- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Whole-genome Identification of Transcriptional Start Sites by Differential RNA-seq in Bacteria

Published: Vol 10, Iss 18, Sep 20, 2020 DOI: 10.21769/BioProtoc.3757 Views: 7477
Reviewed by: Imre GáspárShyam SolankiAdam Idoine
Original research article
The authors used this protocol in:

Apr 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Gene transcription in bacteria often starts some nucleotides upstream of the start codon. Identifying the specific Transcriptional Start Site (TSS) is essential for genetic manipulation, as in many cases upstream of the start codon there are sequence elements that are involved in gene expression regulation. Taken into account the classical gene structure, we are able to identify two kinds of transcriptional start site: primary and secondary. A primary transcriptional start site is located some nucleotides upstream of the translational start site, while a secondary transcriptional start site is located within the gene encoding sequence.
Here, we present a step by step protocol for genome-wide transcriptional start sites determination by differential RNA-sequencing (dRNA-seq) using the enteric pathogen Shigella flexneri serotype 5a strain M90T as model. However, this method can be employed in any other bacterial species of choice. In the first steps, total RNA is purified from bacterial cultures using the hot phenol method. Ribosomal RNA (rRNA) is specifically depleted via hybridization probes using a commercial kit. A 5′-monophosphate-dependent exonuclease (TEX)-treated RNA library enriched in primary transcripts is then prepared for comparison with a library that has not undergone TEX-treatment, followed by ligation of an RNA linker adaptor of known sequence allowing the determination of TSS with single nucleotide precision. Finally, the RNA is processed for Illumina sequencing library preparation and sequenced as purchased service. TSS are identified by in-house bioinformatic analysis.
Our protocol is cost-effective as it minimizes the use of commercial kits and employs freely available software.
Background
Transcription in bacteria is initiated by the RNA polymerase holoenzyme, which recognizes specific sequence elements on the DNA within the promotor region, to which sigma factors are bound (Feklistov et al., 2014). This RNA polymerase holoenzyme binding site defines the Transcriptional Start Site and the direction of transcription. For example, the most common house-keeping sigma factor, named 𝝈70 in Escherichia coli, recognizes two elements centered approximately 10 and 35 bp upstream of the TSS (Feklistov et al., 2014). The RNA polymerase holoenzyme melts the double stranded DNA between 11 nt upstream (position -11) to 3 nt downstream (+3) of the TSS (+1), and the single-stranded DNA can then be used as template for the addition of tri-phosphorylated ribonucleotides. The initiation starts mainly at a specific position, but sometimes “wobbles” of one or more bases up- or downstream are encountered (Murakami and Darst, 2003; Robb et al., 2013; Vvedenskaya et al., 2015). The DNA sequence around TSS have long been recognized as crucial for gene regulation in bacteria (Jacob and Monod, 1961). Depending on the position within the gene structure, which begins with a start codon (usually ATG) and finishes with one of the three stop codons, we can identify two types of transcriptional start sites: primary and secondary. Primary transcriptional start sites (pTSS) are located some nucleotides upstream of the translational start site, while the secondary transcriptional start sites (sTSS) are located within the gene encoding sequence (Figure 1).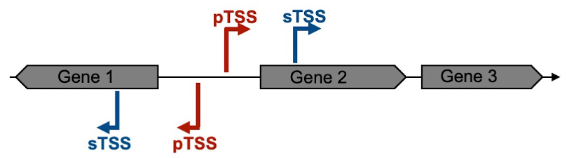
Figure 1. Schematic representation of the Primary and Secondary Transcriptional Start Site definition
Until the advent of next-generation sequencing, in order to locate the TSS of a specific RNA, it was necessary to examine each transcript individually, using either the S1 protection assay, primer extension or a 5’ RACE method (Sharma and Vogel, 2014). Owing to the increasing popularity and a decrease in costs of high-through put sequencing, in 2010 differential RNA-seq (dRNA-seq) was developed to simultaneously map all TSS of a genome using Helicobacter pylori as first model organism (Sharma et al., 2010). Since then, this method has been widely employed to determine the TSS of several bacterial species (Berghoff et al., 2009; Jager et al., 2009; Albrecht et al., 2010 and 2011; Bohn et al., 2010; Irnov et al., 2010; Schluter et al., 2010; Sharma et al., 2010; Beckmann et al., 2011; Deltcheva et al., 2011; Filiatrault et al., 2011; Mitschke et al., 2011a and 2011b; Kroger et al., 2012 and 2013; Madhugiri et al., 2012; Ramachandran et al., 2012 and 2014; Sahr et al., 2012; Schmidtke et al., 2012; Wilms et al., 2012; Cortes et al., 2013; Dugar et al., 2013; Mentz et al., 2013; Nickel et al., 2013; Pfeifer-Sancar et al., 2013; Porcelli et al., 2013; Schluter et al., 2013; Voss et al., 2013; Wiegand et al., 2013; Zhang et al., 2013; Voigt et al., 2014; Cervantes-Rivera et al., 2020).
Primary transcripts of prokaryotes carry a triphosphate at their 5’-ends. In contrast, processed or degraded RNAs only carry a monophosphate at their 5’-ends. This is also the case of ribosomal RNA (rRNA) (Schoenberg, 2007). The dRNA-seq approach used here exploits the properties of a 5’-monophosphate-dependent exonuclease (TEX) to selectively degrade processed transcripts, thereby enriching for unprocessed RNA species carrying a native 5’-triphosphate (Schoenberg, 2007). TSS can then be identified by comparing TEX-treated and untreated RNA-seq libraries, where TSS appear as localized maxima in coverage enriched upon TEX-treatment (Sharma et al., 2010).
Until 2013 TSS annotation was performed manually, but this method is arduous and time-consuming. Nowadays many computational tools are available for automatic TSS annotation using dRNA-seq data. These include TSSPredator (Dugar et al., 2013), TSSAR (Amman et al., 2014), TruHMM (Li et al., 2013), TSSer (Jorjani and Zavolan, 2014) and ReadXplorer2 (Hilker et al., 2016).
Here, we present a step by step protocol for TSS determination through comparison of TEX-treated and untreated RNA libraries in Shigella flexneri serotype 5a strain M90T as originally performed in (Cervantes-Rivera et al., 2020). The overall workflow is illustrated in Figure 2.
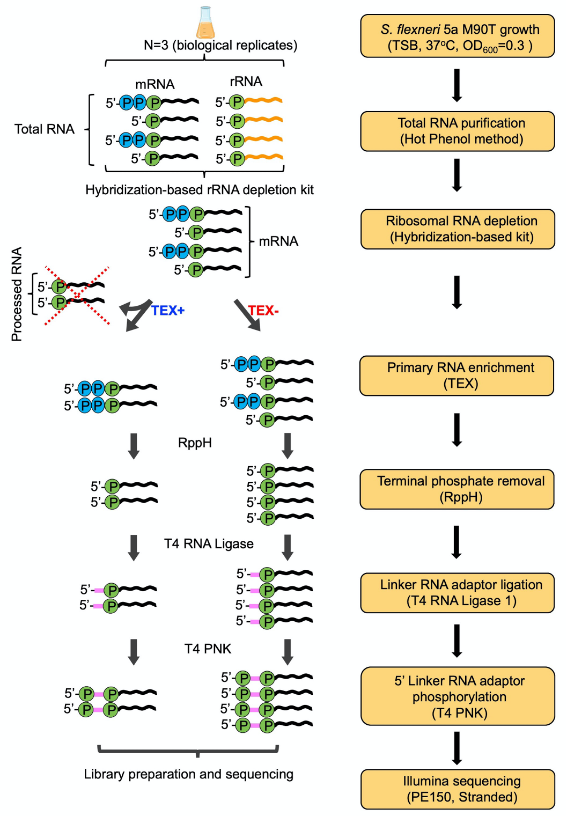
Figure 2. Workflow of dRNA-seq for whole-genome Transcriptional Start Sites identification
Materials and Reagents
- Sterile syringe filter with pore size 0.22 μm (Merck, catalog number: SLGV033RK )
- Nitrile gloves
- Culture tube of 13 ml (TPP, catalog number: 91016 )
- Microfuge tubes 1.5 ml (Eppendorf, Safe-Lock tubes, catalog number: 00 30120086 )
Note: In principle all prepackaged tubes on the market are RNase free and can be used. - Centrifuge tubes of 50 ml (Sarstedt, catalog number: 62.547.254 )
- Pipette tips (VWR, catalog numbers: 89041-404 , 89041-412 , 89041-400 )
Note: In principle all prepackaged tips on the market are RNase free and can be used. - Semi-micro cuvette (Sarstedt, catalog number: 67.742 )
- Shigella flexneri 5a M90T (Sansonetti et al., 1982), can also be purchased from ATCC and available on request from the authors
- Tryptone soy broth (TSB) ready to use powder (Merck, catalog number: 105459 )
- Tryptone soy agar (TSA) ready to use powder (Merck, catalog number: 105458 )
- Congo red (Sigma, catalog number: C6277 )
- Diethyl pyrocarbonate (DEPC) (Sigma, catalog number: 40718 )
- Sodium dodecyl sulfate (SDS) (Sigma, catalog number: L3771 )
- Bromophenol blue sodium salt (Sigma, catalog number: B8026 )
- Xylene cyanol FF (Sigma, catalog number: X4126 )
- Glycerol (Sigma, catalog number: G5516 )
- Formamide (Sigma, catalog number: 47671 )
- Ethanol absolute (VWR Chemicals, catalog number: 20821.558 )
- Sodium acetate 3 M, pH 5.5 (Ambion, catalog number: AM9740 ).
