic columns (Thermo Scientific, catalog number: 29922 )
- Non-sterile 96-well flat bottom transparent plates (Corning, catalog number: 9017 )
- Non-sterile 96-well flat bottom black plates with transparent bottoms (Corning, catalog number: 3631 )
- ZebaTM Spin desalting column (Thermo Fisher Scientific, catalog number: 89882 )
- Purified plasmids containing His-tagged biosensors for lactose/IPTG inducible expression (e.g., the calcium sensor MatryoshCaMP6 (Ast et al., 2017) in the vector pRSET-B) for nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography, as well as an empty vector, or the biosensor backbone protein lacking FPs (negative control) for expression in BL21 GOLD (DE3) cells. Plasmids and strains are described in more detail below
- Strains
Escherichia coli BL21 GOLD (DE3) [fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS; λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5] (New England Biolabs, catalog number: C2527I ) - cOmpleteTM ULTRA Tablets, Mini, EDTA-free protease inhibitor cocktail (Merck, catalog number: 11836170001 )
- Ni-NTA Agarose Beads (Qiagen, catalog number: 30210 )
- Bradford assay kit (Bio-Rad, catalog number: 500-0006 )
- MOPS (3-(N-morpholino)propanesulfonic acid) buffer (Carl Roth, catalog number: 6979.3 )
- Imidazole (AppliChem, catalog number: A1073 ,0500; UN3263)
- D-(+) glucose monohydrate (Sigma-Aldrich, catalog number: 14431-43-7 )
- Lactose (Merk, catalog number: 7660.025 0)
- BSA (Bovine Serum Albumin; Carl Roth, catalog number: 0 163.3 )
- BactoTM Tryptone (BD BioSciences, catalog number: 211705 )
- BactoTM Yeast Extract (BD BioSciences, catalog number: 212750 )
- Sodium chloride (Carl Roth, catalog number: 3957.1 )
- BactoTM Agar (BD Biosciences, catalog number: 214030 )
- Antibiotic(s) required for plasmid selection. For example: ampicillin for vector pRSET-B containing MatryoshCaMP6 (Sigma-Aldrich, catalog number: 69-53-4 )
- Sodium hydroxide (Sigma-Aldrich, catalog number: 1310-73-2 )
- Calcium chloride (Sigma-Aldrich, catalog number: 10043-52-4 )
- 4-20% Mini-PROTEAN® TGXTM Precast Protein Gels, 10-well, 50 µl (Bio-Rad, catalog number: 4561094 )
- 4x SDS Sample buffer (MERK, catalog number: 70607-3 )
- PageRuler Prestained Protein Ladder (Thermo Fisher Scientific, catalog number: 26616 )
- PierceTM 6xHis Protein Tag Stain Reagent Set (Thermo Fisher Scientific, catalog number: 24570 )
- Lysogeny broth (LB) liquid medium with antibiotics (see Recipes)
- LB solid medium (see Recipes)
- Auto-induction medium (for composition and explanations, see Recipes)
- Lysis buffer (see Recipes)
- Wash buffer (see Recipes)
- Elution buffer (see Recipes)
- Final buffer (see Recipes)
- Ligand binding assay buffer (see Recipes)
- Coomassie stain buffer (see Recipes)
- Stock solution of carbenicillin (see Recipes)
- Running Buffer (see Recipes)
Equipment
- Standard micropipette or multichannel (8 or 12 channel) pipette (for 100 µl) (e.g., Sartorius AG, catalog number: 725240 )
- pH meter (InoLab® pH Level1, catalog number: 72.690.001 )
- Sonicator (Branson Sonifier cell disruptor B15)
- Centrifuge (Hettich Rotanta 460/460R )
- Microplate reader with adjustable bandwidths (Spark®, Tecan)
- Fluorescence stereomicroscope for large fields observations (ZEISS, model: Axio Zoom.V16 )
- Eppendorf ThermoMixer® C (Eppendorf, catalog number: 5382000015 )
- Autoclave (Systec GmbH, model: Systec V-150 )
- Mini-PROTEAN® Tetra Vertical Electrophoresis Cell (Bio-Rad, catalog number: 1658004 )
- NanoDrop 2000 (LabX, catalog number: LV40609601 )
- Eppendorf ThermoMixer® C (Eppendorf, catalog number: 5382000015 )
- Eppendorf Thermoblock SmartBlockTM 1.5 ml (Eppendorf, catalog number: 5360000038 )
- Gel Doc XR+ Gel Documentation System (Bio-Rad, catalog number: 1708195 )
Software
- ecan-related software (Spark®, Tecan Life Sciences)
- Excel (Microsoft, Microsoft Office Professional 2016)
- MyCurveFit 2019 (MyCurveFit, MyAssays Limited)
Procedure
- Preparation of purified ratiometric fluorescent biosensors
- Transformation of E. coli
- Thaw the DNA (minipreps of plasmids carrying the biosensors and negative controls. e.g., MatryoshCaMP6 (Ast et al., 2017) and the vector pRSET-B) and aliquots of BL21 GOLD (DE3) competent cells on ice for 10 min.