- EDTA, disodium salt, dihydrate (Sigma, catalog number: 324503 )
- Sodium hydroxide (Sigma, catalog number: S8045 )
- Trizma base (Sigma, catalog number: T1503 )
- Acetic acid (Sigma, catalog number: 695092 )
- Agarose (VWR Life Science, catalog number: 35-1020 )
- DNase I 10 U/ml (Roche, catalog number: 0 4716728001 )
- RNaseOUT 40 U/ml (Invitrogen, catalog number: 10777019 )
- Chloroform (Sigma, Catalog number: C2432 )
- Phenol solution pH 4.3 ± 0.2 (Sigma, catalog number: P4682-400ML )
- Isoamyl alcohol (Sigma, catalog number: W205702 )
- Sodium acetate pH 5.5 (Ambion, catalog number: AM9740 )
- Glycogen 20 mg/ml (Invitrogen, catalog number: R0551 )
- Oligos SF-Hfq-F 5′-ACGATGAAATGGTTTATCGAG-3′ and SF-Hfq-R 5′-ACTGCTTTACCTTCACCTACA-3′ (Sigma, custom order)
- GeneRuler 1 kb DNA ladder (Thermo Scientific, catalog number: SM0211 )
- Linker-RNA-adaptor 5′-GACCUUGGCUGUCACUCA-3′ (Sigma, custom order)
- Ribo-Zero rRNA Removal Kit for bacteria (Illumina, catalog number: MRZB12424 )
Note: This product is not available anymore on the market, but can be replaced with Pan-Prokaryote riboPOOL kit (siTOOLS BIOTECH, Pan-Prokaryote v2), RiboCop rRNA Depletion Kit for Bacteria (Lexogen, catalog number: 125-127 ) or any other rRNA depletion method based on the use of hybridization probes. Replacement products relying on enzymatic-based rRNA depletion such as the Ribo-Zero Plus rRNA Depletion Kit (Illumina, catalog number: 20037125 ) are not suitable. - Terminator-5′-Phosphate-Dependent Exonuclease (TEX) (Lucigene, catalog number: TER51020 )
- 5’-Pyrophosphohydrolase 5,000 units/ml (RppH) (New England BioLabs, catalog number: M0356S )
- T4 RNA ligase 10,000 U/ml (New England BioLabs, catalog number: M0204L )
- T4 polynucleotide kinase (PNK) 10,000 U/ml (New England BioLabs, catalog number: M0201L )
- RNaseZap (Ambion, catalog number: AM9780 )
- Dream Taq DNA polymerase (Thermo Scientific, catalog number: EP0701 )
- dNTPs mix, 2 mM each (Thermo Scientific, catalog number: R0241 )
- GelRed nucleic acid gel stain (Biotium, catalog number: 41003 )
- ATP solution (100 mM) (Thermo Scientific, catalog number: R0441 )
- Tryptone soy broth (TSB) medium (see Recipes)
- Tryptone soy agar (TSA) plates (see Recipes)
- Congo red solution (see Recipes)
- Nuclease-free water (see Recipes)
- Sodium dodecyl sulfate (SDS) 10% solution (see Recipes)
- EDTA 0.5 M, pH 8 (see Recipes)
- 6x DNA Loading buffer (see Recipes)
- 2x RNA Loading buffer (see Recipes)
- Stop Solution (see Recipes)
- Ethanol 80% (see Recipes)
- Lysis solution (see Recipes)
- 10x TAE (see Recipes)
- 1% Agarose (see Recipes)
- Phe-CHISAM solution (see Recipes)
- CHISAM (see Recipes)
Equipment
- Microcentrifuge, refrigerated (VWR, model: Micro Star 17R )
- Spectrophotometer (Amersham, model: Ultrospec 2100 pro )
- Pipettes (Eppendorf Research plus)
- Microwave oven (Whirlpool, model: MD101 )
- Centrifuge (Eppendorf, model: 5810R )
- NanoDrop ND-1000 spectrophotometer (Saveen & Werner AB)
- Orbital Shaker (Edmund Bühler, model: Swip SM25 )
- Block heater (VWR, model: 460-0353 )
- DynaMag-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D )
- T100 Thermo cycler (Bio-Rad, catalog number: 186-1096 )
- Chemical hood
- Horizontal gel electrophoresis system (VWR, model: 700-0015 )
- Electrophoresis power supply (VWR, model: E0202 )
Software
Programs
All programs used in this protocol are freely available
- bcl2fastq (https://emea.support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html)
- FastQC/0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc) (Wingett and Andrews, 2018)
- MultiQC/1.8 (https://multiqc.info/) (Ewels et al., 2016)
- trimmomatic/0.36 (http://www.usadellab.org/cms/?page=trimmomatic) (Bolger et al., 2014)
- samtools/1.9 (http://www.htslib.org/doc/samtools.html) (Li et al., 2009)
- bowtie2/2.3.5.1 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) (Langmead and Salzberg, 2012)
- IGV/2.3.82 (https://software.broadinstitute.org/software/igv/) (Thorvaldsdottir et al., 2013)
- ReadXplorer/2.2.3 (https://www.uni-giessen.de/fbz/fb08/Inst/bioinformatik/software/ReadXplorer) (Hilker et al., 2014; Hilker et al., 2016)
Databases
- GenBank (https://www.ncbi.nlm.nih.gov/genome/182?genome_assembly_id=493862)
- RefSeq (https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP037923.1, https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP037924.1)
- RegulonDB (http://regulondb.ccg.unam.mx/)
- dRNA-seq raw data are available on the SRA database under the accession numbers: SRR8921222(dRNA-Seq_TEX_Positive), SRR8921223 (dRNA-Seq_TEX_Negative) (https://dataview.ncbi.nlm.nih.gov/)
Procedure
Before starting the RNA purification be sure that you have cleaned pipets, centrifuges, bench, the chemical hood, equipment for gels and the marker pen with RNaseZap to ensure that everything is RNase free. At every step of the protocol you should wear nitrile/latex gloves to avoid RNase contamination.
- Total RNA purification using the hot phenol method and RNA quality control by electrophoresis
- Prepare a pre-culture by inoculating 5 ml of TSB medium with a single colony of S. flexneri 5a M90T from a Congo red tryptone soy agar plate and grow overnight (~16 h) at 37 °C with agitation (150 RPM).
Note: Ensure that the colonies used are secretion-competent (forming red colonies on the plate due to Congo red absorption) (Sharma and Puhar, 2019). - Subculture by diluting 100 μl of the pre-culture in 10 ml of TSB (1:100 dilution).
- Grow to OD600 = 0.3 with shaking. This will take approximately 2 h if the medium is pre-warmed.
- Mix the culture with 2 ml of cold stop solution.
- Incubate the mixed culture with the stop solution on ice for 30 min.
Note: To stabilize the RNA and prevent degradation, incubate it for at least 30 min, but not longer than 2 h on ice. - Transfer the culture into a 15 ml tube.
- Centrifuge for 5 min at 16,200 x g at 4 °C.
- Remove the supernatant by decanting. Keep ~2 ml of supernatant to resuspend the pellet in it.
- Transfer the resuspended cells into two 1.5 ml tubes.
- Centrifuge for 5 min at 16,200 x g at 4 °C.
- Discard the supernatant by decanting.
Note: At this point the samples could be frozen with liquid nitrogen and stored at -80 °C. Samples are stable for months. - Purify the total RNA using the hot phenol method as follows.
- Resuspend cell pellets in 500 μl of lysis solution at room temperature.
- Under the chemical hood add 500 μl of phenol pH 4 pre-warmed to 65 °C. Mix by inversion at least 20 times.
Note: Pre-warm in a heat block the phenol before starting the RNA purification. - Vortex vigorously for 20 s.
- Incubate 5 min at 65 °C in the heat block.
- Centrifuge 3 min at 13,800 x g at room temperature.
Note: If the centrifugation step is done at 4 °C the SDS is going to precipitate and the solution turns white, which does not affect the efficacy of the process but makes it difficult to differentiate the two phases. After this centrifugation time you should see two phases, the upper one should be whitish (when the SDS is cooling down it becomes insoluble). RNA is in the aqueous phase (upper phase), while in the clearly white disk interphase there are protein layers and DNA, whereas lipids and some other cellular components are dissolved in the lower transparent phase that is the organic phase. - Transfer the upper phase (~500 μl) to a new RNase-free tube under the chemical hood.
- Add 1 ml of cold (4 °C) absolute ethanol.
- Mix thoroughly by vortexing for 5 s until you see a homogeneous mixture.
- Centrifuge at 16,200 x g for 5 min at 4 °C.
- Discard supernatant that contains the ethanol solution by decanting.
Note: At this step you can see a white/transparent pellet on the bottom. - Add 1 ml of 80% ethanol at room temperature. Shake the tube vigorously for 10 s.
Note: Do not use the vortex, it can detach the pellet from the bottom. - Centrifuge at 16,200 x g for 2 min at 4 °C.
- Discard the supernatant by decanting.
- Centrifuge (short spin) the tubes and discard the remaining liquid by pipetting.
- Dry samples at room temperature by leaving the tubes uncapped on the bench.
Note: Once that the sample is completely dry it turns transparent. It can take 10 min to obtain a completely dry sample. - Resuspend the RNA sample in 100 μl of nuclease free water.
- Check the quality of the purified RNA in a 1% agarose gel in TAE 1x as follows.
- Prepare the sample to be analyzed in a 1.5 ml tube. Add 1 μl of sample plus 5 μl of RNA loading buffer 2x and 4 μl of nuclease free water.
- Heat the sample for 5 min at 65 °C for secondary and tertiary structures denaturation.
- Cool down the tube in ice for 5 min.
- Spin down for 2 s and load the gel.
- Run the gel in TAE 1x buffer for 40 min at 100 V.
- Stain the gel with 3x-concentrated Gel Red for 10 min.
- Wash the gel with 20 ml of nuclease free water for 10 min shaking at room temperature.
- Visualize the gel under UV light (Figure 3A). If the total RNA purification was successful, the 23S and 16S are visible as clear, heavy bands, while in the lower part of the gel weaker tRNA bands are observed. It is not possible to see mRNAs. In case of RNA degradation, bands appear as smear.
- Quantify the RNA in a NanoDrop spectrophotometer (Figure 3B).