- Add 10-50 ng of plasmid to the cells and keep on ice for 30 min.
Note: DNA quantification can be performed by using a NanoDrop device. - Perform heat-shock by using a heat block at 42 °C for 45-60 s.
- Keep on ice for 2 min.
- Recover cells for 1 h with 800 µl of LB medium by shaking at 200 rpm at 37 °C.
Note: SOC (Super optimal broth) medium, a nutrient-rich bacterial growth medium, can be used instead of LB medium for higher recovery after transformation. - Centrifuge at 11,000 x g for 5-10 s.
- Remove most of the supernatant and keep about 100-200 µl.
- Resuspend sedimented cells in remaining LB medium and plate dilutions onto solid LB plates (dilutions depend on transformation efficacy) containing the respective antibiotic, here carbenicillin at 100 μg/ml (see Recipes).
Note: To optimize colony density, plate different volumes (i.e., 30 and 170 µl) or dilute. - Allow bacteria to grow on solid agar overnight at 37 °C.
- Store plates at 4 °C overnight to achieve fluorophore maturation.
- Thaw the DNA (minipreps of plasmids carrying the biosensors and negative controls. e.g., MatryoshCaMP6 (Ast et al., 2017) and the vector pRSET-B) and aliquots of BL21 GOLD (DE3) competent cells on ice for 10 min.
- Clone selection and cell culture
- Visualize cpGFP fluorescence under a fluorescence stereomicroscope and pick bright colonies (Figure 1).
Note: Leaky expression is observed on LB solid plates. Brightness of colonies can be used as a criterion to evaluate the sensor expression levels.
Figure 1. E. coli cells transformed with empty vector and GO-MatryoshCaMP6s on solid LB plates. A-B. Colonies obtained after transformation with empty vector. C-D. Cells transformed with GO-MatryoshCaMP6s sensor. Fluorescence of the E. coli cells was recorded using a ZEISS Axio Zoom.V16 fluorescence stereo zoom microscope and GFP filter settings (λex 470/40 nm and λem 525/50 nm, with a beam splitter at λem 495 nm). Colonies were visualized using bright-field illumination (A, C) and GFP excitation for fluorescence detection (B, D). Scale bars = 1 mm. - For each of the 3 biological replicate, inoculate 5 ml of LB liquid medium (with antibiotics for plasmid selection) with 1 independent bright colony and grow overnight in LB plus antibiotic at 37 °C and at 200 rpm shaking (Figure 2).
- For each biological replicate, inoculate 200 ml of Auto-induction medium containing antibiotics (see Recipes) with the 5 ml preculture in a large flask and grow at 37 °C for 2 h in the dark on a shaker at 220 rpm (Figure 2). Use sample from the remaining preculture for generating glycerol stocks.
Notes:- To get optimal protein yield and avoid toxicity, Auto-induction media (details available here) should be used instead of IPTG, and cells expressing biosensors should be grown at 20 °C rather than 37 °C. The Auto-induction medium is used for obtaining high-levels of recombinant protein expression of lactose-inducible expression-systems. It does not require the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) and it avoids the constraints of monitoring optical density (O.D.) for the determination of the IPTG supplementation time. The medium contains glucose and lactose that are metabolized at different rates. Glucose is favored initially and represses promoter activity. Once glucose is consumed, repression will be relieved and lactose leads to induction of transcription.
- Auto-induction media are used for lactose/IPTG inducible expression systems otherwise this part of the protocol has to be customized.
- Glycerol stocks are generated by mixing cell culture 1:1 with LB medium containing 50% w/v glycerol.
- When growing 200 ml cultures, flasks should have a volume of 1 or 2 L. Do not fill flasks beyond 20% of total volume to ensure optimal oxygenation.
- The initial incubation at 37 °C is important to initiate faster the growth of large cultures from a single colony. Do not extend the incubation time, as it may later lead to a lower amount of soluble biosensors and an increased occurrence of inclusion bodies.
- To get optimal protein yield and avoid toxicity, Auto-induction media (details available here) should be used instead of IPTG, and cells expressing biosensors should be grown at 20 °C rather than 37 °C. The Auto-induction medium is used for obtaining high-levels of recombinant protein expression of lactose-inducible expression-systems. It does not require the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) and it avoids the constraints of monitoring optical density (O.D.) for the determination of the IPTG supplementation time. The medium contains glucose and lactose that are metabolized at different rates. Glucose is favored initially and represses promoter activity. Once glucose is consumed, repression will be relieved and lactose leads to induction of transcription.