Note: At least 20 μg (10 μg for each of two conditions) of total RNA are necessary to continue with the protocol. Typical concentrations should be around 500-900 ng/μl. Very successful purifications yield up to 1,500 ng/μl RNA.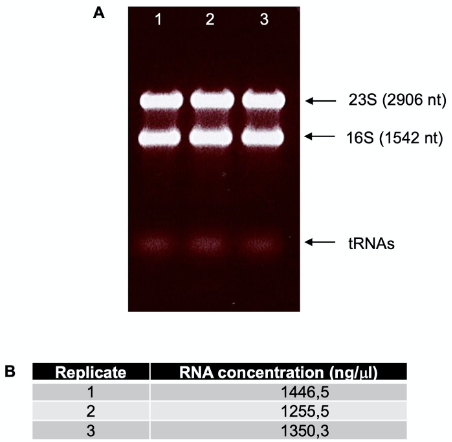
Figure 3. Total RNA quality control. A. Separation of total RNA from three replicates in a 1% agarose gel in TAE of Shigella flexneri 5a M90T as total RNA quality control. B. Examples of typical values obtained from total RNA quantification in a NanoDrop. The 260/280 ratio should be around 1.88-2.00. - Freeze the RNA samples at -20 °C. The RNA samples are stable for months.
- Prepare a pre-culture by inoculating 5 ml of TSB medium with a single colony of S. flexneri 5a M90T from a Congo red tryptone soy agar plate and grow overnight (~16 h) at 37 °C with agitation (150 RPM).
- DNase treatment to remove contaminating genomic DNA from total RNA
Protocol volumes are for one reaction, but every step is scalable to two or more reactions.- Dilute 10 μg of total RNA to a final volume of 30 μl using nuclease free water.
- Denature RNA in nuclease free water for 5 min at 65 °C in the heat block.
- Cool down in ice for 5 min.
- Prepare a reaction mix containing the components listed in Table 1. The indicated quantities are sufficient for one sample containing 10 μg of RNA.
Table 1. DNase reaction mix for one reaction
Note: 1 Unit of DNase I for 1 μg of total RNA.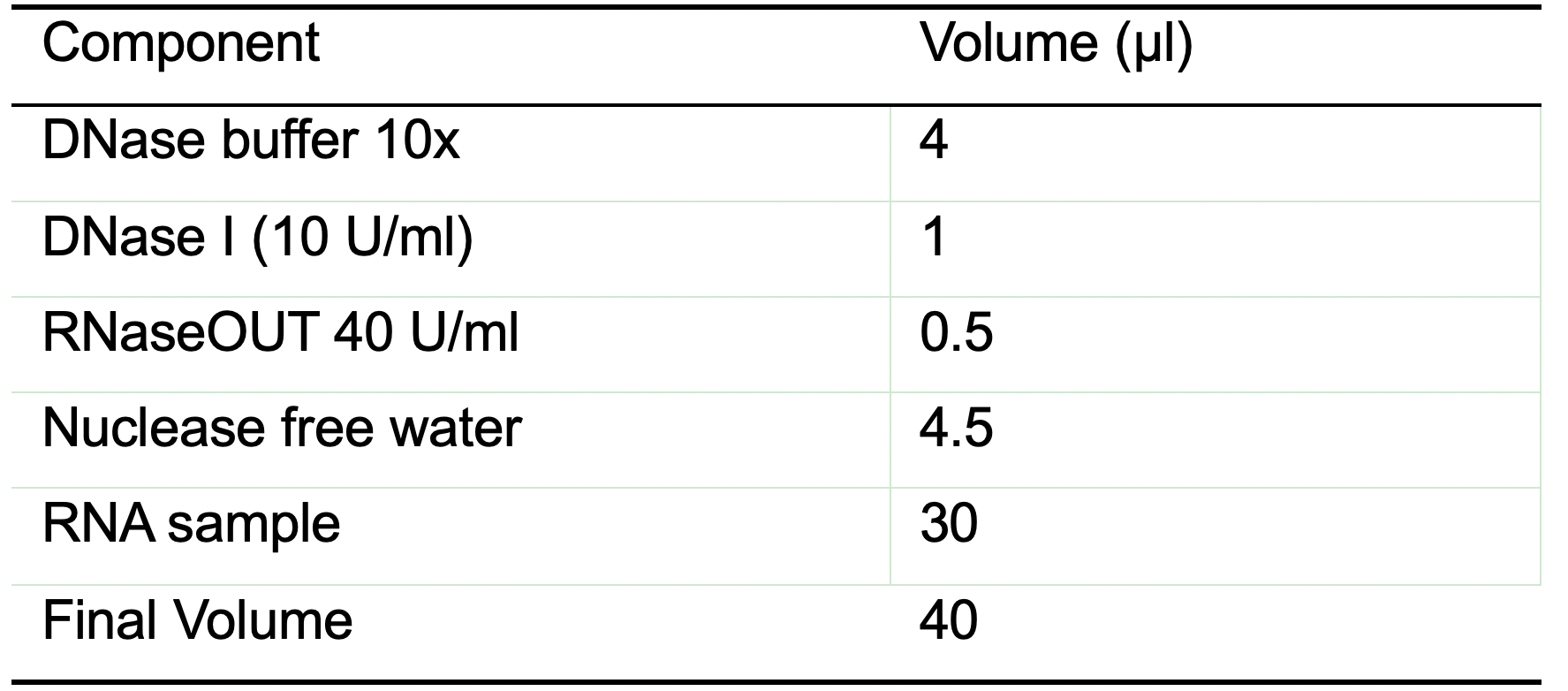
- Add the reaction mix to the RNA sample tube on ice.
- Incubate the reaction mix for 60 min at 37 °C in the heat block.
Note: Be sure that the heat block is at the correct temperature, DNase works efficiently at 37 °C. - Add to the reaction tube 60 μl of nuclease free water to obtain 100 μl final volume.
- RNA clean-up by chloroform:isoamyl alcohol extraction and ethanol precipitation
- Add 100 μl of CHISAM to the reaction tube under the chemical hood.
- Mix with the vortex vigorously for 15 s.
- Centrifuge the tube for 2 min at 13,800 x g, 4 °C.
- Under the chemical hood transfer ~100 μl from the aqueous phase (upper) that contains the RNA to a new 1.5 ml tube. Discard the organic phase (lower), proteins and salts from the buffer are dissolved in this phase.
- Repeat Steps C1-C4.
- Add 300 μl of cold absolute ethanol (3 sample volumes), 10 μl of sodium acetate 3 M pH 5.5 (1/10 of sample volume) and 1 μl of glycogen (20 mg/ml).
- Vortex the mix vigorously for 5 s.
- Incubate the tube at -20 °C for 30 min.
Note: This step is optional, but it is good to do it to decrease the loss of RNA in the next steps. - Centrifuge at 16,200 x g (or maximum speed) for 30 min at 4 °C.
Note: Centrifugation step can be for longer time. The minimum is 30 min, but it could be for 2 to 4 h. - Discard the supernatant carefully by decanting.
Note: On the bottom of the tube there should be a white pellet. Discard the supernatant without detaching the pellet. - Add 1 ml of 80% ethanol at room temperature to wash the RNA.
- Mix the tube by inverting 20 times.
- Centrifuge the tube for 5 min at 13,800 x g, 4 °C.
- Discard the supernatant by decanting.
- Centrifuge (short spin) and discard the remaining liquid with a 100 μl pipette.
- Keep the tubes open on ice to dry the samples.
Note: The time required to dry the samples depends on how well the liquid phase was removed (usually it takes 10-15 min). The pellet is completely transparent once it is completely dry. - Resuspend the total RNA in 23 μl of nuclease free water.
- Check the integrity of the DNase-free RNA by visual inspection of the 23S and 16S band on 1% agarose gel as performed after the total RNA extraction.
- Prepare the running sample as in Steps A30-A37.
- Visualize the gel under UV light (Figure 4A).
- Quantify the RNA in the NanoDrop spectrophotometer (the final RNA concentration should be ~600-1,000 ng/ml) (Figure 4B). It is normal to detect lower concentrations after the process, as the presence of DNA increase the absorbance of the sample.
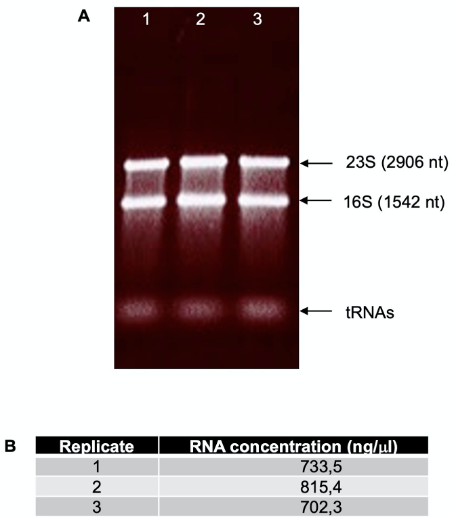
Figure 4. Total RNA quality control after DNase treatment. A. Separation of total RNA from three replicates in a 1% agarose gel in TAE of Shigella flexneri 5a M90T as total RNA quality control. B. Examples of typical values obtained from total RNA quantification after DNase treatment in a NanoDrop. - Freeze the RNA samples at -20 °C. The RNA samples are stables for months.
- PCR to verify the removal of genomic DNA
To be sure that there is no trace of DNA in the RNA samples it is extremely important to check the absence of genomic DNA by PCR.- Use 1 μl (~1 μg) of the DNase treated RNA sample as a template to check for the absence of genomic DNA.
- As a positive control use ~10 ng of genomic DNA from S. flexneri 5a M90T. Genomic DNA can be extracted using the phenol-chloroform protocol as described in (He, 2011).
- As a negative control use a reaction without any template.
- Prepare the PCR mix (Table 2). The indicated quantities are sufficient for one sample.