- Transfer the culture to 20 °C and grow for 48 h at 220 rpm in the dark to avoid any photo-bleaching of FPs (Figure 2).
- Centrifuge the culture by using high-speed 50 ml centrifuge tubes for 30 min at 20,000 x g at 4 °C, discard the supernatant and freeze the sedimented cells at -20 °C overnight (Figure 2). Do not skip this freezing step, as it helps the purification process. A yellowish sediment of cells expressing the sensor should be visible.
Notes:- The yield of purified proteins may be increased by freezing and thawing the pellet, which helps to disrupt the cells.
- High-speed centrifuge tubes for 20,000 x g should be used to avoid breaking of the tubes during centrifugation. Alternatively: Normal falcon tubes can be used by reducing the speed to 17,000 x g in a way to prevent any leakage.
- The yield of purified proteins may be increased by freezing and thawing the pellet, which helps to disrupt the cells.
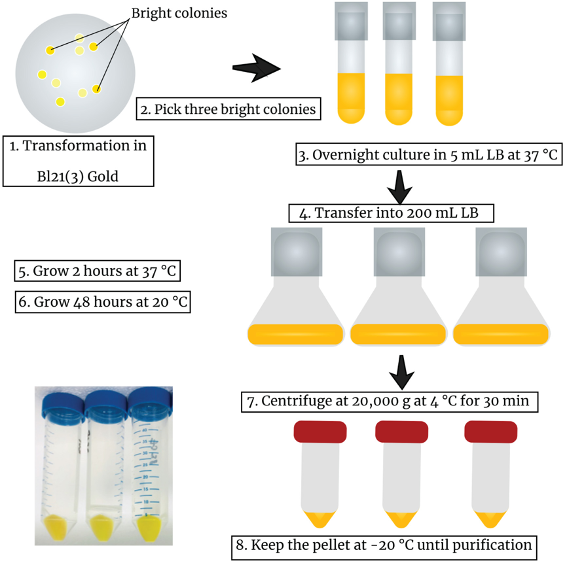
Figure 2. Expression of GO-Matryoshka biosensors in E. coli. Workflow of the experiment starting from the screening of the colonies until the harvesting of the expressed biosensor as a bright yellow sediment. - Visualize cpGFP fluorescence under a fluorescence stereomicroscope and pick bright colonies (Figure 1).
- Biosensor purification
- Thaw the pellet on ice.
- Immediately resuspend the cells by pipetting up and down in 5 ml of lysis buffer containing protease inhibitor cocktail.
- Sonicate the cell suspension into the tubes on ice (10-15 cycles, with each cycle consisting of 10 s of sonication and 10-15 s of rest). Be careful with this step since it is important to optimally break the cells while at the same time preventing overheating samples, which may lead to protein denaturation. This step is critical for the quality of the protein sample.
Note: Do not heat the sample (e.g., during sonication), which could lead to undesired protein denaturation and aggregation. - Centrifuge the lysate by using high-speed 15 ml centrifuge tubes (4 °C; 20,000 x g) for at least 30 min.
- During centrifugation, prepare the column by adding 1-2 ml final volume of Ni-NTA agarose bead material into a column (placed on its holder).
- At room temperature, wash the Ni-NTA beads three times by adding 5-10 ml wash buffer to the column, then let it flow through by gravity.
- Add the lysate to the column. Allow the lysate to flow through by gravity. The His-tagged biosensors will bind to the Ni-NTA beads.
- Wash three times by gravity flow with 5-10 ml wash buffer.
- Elute the sensors by gravity flow with up to 5 successive fractions of 300 µl of elution buffer and store in the dark at 4 °C for at least 24 h before analysis to allow the maturation of all purified biosensors. Alternatively: incubate the elution fraction for 3 h at 37 °C.
Note: Due to the longer maturation time of the LSSmOrange compared to derivatives (Shcherbakova et al., 2012), incubation at 4 degrees is crucial to obtain fully matured FPs. - To remove imidazole, salt contaminants and bound ligands, the eluate can be further purified with a ZebaTM Spin desalting column in a final volume of 130 µl according to the manufacturer’s instructions.
- Thaw the pellet on ice.
- Quality control of purified biosensor
Perform a Bradford assay to determine protein concentration according to manufacturer’s protocol. It is recommended that the concentration is at least about 1 mg/ml otherwise it is recommended to repeat the purification with a reduced elution volume.
Note: Biosensors can be stored at 4 °C in the dark for a couple of weeks.
- Transformation of E. coli
- Load 10-20 µg of protein samples into the SDS-Polyacrylamide gel (SDS-PAGE) (Figure 3).