Table 2. Reaction mix for the PCR control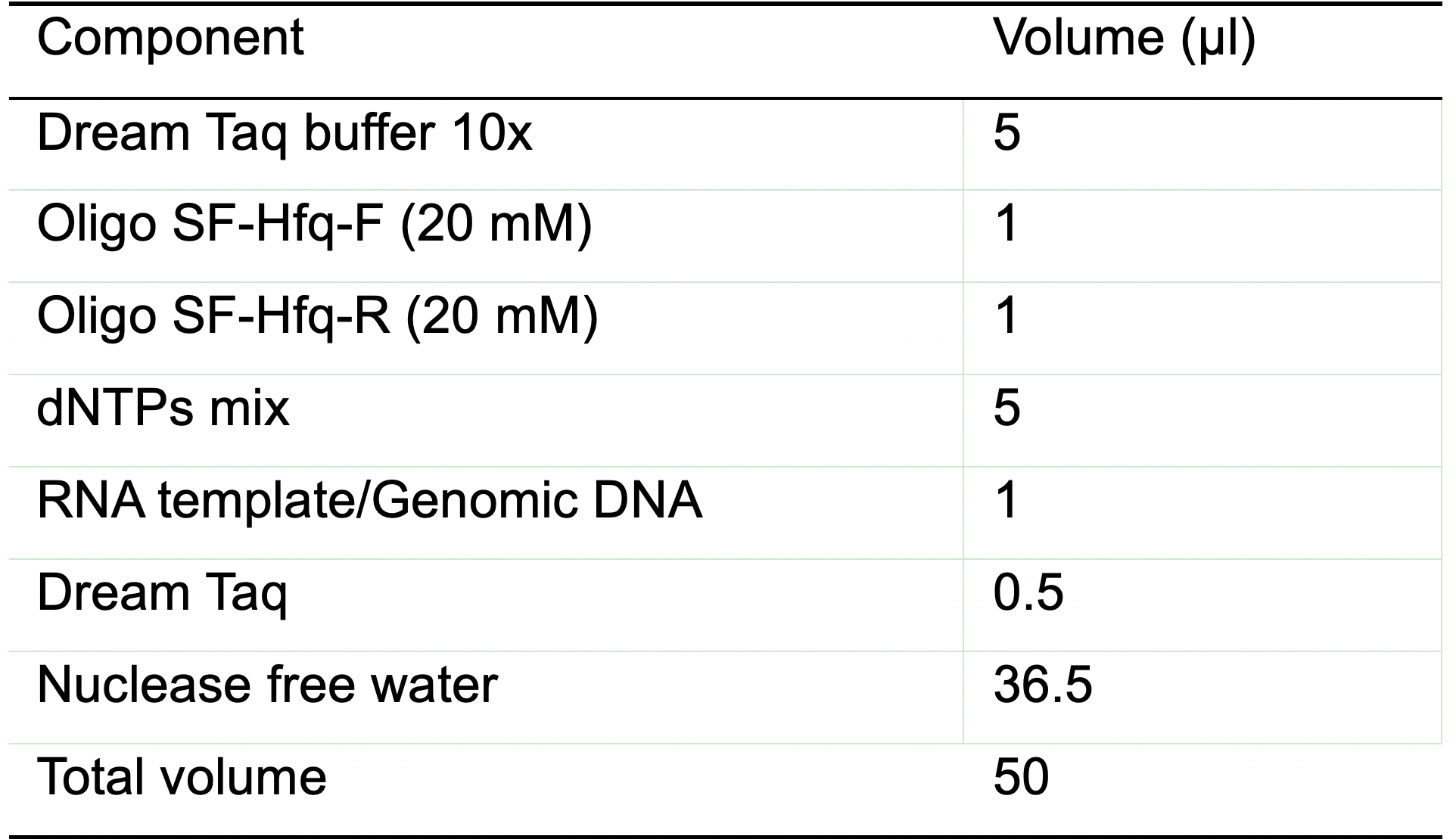
Note: The oligos used here amplify a ~909 bp long product of the hfq gene (303 nt) from S. flexneri 5a M90T including 300 bp upstream and 300 bp downstream of the hfq gene. - PCR reaction settings (Table 3).
Table 3. Settings for control PCR
- Check the product in a 1% agarose gel as follows.
- Prepare the sample to be analyzed. Add 6 μl of sample plus 2 μl of DNA loading buffer 6x.
- Run the 1% agarose gel in TAE 1x buffer for 40 min at 100 V.
- Stain the gel with Gel Red 3x for 10 min.
- Wash the gel with nuclease free water.
- Visualize the gel under UV light (Figure 5).
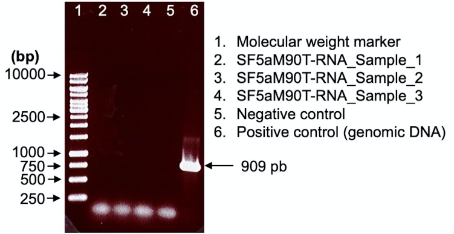
Figure 5. Control PCR to verify the absence of genomic DNA with the hfq gene plus 300 pb upstream and downstream of Shigella flexneri 5a M90T as target using genomic DNA as a positive control. Agarose gel 1% in TAE 1x. The absence of a PCR product seen as a 909 bp band in lanes 2-4 indicates that the purified RNA is devoid of DNA contaminations.
- rRNA depletion
Ribosomal RNA is the most abundant RNA species. If rRNA is not removed, most reads from sequencing will originate from rRNA. As a consequence, samples which are compared need to all either have or be devoid of rRNA, or else the sequencing depth will be very different and statistical analysis cannot be performed. The TEX treatment described in the next section to enrich for primary RNA removes all RNA that is not tri-phosphorylated including rRNA, making this library rRNA-depleted. Therefore, to allow the comparison of TEX-treated with mock-treated samples, rRNA needs to be specifically depleted first from total RNA before splitting the RNA into two for further treatment.
rRNA depletion is carried out using a commercial kit, which specifically depletes bacterial rRNA, according to manufacturer’s instructions. Specific depletion of bacterial rRNA generally relies on hybridization with tagged pan-bacterial probes that recognize conserved sequences in rRNA, followed by removal of captured rRNAs, for example by precipitation with magnetic beads that bind the tag of the probes.
We have used the Ribo-Zero rRNA Removal Kit for Bacteria from Illumina, but unfortunately it is not available anymore on the market. However, it can be replaced with any hybridization-based kit of your choice. We provided some suggestions in the Material section. - Terminator exonuclease (TEX) treatment to enrich for primary transcripts
- Prepare two (TEX- and TEX+) 1.5 ml reaction tubes with 10 μg (10 μl) each of DNase I treated RNA (from the same sample) in nuclease free water.
- Denature RNAs for 2 min at 90 °C.
- Cool down on ice for 5 min.
- Add to each tube the reaction mix described in Table 4. The indicated quantities are sufficient for one sample containing 10 μg of RNA:
Table 4. Reaction mix for TEX treatment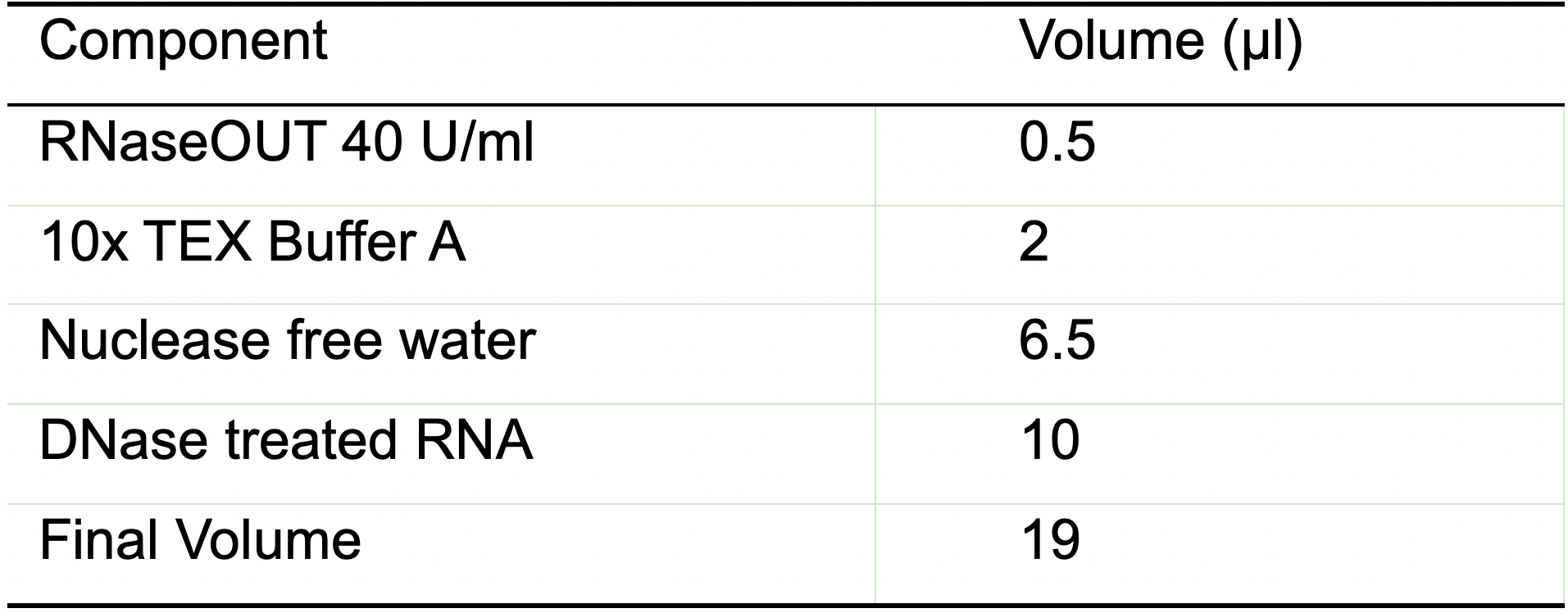
Note: The final reaction volume is going to be 20 μl after adding the enzyme for the TEX+ sample or water for the TEX- sample.- Add 1 μl of nuclease free water to the TEX- sample.
- Add 1 μl of TEX (1 U/ml) to the TEX+ sample.
- Incubate for 60 min at 30 °C in the thermo cycler.
Note: This step could be carried in a heat block, but the temperature variation and liquid evaporation are important factors that hamper good results, and these effects are stronger in a heat block. - Add 80 μl of nuclease free water to obtain 100 μl final volume.
- Add 100 μl of Phe-CHISAM to the reaction tube under the chemical hood.
- Mix with the vortex vigorously for 15 s.
- Centrifuge the tube for 2 min at 13,800 x g, 4 °C.
- Transfer ~100 μl of the upper phase that contains the RNA to a new tube under the chemical hood.
- Repeat Steps F9 to F12.
- Add 300 μl of cold absolute ethanol (3 sample volumes), 10 μl of sodium acetate 3M pH 5.5 (1/10 of sample volume) and 2 μl of glycogen (20 mg/ml).
- Vortex the sample vigorously.
- Incubate the tube at -20 °C overnight (around 16 h), but could be longer, for example over the weekend.
Note: This step is very important. It helps complete recovery of the RNA. - Centrifuge at 16,200 x g for 1 h at 4 °C.
Note: The centrifugation step could be for longer time. Minimum 1 h, but could be for 2-4 h. - Discard the aqueous phase carefully by decanting.
Note: On the bottom of the tube there should be a white pellet. Discard the aqueous phase without detaching the pellet. In the case that you do not see any pellet before discarding the supernatant, just add 10 μl of sodium acetate 3 M pH 5.5 (1/10 of sample volume) and 2 μl of glycogen (20 mg/ml) and repeat Steps F16 to F18). - Add 1 ml of cold 80% ethanol at room temperature to wash the RNA.
- Centrifuge for 5 min at 13,800 x g, 4 °C.
- Discard the aqueous phase by decanting.
- Centrifuge (short spin) and discard the remaining liquid with a 100 μl pipette.
- Keep the tubes open on ice to dry the samples.
Note: The time required to dry the samples depends on how well the liquid phase was removed. It can take around 10-15 min. The pellet will be completely transparent once it is completely dry. - Resuspend the RNA in 13 μl of nuclease free water.
- Check the integrity of the TEX- and TEX+ treated RNA by visual inspection on 1% agarose gel.
- Prepare the running sample as in Steps A29-A36.
- Visualize the gel under UV light (Figure 6).
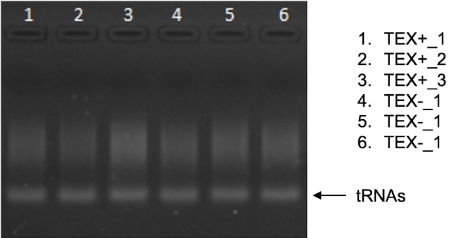
Figure 6. Shigella flexneri 5a M90T RNA treated (TEX+) and untreated (TEX-) with TEX
Note: It is normal not to see clear bands in this gel for the mRNA, which is a mix of different transcripts of variable length. To see a smear in the gel means that the treatment was successful. If you do not have a high quantity of RNA maybe you will see only tRNAs. - Quantify the RNA in the NanoDrop spectrophotometer (final RNA concentration should be ~20 ng/ml).
- Freeze the RNA samples at -20 °C. The RNA samples are stables for months.
- Prepare two (TEX- and TEX+) 1.5 ml reaction tubes with 10 μg (10 μl) each of DNase I treated RNA (from the same sample) in nuclease free water.
- RNA 5’ Pyrophosphohydrolase (RppH) treatment of RNA to remove terminal phosphates
In the following steps, the 5′-ends of the RNA prepared in Procedure F is modified with a linker-RNA-adaptor of known sequence, which allows the identification of TSS with single nucleotide precision. This is necessary because the sequence of adaptors added during library preparation for sequencing can be a trade secret (the full sequence of Illumina adaptors was only revealed recently and other platforms using adaptors of unknown sequence exist). During bioinformatic analysis of sequencing results, the sequence of adaptors is occasionally incompletely removed from reads and the remaining nucleotides could be confused for transcribed bases if they happened to align with the DNA sequence. However, this problem can be avoided by introducing the RNA adaptor of known sequence between the adaptors and the TSS. Moreover, the linker-RNA-adaptor allows to univocally identify 5′ ends present before RNA shearing during library preparation. To allow the ligation of adaptors to the 5’ end of the linker-RNA-adaptor during library preparation, the linker-RNA-adaptor must have a triphosphate in the 5′ end.- Denature remaining 10 μl of TEX+ and TEX- treated RNA for 1 min at 90 °C.
- Cool down on ice for 5 min.
- Prepare RppH mix as described in the Table 5. The indicated quantities are sufficient for one sample:
Table 5. Reaction mix for RppH reaction of TEX treated RNA
- Incubate at 37 °C for 30 min in the thermocycler.
- Add 80 μl of nuclease free water and follow the Steps F8-F23.
- Resuspend the RNA sample in 13 μl of nuclease free water.
- Check the integrity of the RppH treated RNA by visual inspection on 1% agarose gel.
Note: This step it not strictly necessary, and with every step you are losing some RNA. The gel should look similar as the one shown in Figure 6. - Quantify the RNA in the NanoDrop spectrophotometer.
- Linker-RNA-adaptor phosphorylation prior to ligation to TEX+ and TEX- libraries
- Denature 100 pmol (10 μl) of the linker-RNA-adaptor per sample dissolved in nuclease free water for 5 min at 90 °C.
- Cool down on ice for 5 min.
Note: You can keep the oligo on ice for longer time while you prepare the reaction. - Prepare the T4 PNK mix according the Table 6.
Table 6. Reaction mix for T4 PNK phosphorylation of the linker-RNA-adaptor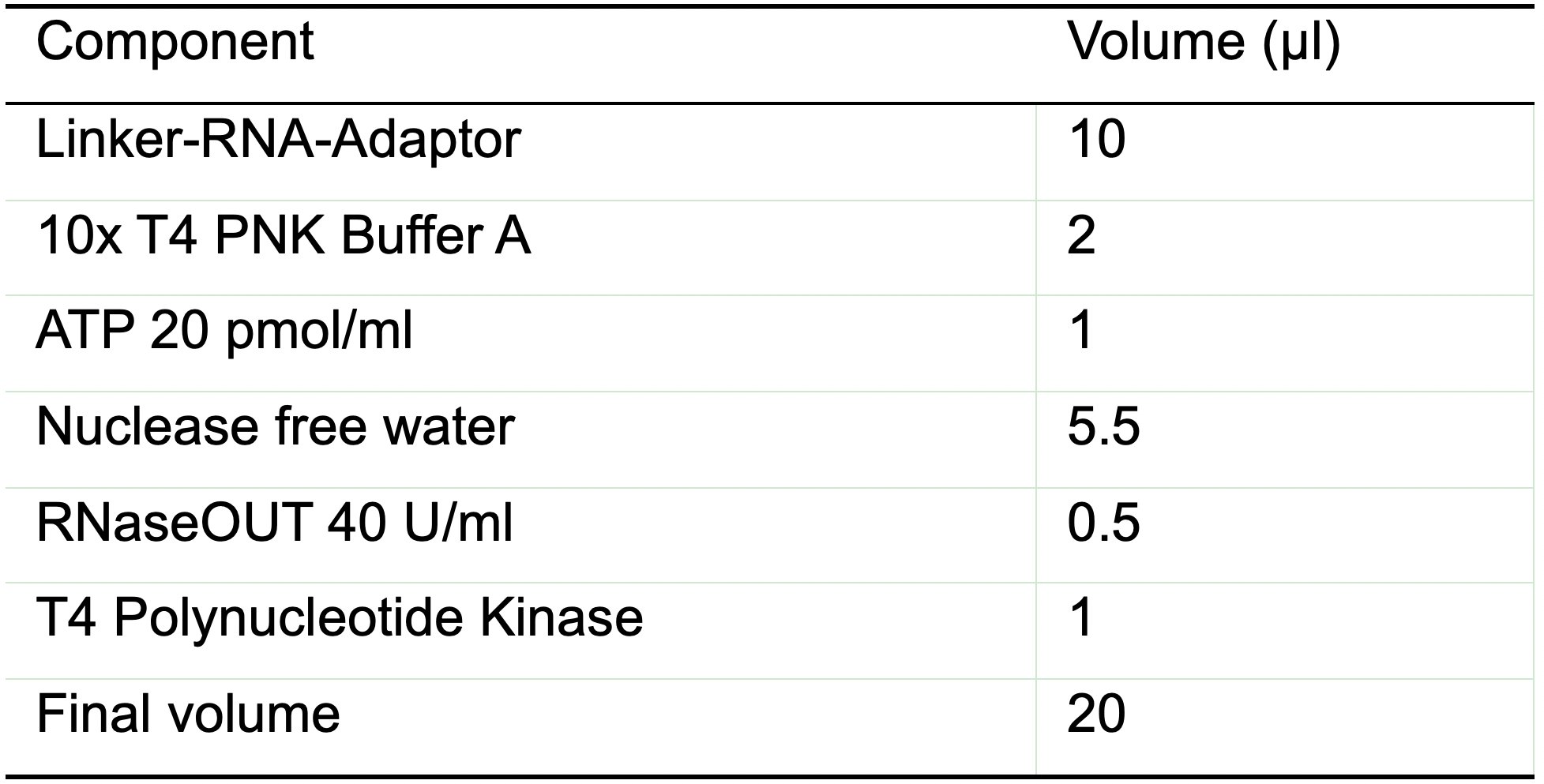
- Incubate at 37 °C for 30 min in the thermo cycler.
- Add 80 μl of nuclease free water, follow the Steps F8-F23.
- Resuspend the RNA sample in 10 μl of nuclease free water.
Note: After this step the RNA adaptor can be frozen at -20 °C and used later.
- Linker-RNA-adaptor ligation to TEX+ and TEX- samples
The efficiency of linker-RNA-adaptor ligation is around 80-90%.- Denature the phosphorylated RNA adaptor (10 μl) and RNAs treated with RppH (10 μl) at 65 °C for 5 min.
- Cool down on ice for 5 min.
- Prepare the T4 RNA ligase mix as described in Table 7 (quantities described on the table are for only one sample, if you are processing more samples just scale them up).