- Mix the samples with 1x SDS Sample buffer and add water to a final volume of 20-40 µl.
- Boil the sample at 90 °C during 5 min to speed up the process of denaturation.
Note: Once denatured, the samples can be kept at room temperature until loading. - Load the sample into the wells of SDS-PAGE gel.
- After electrophoresis, proceed to the staining of 6xHis affinity-tagged protein (Pierce 6xHis Stain Reagent Set) according to the manufacturer’s instructions and to Coomassie staining for total protein visualization.
- Perform protein detection by using visible light (Coomassie staining) and UV-light excitation at a wavelength 280-310 nm (6xHis affinity-tagged staining).
Note: The staining of 6xHis affinity-tagged proteins requires 5.6 pmol of His-tagged proteins per band for detection with a CCD camera or 57 pmol of His-tagged proteins per band for detection with a UV transilluminator.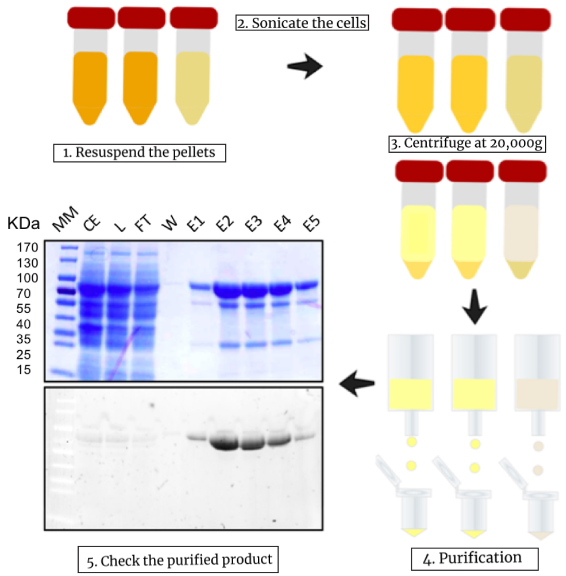
Figure 3. Isolation of GO-MatryoshCaMP6s sensors expressed in E. coli. Timeline of the experiment starting from the sedimented cells until the elution of the biosensor by affinity chromatography. Samples loaded into the SDS-PAGE gel are: MW (prestained molecular mass marker), CE (crude extract), L (lysate), FT (flow-through), W (wash), and E1-E5 (eluates from fractions 1 to 5). Upper gel panel was stained for total protein visualization using Coomassie stain buffer and lower gel panel was stained with 6xHis Protein Tag Stain Reagent Set (Thermo Fisher Scientific). Detection was performed by using UV-light excitation at a wavelength 280-310 nm.
- In vitro binding assay with purified biosensors
- Optimization of the sample dilution in final buffer (i.e., 20 mM MOPS, pH 7.0, see Recipes) and fluorescence reading parameters
In this section, protocol users will identify the proper dilutions required for each purified biosensor solution based on individual excitation of cpGFP and LSSmOrange using their respective maximum excitation and emission wavelengths. Users will also identify a single wavelength that provides the greatest simultaneous excitation of both cpGFP and LSSmOrange.- Select the fraction with the highest protein content and make a predilution of about 40 times to obtain a final concentration of about 5-10 µM.
- From the pre-diluted sample, prepare a dilution series (1; 1/2; 1/3; 1/4; 1/5; 1/10; 1/100) in a final volume of 200 µl and load into a 96-well plate suitable for reading fluorescence intensities and spectra in a microplate reader (see note).
Note: Although best results are obtained with black transparent bottom 96-well plates, their cost is comparatively high. The use of this black plastic helps preventing signal contamination between wells and is therefore favored when precise measurements are needed. However, for preliminary sensor characterization, we use regular non-sterile 96-well plates to minimize costs. Alternatively, to avoid light emission contamination between wells, empty wells can be placed systematically in-between samples. - Measure cpGFP and LSSmOrange fluorescence intensities of dilution series in the plate reader using the SPARKControl software and the following parameters: (1) cpGFP: Excitation at 480 nm with a band width of 20 nm, emission at 515 nm with a band width of 15 nm and (2) LSSmOrange: Excitation at 440 nm with a band width of 10 nm, emission at 570 nm with a band width of 15 nm. Adjust the gain and z-position of the reading so that the measurements fall roughly within the middle of the detector range. The gain is expected between 50-90 and should never exceed 130 while the Z position, when using 200 µl, is expected at about 20,000 μm.
Note: While it is possible to find a compromise by using a single intermediate wavelength to excite both FPs, their excitation maxima are distinct, thus for in vitro characterization in a fast fluorimeter, it is better to excite the two FPs separately. - Perform an excitation scan with 2 nm intervals between 440 to 480 nm to verify which excitation wavelength provides the highest amplitude for both GFP and the LSSmOrange emission in your conditions (Figure 4). This is typically around 455 nm for GO-Matryoshka biosensors.
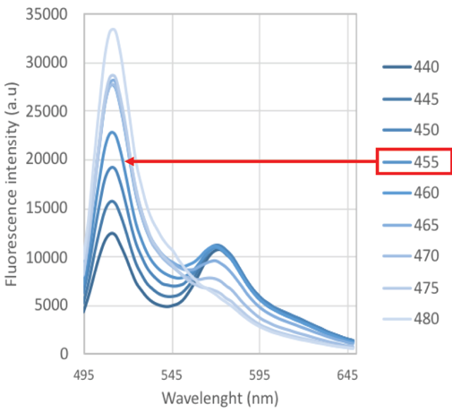
Figure 4. Excitation parameters for the detection of GO-Matryoshka sensors. Emission spectra of MatryoshCaMP6 reveal that increasing the excitation wavelength from 440 nm to 480 nm results in a rise in fluorescence intensity of cpGFP emission but a decrease in the emission of the stable reference LSSmOrange. A.u.: arbitrary units. - Select the optimal dilution factor based on emission intensities obtained in Steps C1c and C1d. The dilution factor depends on each instrument, here the targeted range of intensity is of about 20,000 arbitrary units (a.u.) to stay within the optimal range of the detectors without leading to saturation. Lower gain factors of the instrument will yield higher quality data (less noise).
- Select the fraction with the highest protein content and make a predilution of about 40 times to obtain a final concentration of about 5-10 µM.
- Binding assays
- Design the sample loading pattern of your plate by including technical triplicates and purifications of lysate from 3 biological replicates for each biosensor. Depending on the expected Kd, include suitable concentrations of the ligand (e.g., dilution series from 0 mM up to 5 times the Kd; use sufficient data points to obtain reliable apo and saturated states as well as linear range of binding isotherm). Make sure that the previously selected dilution factor is compatible with this design, and include control wells that only contain buffer to measure background.
Note: The order of components added does not matter. However, we recommend to add the component with the highest volume first, the buffer and end with the lowest volume, the biosensors. Load the 96-well plate according to your loading pattern. Technical triplicates for each of the three colonies will be loaded from a single Master Mix (see notes). Make sure to scale Master Mix for 4 reactions for technical triplicates, to maintain a margin of 1 reaction when pipetting triplicates. In addition, make sure that the final volume in each well remains identical, even with different ligand concentrations. Bubbles in the wells should be avoided, as they might affect the reading.
Notes:- Since the analysis is quantitative, accurate pipetting is crucial. Use high precision, use best practices when pipetting and handling pipettes, ensure that pipettes are in good state, ensure proper calibration of pipettes, prepare master mix (i.e., to avoid pipetting low volumes) and pipet slowly in order to reduce pipetting errors. Do not snap plunger, never allow any liquid to enter the shaft.
- Incubation time is recommended to be minimum 5 min. It is necessary to ensure to have reached binding equilibrium before performing the reading. The preparation of the 96-well plate is ususally taking a couple of minutes which is sufficient.
- It is recommended to include buffer-only wells as blanks.
- Since the analysis is quantitative, accurate pipetting is crucial. Use high precision, use best practices when pipetting and handling pipettes, ensure that pipettes are in good state, ensure proper calibration of pipettes, prepare master mix (i.e., to avoid pipetting low volumes) and pipet slowly in order to reduce pipetting errors. Do not snap plunger, never allow any liquid to enter the shaft.
- Read fluorescence using the plate reader as described in Step C1 to obtain three sets of data for each well: Emission spectrum and point measurements for cpGFP and LSSmOrange of maximum intensities of the GO-Matryoshka sensor (Figure 5).