Table 7. Reaction mix for linker-RNA-adaptor ligation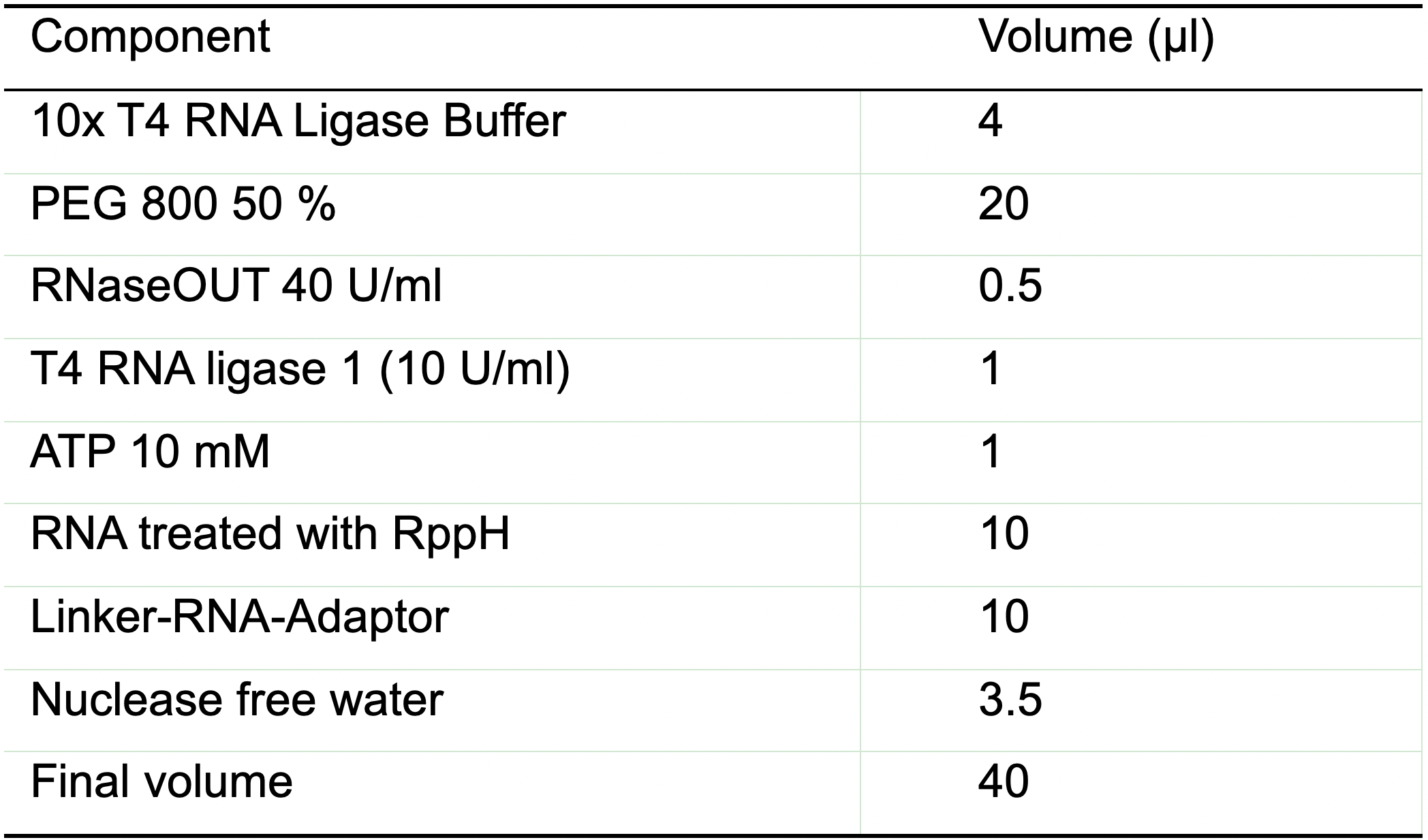
- Incubate at 25 °C for 16 h in the thermo cycler.
- Add 60 μl of nuclease free water.
- Stop the reaction and precipitate the reaction following Steps F9-F23.
- Resuspend the RNA sample in 23 μl of nuclease free water.
Note: Resuspension volume is defined by the sequencing facilities that you have decided to use. The RNA concentration in our case was 150 ng in 23 μl. - Quantify the RNA adaptor by NanoDrop spectrophotometer.
- Library preparation for Illumina sequencing
Library preparation for sequencing and RNA-sequencing is performed at the service platform or company of your choice. Stranded libraries must be prepared to identify the coding DNA strand. Paired-end sequencing with 150 nt read length should be performed. We used a TrueSeq library on a HiSeq 2000 platform. For microbial genomes, 10-20 M reads per sample will provide appropriate sequencing depth, but 5 M it is enough for an acceptable coverage.
Data analysis
All the programs used in this protocol are based on Linux and freely available on the web. We perform all analyses remotely on a high-performance computer cluster. However, if no access to high-speed remote analysis is available, it is possible to run all the analysis locally after downloading the programs, but this may take a long time if the computing power is low.
Download all the programs, raw data and the reference genome into the working directory. In this protocol as example we have called the working directory dRNA_seq.
Note: If you use Windows system you should install Ubuntu (https://ubuntu.com/download/desktop) and run all the programs in this environment.
- Demultiplexing of Illumina reads
Note: This step is only necessary in case you perform the sequencing in your own laboratory. If it is the case you will get a file called *.bcl.gz, which first needs demultiplexing for downstream analysis. This step is not necessary if you use a sequencing service, because you will get the already processed *.fastq.gz file.
bcl2fastq- Go to your working directory where you have your *.bcl.gz file.
- Execute bcl2fastq
$ bcl2fastq [options]
- Go to your working directory where you have your *.bcl.gz file.
- Quality control of raw data
- FastQC (Figure 7)
- Go to your working directory. In this example the working directory:
$ cd dRNA_seq - Create a new directory for the output of FastQC analysis.
$ mkdir FastQC_1 - Execute FastQC.
$ fastqc /dRNA_seq/your_file.fq.gz -o /dRNA_seq/ FastQC_1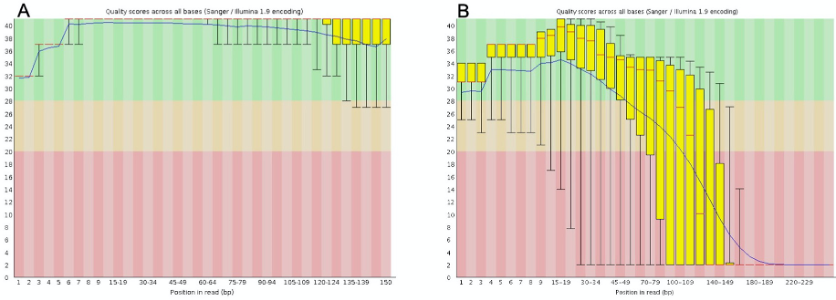
Figure 7. FastQC report of quality control. A. Successful quality control. The quality scores should be in the green area for the entire length of the sequenced fragment. B. Unsuccessful quality control. The quality scores for part of the reads are low (found in the orange or red areas).
- Go to your working directory. In this example the working directory:
- MultiQC
Run MultiQC to merge all the FastQC outputs.
$ multiqc /FastQC_1
- FastQC (Figure 7)
- Cleaning raw data from low quality sequences and Illumina adaptors
Trimmomatic
Run Trimmomatic in your working directory.
$ java -jar trimmomatic.jar PE /your_raw_data_1.fq.gz your_raw_data_2.fq.gz your_raw_data_paired_1.fq.gz your_raw_data_unpaired_1.fq.gz your_raw_data_paured_2.fq.gz your_raw_data_unpaired_2.fq.gz ILLUMINACLIP:/TruSeq3-PE-2.fa:2:30:10 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 MINLEN:100 - Select the fastq files that contains the RNA adaptor sequence
The easiest way to perform this step is using the command grep in your terminal. The linker-RNA-adaptor at this step should be in DNA format.
$ zgrep -B 1 -A 2 GACCTTGGCTGTCACTCA your_raw_data_paired_1.fq.gz > Picked_Up.fastq
- Genome reference index
- It is quite likely that your reference genome has more than one replicon. We would recommend to merge all of them into one single file to be used as a refence genome.
$ cat chromosome.fasta plasmid.fasta > reference_genome.fasta - Create the index with bowtie2.
$ bowtie2-build reference_genome.fasta index_base_name
- It is quite likely that your reference genome has more than one replicon. We would recommend to merge all of them into one single file to be used as a refence genome.
- Genome reference mapping
Map the selected reads with the reference genome.
$ bowtie2 -q -N 0 –no-unal -x index_base_name -U your_raw_data_paured_1.fq.gz -S mapping_1.sam - Sort by position and build an index of BAM file
The output file from Bowtie2 should be sorted and indexed for use in IGV.
$ samtools view -Sb -o mapping_1.bam mapping_1.sam
$ samtools sort -o mapping_Sorted_1.bam mapping_1.bam - Visualize the mapping files in IGV (Figure 8)
- Create a genome reference index.
$ samtools index reference_genome.fasta reference_genome - Create your genome file in IGV.
IGV>Genomes>Create .genome File
Load your reference_genome.fasta and reference_genome.gbk files - Load your mapping file.
IGV>File>Load from File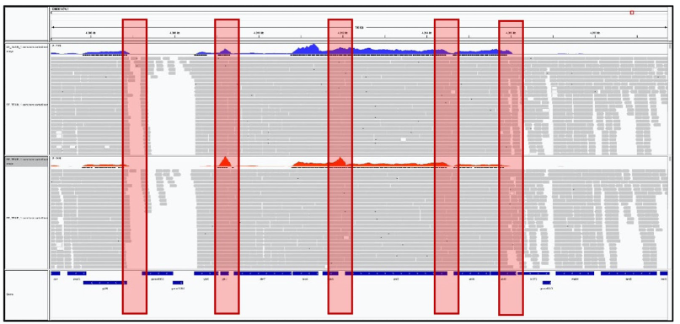
Figure 8. Screenshot of the IGV browser for alignment visualization. Alignment of TEX+ and TEX- libraries with the reference genome of S. flexneri 5a M90T. The blue histogram shows the coverage of the TEX- sample, the red histogram shows the TEX+ sample coverage. The red highlight shows the putative transcriptional start site.
- Create a genome reference index.
- Identify the transcriptional start sites
- Merge the three replicates in one file.
$ samtools merge output.sam mapping_Sorted_1.bam mapping_Sorted_2.bam mapping_Sorted_3.bam - Load the merged file in ReadXplorer.