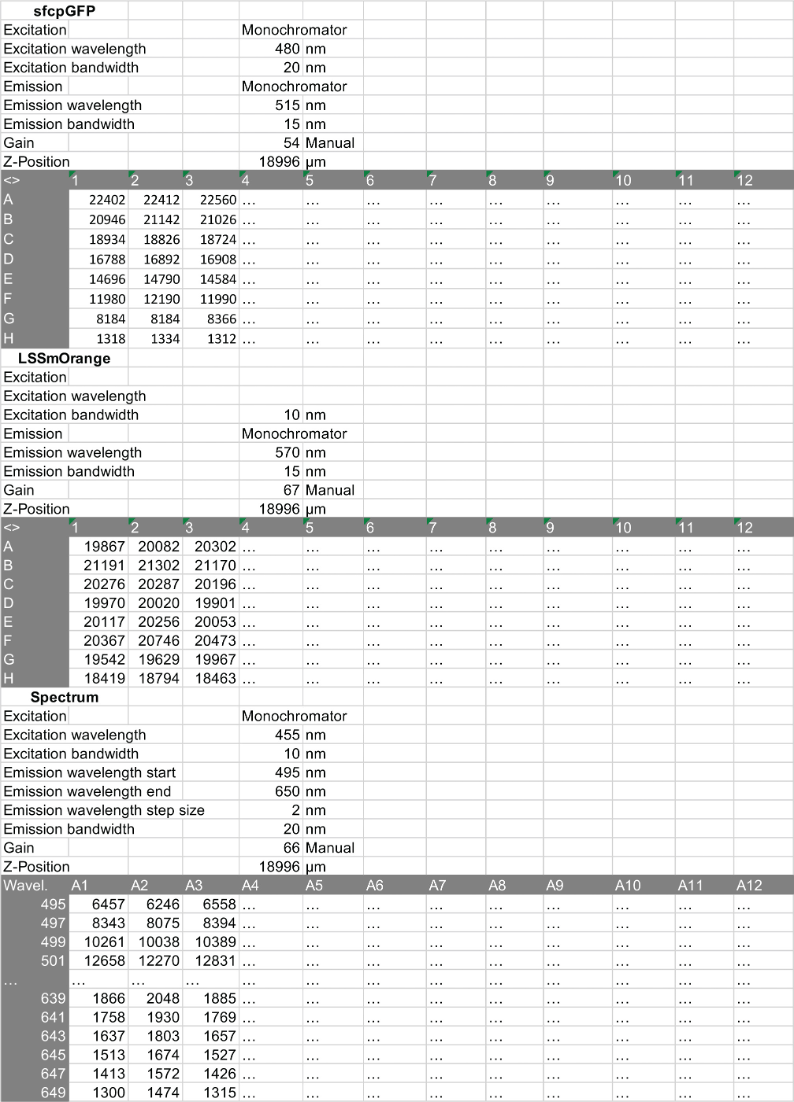
Figure 5. Screenshot of an excel worksheet displaying an example of data and parameters from one binding assay experiments. Three set of data are obtained after performing point measurements and emission spectrum scan of GO-MatryoshCaMP6s in biosensor binding assay. - Design the sample loading pattern of your plate by including technical triplicates and purifications of lysate from 3 biological replicates for each biosensor. Depending on the expected Kd, include suitable concentrations of the ligand (e.g., dilution series from 0 mM up to 5 times the Kd; use sufficient data points to obtain reliable apo and saturated states as well as linear range of binding isotherm). Make sure that the previously selected dilution factor is compatible with this design, and include control wells that only contain buffer to measure background.
- Analysis of the data for each biological replicate
- Set a default Excel worksheet, which can be used as template for automated analysis of data obtained in the previous section (Figure 5). This should be done by entering the required formula for processing data and generating graphs from any experiment by simply pasting its raw values. This template has to include:
- Calculation of the average value for the technical triplicates.
- Subtraction of the background fluorescence of the buffer for each condition and each data type (cpGFP and LSSmOrange intensities as well as the emission spectrum).
- Plotting the data by graphically representing the fluorescence intensity as a function of wavelength and generate emission spectra recorded at different ligand concentrations to quantify intensity changes responding to analyte concentration (Bermejo et al., 2011) (Figure 6).
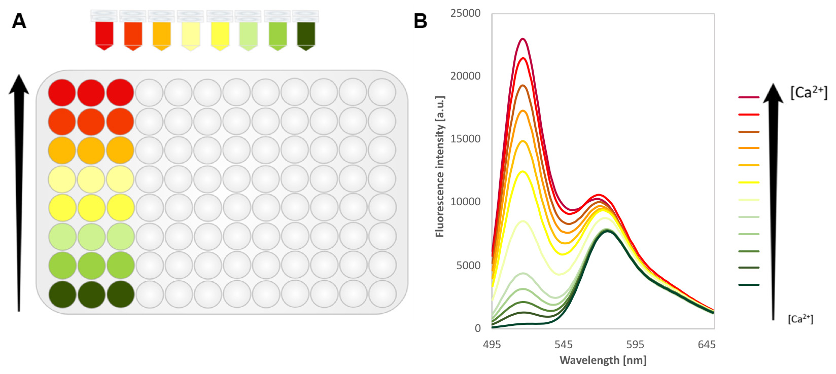
Figure 6. Binding assay using purified Matryoshka (here GO-MatryoshCaMP6s) biosensors. A. 96-well plate with GO-MatryoshCaMP6s plus increasing calcium concentrations, color code from green (without calcium) to red (10 µM of calcium), and in triplicate. B. The spectrum of MatryoshCaMP6, with excitation at 455 nm. Increasing concentrations of calcium result in a rise in the fluorescence intensity of cpGFP emission while the reference LSSmOrange emission stay stable (slight increase due to bleedthrough). [a.u.]: arbitrary units. Data are presented as the average of three technical replicates.
Note: It is observed that bleed-through occurs and leads to a slight but significant apparent increase in fluorescence intensity of the reference FP LSSmOrange. However, separate point measurements by using two excitation wavelengths should get rid of this effect. - Calculation of the average value for the technical triplicates.
- Measure the affinity by using three biological replicates:
- Calculate the ratio (R) of intensities at 520 nm/575 nm emission for each ligand concentration by using the following equation:
R = FI520nm/FI575nm - Calculate R0 as the value of R without substrate.
- Calculate ∆R the value of R at a specific calcium concentration subtracted by R0 as follows:
∆R = R - R0 - Calculate ∆R/R0.
- Plot the data against the concentration by using e.g., MyCurveFit.
- Use a logarithmic scale for the X axis.
- Apply a sigmoidal fit by using, e.g., MyCurveFit, and calculate the Kd (Figure 7).
- Calculate the ratio (R) of intensities at 520 nm/575 nm emission for each ligand concentration by using the following equation:
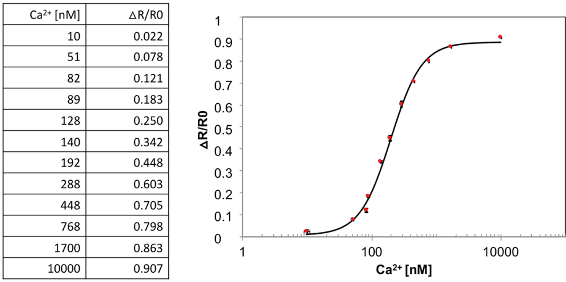
Figure 7. In vitro saturation curves of purified GO-Matryoshka. Calcium binding isotherms of MatryoshCaMP6s. Analyses of sensor output at different calcium concentrations was performed using purified sensor. Calcium binding was determined as previously described (Ast et al., 2017). Data are presented as the average of three biological replicates (mean ± S.E.M. from biological replicates with n = 3). - Set a default Excel worksheet, which can be used as template for automated analysis of data obtained in the previous section (Figure 5). This should be done by entering the required formula for processing data and generating graphs from any experiment by simply pasting its raw values. This template has to include:
- Optimization of the sample dilution in final buffer (i.e., 20 mM MOPS, pH 7.0, see Recipes) and fluorescence reading parameters
Recipes
- LB liquid medium with antibiotics
- Dissolve 10 g BactoTM tryptone, 10 g sodium chloride and 5 g BactoTM yeast extract in 800 ml MilliQ water
- Adjust to pH 7.0 with NaOH
- Adjust final volume to 1 L with MilliQ water
- Autoclave at 121 °C for 20 min
- Before use, add the antibiotics corresponding to the required antibiotic resistance used for selection
- Dissolve 10 g BactoTM tryptone, 10 g sodium chloride and 5 g BactoTM yeast extract in 800 ml MilliQ water
- LB solid medium (pH 7.0)
- Dissolve 10 g BactoTM tryptone, 10 g sodium chloride and 5 g BactoTM yeast extract and 15 g of agar in 800 ml MilliQ water
- Adjust to pH 7.0 with NaOH
- Adjust final volume to 1 L with MilliQ water
- Autoclave at 121 °C for 20 min
- Allow to cool down to about 50-60 °C, then add the antibiotics corresponding to the required antibiotic resistance used for selection
- Dissolve 10 g BactoTM tryptone, 10 g sodium chloride and 5 g BactoTM yeast extract and 15 g of agar in 800 ml MilliQ water
- Auto-induction medium (pH 7.0)
LB liquid medium
0.05% w/v D-glucose
0.2% w/v lactose - Lysis buffer (pH 7.0)
20 mM MOPS
1 cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail per 50 ml of solution
Note: The buffer can be stored at 4 degrees for months. 1 tablet of cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail is dissolved by extensive vortexing. - Wash buffer (pH 7.0)
20 mM MOPS
1 cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail per 50 ml of solution
Note: The buffer can be stored at 4 degrees for months. 1 tablet of cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail is dissolved by extensive vortexing. - Elution buffer (pH 7.0):
20 mM MOPS buffer
250 mM imidazole
1 cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail per 50 ml of solution
Note: The buffer can be stored at 4 degrees. for months 1 tablet of cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail is dissolved by extensive vortexing. - Final buffer (pH 7.0)
20 mM MOPS
Note: The buffer can be stored at 4 degrees for months. 1 tablet of cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail is dissolved by extensive vortexing. - Ligand binding assay buffer (pH 7.0)
20 mM MOPS with 0 to 10-20 mM calcium chloride
Note: The buffer can be stored at 4 degrees for months. 1 tablet of cOmpleteTM ULTRA Tablet Mini protease inhibitor cocktail is dissolved by extensive vortexing. - Coomassie staining buffer
Coomassie Blue R250 staining reagent
10% v/v acetic acid
50% v/v methanol - Stock solution of carbenicillin:
- Dissolve 1 g of disodium carbenicillin in 9.5 ml of MilliQ water
- Adjust final volume to 10 ml
- Filter-sterilize by using a 0.22-μm filter
- Dissolve 1 g of disodium carbenicillin in 9.5 ml of MilliQ water
- Running Buffer
25 mM Tris
192 mM glycine
0.1% (w/v) SDS
Acknowledgments
The authors gratefully acknowledge grant support from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–EXC-2048/1–project ID 390686111, and the Alexander von Humboldt Professorship to WBF. Support from the CEA-Enhanced Eurotalents program and ANR-19-CE13-0007 “PHLOWZ” is gratefully acknowledged (HJ).