- Run the TSS analysis with these parameters (Table 8):
Table 8. Settings for the TSS analysis in ReadXplorer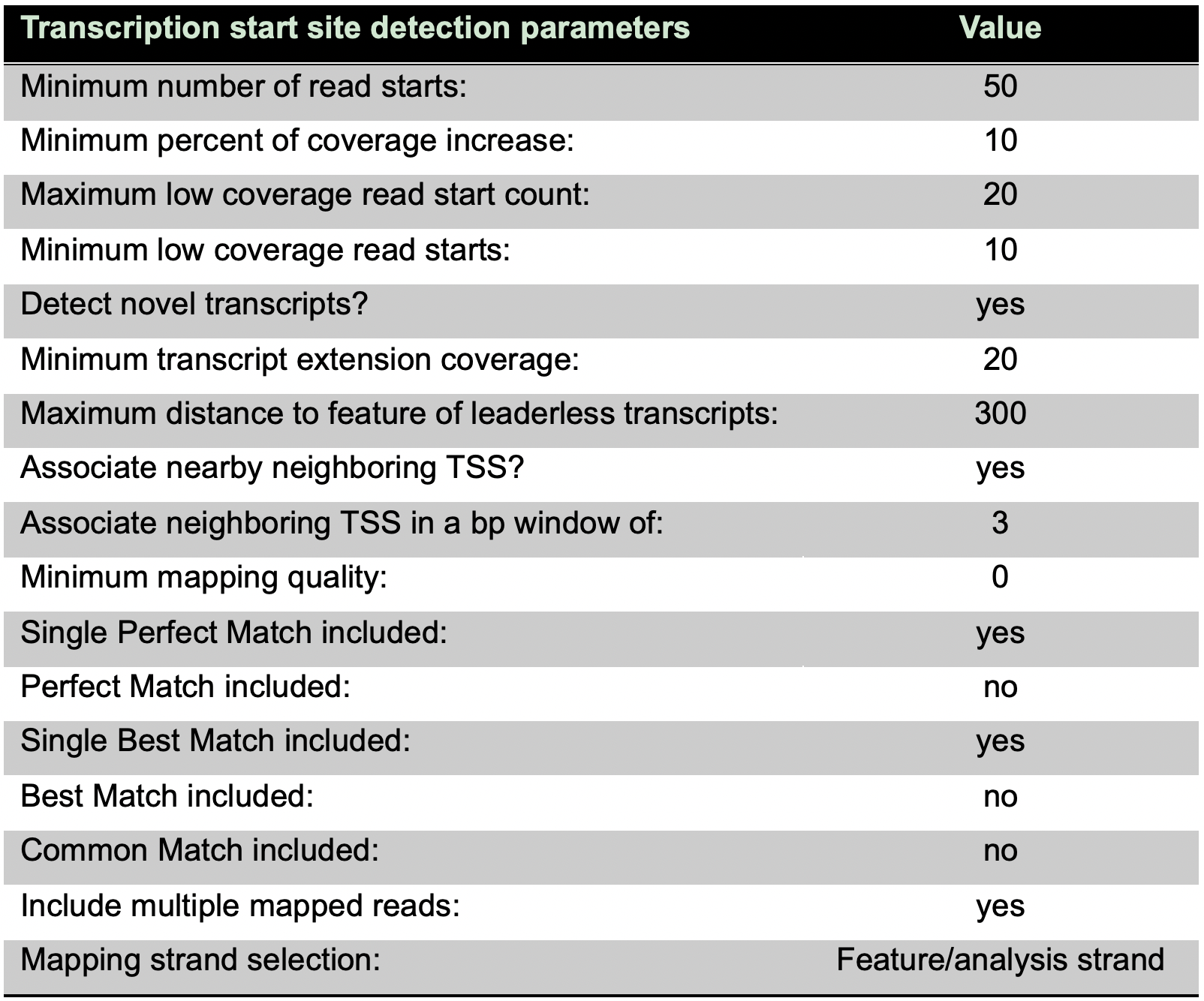
- The output file of ReadXplorer analysis for TSS is a table in plain text with 39 different columns that you can use for a downstream analysis. As an example you can see an extract of some columns of the output file in Figure 9.
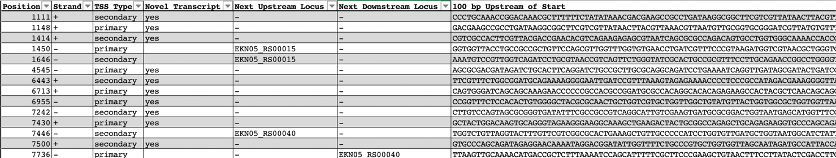
Figure 9. Example of extract from the ReadXplorer output file
- Merge the three replicates in one file.
Recipes
- Tryptone soy broth (TSB)
30 g TSB medium powder
MilliQ water to 1 L
Autoclave the solution - Tryptone soy agar (TSA) plates
40 g Tryptone soy agar powder
MilliQ water to 1 L
Autoclave the solution
Cool to ~50 °C in the water bath, add 10 ml of 1% Congo red solution and pour plates - Congo red solution 1% (w/v)
1 g of Congo red dye
100 ml of MilliQ-water
Filter sterilize the solution, using a 0.2 μm membrane filter.
Note: The Congo red solution is stable for months at room temperature. - Nuclease free water
1 ml of Diethyl Pyrocarbonate (DEPC), (0.1% final concentration)
1 L of MilliQ water
Mix well over night using a magnetic stirring bar
Autoclave
Let cool down to room temperature prior to use - Sodium dodecyl sulfate (SDS) 10% (w/v)
10 g of SDS powder
Nuclease free water 100 ml
Autoclave the solution - EDTA 0.5 M, pH 8
18.61 g disodium ethylenediaminetetraacetate dihydrate (EDTA)
2 g Sodium hydroxide
Nuclease free water 100 ml
Mix well and sterilize
Note: EDTA is solved at pH 8. - DNA loading buffer 6x
0.125 g bromophenol blue (0.025% final concentration)
0.125 g xylene cyanol (0.025% final concentration)
30 ml Glycerol
Nuclease free water to 100 ml
Mix well, it is stable at room temperature for years - RNA loading buffer 2x
0.125 g bromophenol blue (0.025% final concentration)
0.125 g xylene cyanol (0.025% final concentration)
100 ml of formamide
Mix well, it is stable at room temperature for years - Stop Solution
95 ml of ethanol
5 ml of phenol pH 4
Final volume 100 ml
Mix well, store it at 4 °C - Ethanol 80%
80 ml of ethanol
20 ml of nuclease free water
Mix well and store it at 4 °C - Lysis solution
5 ml of SDS 10% (0.5% final concentration)
0.6 ml of sodium acetate pH 5.5 (20 mM final concentration)
2 ml of EDTA pH 8 (10 mM final concentration)
Nuclease free water to 100 ml (92.4 ml)
Mix well, store at room temperature.
Note: At cold temperature the SDS precipitates. If this happens warm up for 15 s at maximum power in the microwave oven. - 10x TAE
11.42 ml of acetic acid
48.4 g of Trizma-base
20 ml of EDTA 0.5 M, pH 8
Nuclease free water to 1 L - Agarose 1%
1 g of agarose powder
100 ml of TAE 1x
Melt the agarose in a microwave oven - Phe-CHISAM solution
25 ml of phenol acid pH 4
24 ml of chloroform
1 ml of isoamyl alcohol
Mix well, store at room temperature in the chemical hood - CHISAM
96 ml of chloroform
4 ml of isoamyl alcohol
Mix well, store at room temperature in the chemical hood
Acknowledgments
A short version of this protocol was published in Cervantes-Rivera et al. (2020). We gratefully acknowledge funding from the Kempe Foundation (JCK-1528), the Knut and Alice Wallenberg Foundation (KAW 2015.0225), Umeå Centre for Microbial Research (UCMR), Umeå University and The Laboratory for Molecular Infection Medicine Sweden (MIMS). We are grateful for travel support and courses organized by The National Doctoral Program in Infections and Antibiotics (NDPIA). The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under Project SNIC 2017-7-258.
Competing interests
The authors declare no conflict of interest.
References
- Albrecht, M., Sharma, C. M., Dittrich, M. T., Muller, T., Reinhardt, R., Vogel, J. and Rudel, T. (2011). The transcriptional landscape of Chlamydia pneumoniae. Genome Biol 12(10): R98.
- Albrecht, M., Sharma, C. M., Reinhardt, R., Vogel, J. and Rudel, T. (2010). Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38(3): 868-877.
- Amman, F., Wolfinger, M. T., Lorenz, R., Hofacker, I. L., Stadler, P. F. and Findeiss, S. (2014). TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinformatics 15: 89.
- Beckmann, B. M., Burenina, O. Y., Hoch, P. G., Kubareva, E. A., Sharma, C. M. and Hartmann, R. K. (2011). In vivo and in vitro analysis of 6S RNA-templated short transcripts in Bacillus subtilis. RNA Biol 8(5): 839-849.
- Berghoff, B. A., Glaeser, J., Sharma, C. M., Vogel, J. and Klug, G. (2009). Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol Microbiol 74(6): 1497-1512.
- Bohn, C., Rigoulay, C., Chabelskaya, S., Sharma, C. M., Marchais, A., Skorski, P., Borezee-Durant, E., Barbet, R., Jacquet, E., Jacq, A., Gautheret, D., Felden, B., Vogel, J. and Bouloc, P. (2010). Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38(19): 6620-6636.
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
- Cervantes-Rivera, R., Tronnet, S. and Puhar, A. (2020). Complete genome sequence and annotation of the laboratory reference strain Shigella flexneri serotype 5a M90T and genome-wide transcriptional start site determination. BMC Genomics 21(1): 285.
- Conway, T., Creecy, J. P., Maddox, S. M., Grissom, J. E., Conkle, T. L., Shadid, T. M., Teramoto, J., San Miguel, P., Shimada, T., Ishihama, A., Mori, H. and Wanner, B. L. (2014). Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing. mBio 5(4): e01442-01414.
- Cortes, T., Schubert, O. T., Rose, G., Arnvig, K. B., Comas, I., Aebersold, R. and Young, D. B. (2013). Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep 5(4): 1121-1131.
- Deltcheva, E., Chylinski, K., Sharma, C. M., Gonzales, K., Chao, Y., Pirzada, Z. A., Eckert, M. R., Vogel, J. and Charpentier, E. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340): 602-607.
- Dugar, G., Herbig, A., Forstner, K. U., Heidrich, N., Reinhardt, R., Nieselt, K. and Sharma, C. M. (2013). High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet 9(5): e1003495.