Competing interests
W.B.F. is author of the patent filed for the Matryoshka technology (U.S. Appl. No. 15/438,078). The remaining authors have no conflict of interest or competing interest to declare.
References
- Ast, C., Foret, J., Oltrogge, L. M., De Michele, R., Kleist, T. J., Ho, C. H. and Frommer, W. B. (2017). Ratiometric Matryoshka biosensors from a nested cassette of green- and orange-emitting fluorescent proteins. Nat Commun 8(1): 431.
- Bermejo, C., Haerizadeh, F., Takanaga, H., Chermak, D. and Frommer, W. B. (2011). Optical sensors for measuring dynamic changes of cytosolic metabolite levels in yeast. Nat Protoc 6(11): 1806-1817.
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W. and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263(5148): 802-805.
- Chudakov, D. M., Matz, M. V., Lukyanov, S. and Lukyanov, K. A. (2010). Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90(3): 1103-1163.
- Deuschle, K., Chaudhuri, B., Okumoto, S., Lager, I., Lalonde, S. and Frommer, W. B. (2006). Rapid metabolism of glucose detected with FRET glucose nanosensors in epidermal cells and intact roots of Arabidopsis RNA-silencing mutants. Plant Cell 18(9): 2314-2325.
- Frommer, W. B., Davidson, M. W. and Campbell, R. E. (2009). Genetically encoded biosensors based on engineered fluorescent proteins. Chem Soc Rev 38(10): 2833-2841.
- Kaper, T., Lager, I., Looger, L. L., Chermak, D. and Frommer, W. B. (2008). Fluorescence resonance energy transfer sensors for quantitative monitoring of pentose and disaccharide accumulation in bacteria. Biotechnol Biofuels 1(1): 11.
- Kuner, T. and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27(3): 447-459.
- March, J. C., Rao, G. and Bentley, W. E. (2003). Biotechnological applications of green fluorescent protein. Appl Microbiol Biotechnol 62(4): 303-315.
- Mehta, S., Zhang, Y., Roth, R. H., Zhang, J. F., Mo, A., Tenner, B., Huganir, R. L. and Zhang, J. (2018). Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat Cell Biol 20(10): 1215-1225.
- Miesenbock, G., De Angelis, D. A. and Rothman, J. E. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394(6689): 192-195.
- Misteli, T. and Spector, D. L. (1997). Applications of the green fluorescent protein in cell biology and biotechnology. Nat Biotechnol 15(10): 961-964.
- Perez Koldenkova, V. and Nagai, T. (2013). Genetically encoded Ca(2+) indicators: properties and evaluation. Biochim Biophys Acta 1833(7): 1787-1797.
- Shimomura, O., Johnson, F. H. and Saiga, Y. (1962). Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol 59: 223-239.
- Valeur, B. and Berberan-Santos, M. N. (2012). Molecular fluorescence: principles and applications. John Wiley & Sons.
- Shcherbakova, D. M., Hink, M. A., Joosen, L., Gadella, T. W. and Verkhusha, V. V. (2012). An orange fluorescent protein with a large Stokes shift for single-excitation multicolor FCCS and FRET imaging. J Am Chem Soc 134(18): 7913-7923.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sadoine, M., Castro-Rodríguez, V., Poloczek, T., Javot, H., Sunal, E., Wudick, M. M. and Frommer, W. B. (2020). Affinity Purification of GO-Matryoshka Biosensors from E. coli for Quantitative Ratiometric Fluorescence Analyses. Bio-protocol 10(19): e3773. DOI: 10.21769/BioProtoc.3773.
Category
Cell Biology > Cell-based analysis > Protein interaction
Molecular Biology > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.