- Ewels, P., Magnusson, M., Lundin, S. and Kaller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32(19): 3047-3048.
- Feklistov, A., Sharon, B. D., Darst, S. A. and Gross, C. A. (2014). Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol 68: 357-376.
- Filiatrault, M. J., Stodghill, P. V., Myers, C. R., Bronstein, P. A., Butcher, B. G., Lam, H., Grills, G., Schweitzer, P., Wang, W., Schneider, D. J. and Cartinhour, S. W. (2011). Genome-wide identification of transcriptional start sites in the plant pathogen Pseudomonas syringae pv. tomato str. DC3000. PLoS One 6(12): e29335.
- He, F. (2011). E. coli Genomic DNA Extraction. Bio-protocol 1(14): e97.
- Hilker, R., Stadermann, K. B., Doppmeier, D., Kalinowski, J., Stoye, J., Straube, J., Winnebald, J. and Goesmann, A. (2014). ReadXplorer--visualization and analysis of mapped sequences. Bioinformatics 30(16): 2247-2254.
- Hilker, R., Stadermann, K. B., Schwengers, O., Anisiforov, E., Jaenicke, S., Weisshaar, B., Zimmermann, T. and Goesmann, A. (2016). ReadXplorer 2-detailed read mapping analysis and visualization from one single source. Bioinformatics 32(24): 3702-3708.
- Irnov, I., Sharma, C. M., Vogel, J. and Winkler, W. C. (2010). Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res 38(19): 6637-6651.
- Jacob, F. and Monod, J. (1961). Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol 3: 318-356.
- Jager, D., Sharma, C. M., Thomsen, J., Ehlers, C., Vogel, J. and Schmitz, R. A. (2009). Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 106(51): 21878-21882.
- Jorjani, H. and Zavolan, M. (2014). TSSer: an automated method to identify transcription start sites in prokaryotic genomes from differential RNA sequencing data. Bioinformatics 30(7): 971-974.
- Kroger, C., Colgan, A., Srikumar, S., Handler, K., Sivasankaran, S. K., Hammarlof, D. L., Canals, R., Grissom, J. E., Conway, T., Hokamp, K. and Hinton, J. C. (2013). An infection-relevant transcriptomic compendium for Salmonella enterica Serovar Typhimurium. Cell Host Microbe 14(6): 683-695.
- Kroger, C., Dillon, S. C., Cameron, A. D., Papenfort, K., Sivasankaran, S. K., Hokamp, K., Chao, Y., Sittka, A., Hebrard, M., Handler, K., Colgan, A., Leekitcharoenphon, P., Langridge, G. C., Lohan, A. J., Loftus, B., Lucchini, S., Ussery, D. W., Dorman, C. J., Thomson, N. R., Vogel, J. and Hinton, J. C. (2012). The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci U S A 109(20): E1277-1286.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G. and Durbin, R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Li, S., Dong, X. and Su, Z. (2013). Directional RNA-seq reveals highly complex condition-dependent transcriptomes in E. coli K12 through accurate full-length transcripts assembling. BMC Genomics 14: 520.
- Madhugiri, R., Pessi, G., Voss, B., Hahn, J., Sharma, C. M., Reinhardt, R., Vogel, J., Hess, W. R., Fischer, H. M. and Evguenieva-Hackenberg, E. (2012). Small RNAs of the Bradyrhizobium/Rhodopseudomonas lineage and their analysis. RNA Biol 9(1): 47-58.
- Mentz, A., Neshat, A., Pfeifer-Sancar, K., Puhler, A., Ruckert, C. and Kalinowski, J. (2013). Comprehensive discovery and characterization of small RNAs in Corynebacterium glutamicum ATCC 13032. BMC Genomics 14: 714.
- Mitschke, J., Georg, J., Scholz, I., Sharma, C. M., Dienst, D., Bantscheff, J., Voss, B., Steglich, C., Wilde, A., Vogel, J. and Hess, W. R. (2011a). An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A 108(5): 2124-2129.
- Mitschke, J., Vioque, A., Haas, F., Hess, W. R. and Muro-Pastor, A. M. (2011b). Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc Natl Acad Sci U S A 108(50): 20130-20135.
- Murakami, K. S. and Darst, S. A. (2003). Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol 13(1): 31-39.
- Nickel, L., Weidenbach, K., Jager, D., Backofen, R., Lange, S. J., Heidrich, N. and Schmitz, R. A. (2013). Two CRISPR-Cas systems in Methanosarcina mazei strain Go1 display common processing features despite belonging to different types I and III. RNA Biol 10(5): 779-791.
- Pfeifer-Sancar, K., Mentz, A., Ruckert, C. and Kalinowski, J. (2013). Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics 14: 888.
- Porcelli, I., Reuter, M., Pearson, B. M., Wilhelm, T. and van Vliet, A. H. (2013). Parallel evolution of genome structure and transcriptional landscape in the Epsilonproteobacteria. BMC Genomics 14: 616.
- Ramachandran, V. K., Shearer, N., Jacob, J. J., Sharma, C. M. and Thompson, A. (2012). The architecture and ppGpp-dependent expression of the primary transcriptome of Salmonella Typhimurium during invasion gene expression. BMC Genomics 13: 25.
- Ramachandran, V. K., Shearer, N. and Thompson, A. (2014). The primary transcriptome of Salmonella enterica Serovar Typhimurium and its dependence on ppGpp during late stationary phase. PLoS One 9(3): e92690.
- Robb, N. C., Cordes, T., Hwang, L. C., Gryte, K., Duchi, D., Craggs, T. D., Santoso, Y., Weiss, S., Ebright, R. H. and Kapanidis, A. N. (2013). The transcription bubble of the RNA polymerase-promoter open complex exhibits conformational heterogeneity and millisecond-scale dynamics: implications for transcription start-site selection. J Mol Biol 425(5): 875-885.
- Sahr, T., Rusniok, C., Dervins-Ravault, D., Sismeiro, O., Coppee, J. Y. and Buchrieser, C. (2012). Deep sequencing defines the transcriptional map of L. pneumophila and identifies growth phase-dependent regulated ncRNAs implicated in virulence. RNA Biol 9(4): 503-519.
- Sansonetti, P. J., Kopecko, D. J. and Formal, S. B. (1982). Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect Immun 35(3): 852-860.
- Schluter, J. P., Reinkensmeier, J., Barnett, M. J., Lang, C., Krol, E., Giegerich, R., Long, S. R. and Becker, A. (2013). Global mapping of transcription start sites and promoter motifs in the symbiotic alpha-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 14: 156.
- Schluter, J. P., Reinkensmeier, J., Daschkey, S., Evguenieva-Hackenberg, E., Janssen, S., Janicke, S., Becker, J. D., Giegerich, R. and Becker, A. (2010). A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 11: 245.
- Schmidtke, C., Findeiss, S., Sharma, C. M., Kuhfuss, J., Hoffmann, S., Vogel, J., Stadler, P. F. and Bonas, U. (2012). Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res 40(5): 2020-2031.
- Schoenberg, D. R. (2007). The end defines the means in bacterial mRNA decay. Nat Chem Biol 3(9): 535-536.
- Sharma, A. and Puhar, A. (2019). Gentamicin protection assay to determine the number of intracellular bacteria during infection of human tc7 intestinal epithelial cells by Shigella flexneri. Bio-protocol 9(13): e3292.
- Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., Chabas, S., Reiche, K., Hackermuller, J., Reinhardt, R., Stadler, P. F. and Vogel, J. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464(7286): 250-255.
- Sharma, C. M. and Vogel, J. (2014). Differential RNA-seq: the approach behind and the biological insight gained. Curr Opin Microbiol 19: 97-105.
- Thomason, M. K., Bischler, T., Eisenbart, S. K., Forstner, K. U., Zhang, A., Herbig, A., Nieselt, K., Sharma, C. M. and Storz, G. (2015). Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197(1): 18-28.
- Thorvaldsdottir, H., Robinson, J. T. and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2): 178-192.
- Voigt, K., Sharma, C. M., Mitschke, J., Lambrecht, S. J., Voss, B., Hess, W. R. and Steglich, C. (2014). Comparative transcriptomics of two environmentally relevant cyanobacteria reveals unexpected transcriptome diversity. ISME J 8(10): 2056-2068.
- Voss, B., Bolhuis, H., Fewer, D. P., Kopf, M., Moke, F., Haas, F., El-Shehawy, R., Hayes, P., Bergman, B., Sivonen, K., Dittmann, E., Scanlan, D. J., Hagemann, M., Stal, L. J. and Hess, W. R. (2013). Insights into the physiology and ecology of the brackish-water-adapted Cyanobacterium Nodularia spumigena CCY9414 based on a genome-transcriptome analysis. PLoS One 8(3): e60224.
- Vvedenskaya, I. O., Zhang, Y., Goldman, S. R., Valenti, A., Visone, V., Taylor, D. M., Ebright, R. H. and Nickels, B. E. (2015). Massively systematic transcript end readout, "MASTER": transcription start site selection, transcriptional slippage, and transcript yields. Mol Cell 60(6): 953-965.
- Wiegand, S., Dietrich, S., Hertel, R., Bongaerts, J., Evers, S., Volland, S., Daniel, R. and Liesegang, H. (2013). RNA-Seq of Bacillus licheniformis: active regulatory RNA features expressed within a productive fermentation. BMC Genomics 14: 667.
- Wilms, I., Overloper, A., Nowrousian, M., Sharma, C. M. and Narberhaus, F. (2012). Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol 9(4): 446-457.
- Wingett, S. W. and Andrews, S. (2018). FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 7: 1338.
- Zhang, Y., Heidrich, N., Ampattu, B. J., Gunderson, C. W., Seifert, H. S., Schoen, C., Vogel, J. and Sontheimer, E. J. (2013). Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50(4): 488-503.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Cervantes-Rivera, R. and Puhar, A. (2020). Whole-genome Identification of Transcriptional Start Sites by Differential RNA-seq in Bacteria. Bio-protocol 10(18): e3757. DOI: 10.21769/BioProtoc.3757.
Category
Microbiology > Microbial genetics > Gene expression
Microbiology > Microbial genetics > RNA > Sequencing
Systems Biology > Genomics > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.