- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Optogenetic Tuning of Protein-protein Binding in Bilayers Using LOVTRAP
Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3745 Views: 6192
Reviewed by: Samantha E. R. DundonSunanda MarellaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2019


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Modern microscopy methods are powerful tools for studying live cell signaling and biochemical reactions, enabling us to observe when and where these reactions take place from the level of a cell down to single molecules. With microscopy, each cell or molecule can be observed both before and after a given perturbation, facilitating better inference of cause and effect than is possible with destructive modes of signaling quantitation. As many inputs to cell signaling and biochemical systems originate as protein-protein interactions near the cell membrane, an outstanding challenge lies in controlling the timing, location and the magnitude of protein-protein interactions in these unique environments. Here, we detail our procedure for manipulating such spatial and temporal protein-protein interactions in a closed microscopy system using a LOVTRAP-based light-responsive protein-protein interaction system on a supported lipid bilayer. The system responds in seconds and can pattern details down to the one micron level. We used this technique to unlock fundamental aspects of T cell signaling, and this approach is generalizable to many other cell signaling and biochemical contexts.
Keywords: OptogeneticsBackground
Questions in cell signaling and cell biology commonly focus on how cells sense and respond to their environment. The signaling cascades that carry out these cellular decisions contain proteins that can move nanometers to microns in a matter of seconds to minutes. Some common methods, such as western blots, qPCR and deep sequencing, give us insight into the levels and activities of signaling proteins. However, because these approaches require destruction of the sample to make their measurements, they only give snapshots of the underlying biology. Light microscopy is an attractive complementary technique to fill in these gaps of knowledge by measuring how cells process and propagate signals in space and time.
Existing microscopy techniques can measure cell signaling on a variety of spatial and temporal scales. At the nanometer scale, fluorescence correlation spectroscopy can be used to monitor protein complex formation, and live cell FRET sensors can be used to measure protein conformation and protein-protein interactions. At around 10-100 nm, live cell super-resolution techniques like structured illumination (SIM) and stimulated emission depletion (STED) microscopy can map cellular sub-structures such as the cytoskeleton. Common wide-field and confocal techniques remain a standby to measure organelle-level localization of proteins and overall morphology of the cell. Images can be taken every millisecond to many minutes, depending on the technique and field of view.
Both conventional and super-resolution microscopy techniques are powerful because they capture the diverse temporal and spatial responses cells make. Although we can image cellular signals dynamically in space and time, our ability to manipulate these signals is much more limited. Stamps with micron-sized patterns can deposit molecules of interest onto a coverslip or other flat surface with near arbitrary shapes. Nanolithography techniques can take this further by carving complex 3-dimensional structures into a surface. While these techniques can create a variety of shapes, the shapes themselves cannot be readily manipulated in time and space to mimic the dynamics of actual stimuli. Micropipettes and/or microfluidics can deliver pulses of an extracellular ligand, but they are limited to controlling spatial distribution and are less suited to manipulate surface-presented ligands. For a better understanding of how cells sense and respond to their environments, we need new techniques to control stimuli in both space and time.
Cells commonly sense their environment through protein-protein interactions at the cell membrane. In this protocol, we mimic these cell-presented ligands using a light-gated protein-protein interaction system on a supported lipid bilayer (Figure 1). Light controllable protein-protein interactions are also powerful for studying biochemical reactions that occur close to a membrane, such as the phospho-inositol cycle or Ras signaling (Toettcher et al., 2011 and 2013).
Our approach for manipulating signals in bilayers is based on LOVTRAP, a light-induced protein dissociation technique (Wang et al., 2016) that consists of a naturally light-sensitive protein (the LOV2 domain of Avena sativa phototropin I) and an engineered binding partner, Zdk. In the ground state, Zdk binds to LOV2 with high affinity. When LOV2 is excited by blue light, it changes conformation, causing Zdk to dissociate. Active LOV2 slowly relaxes to the ground state, allowing Zdk to rebind and reset the system. By controlling when and where blue light is delivered, micron-sized patterns of a protein-protein interaction can be generated and altered in seconds. Because LOV2 is insensitive to red and infra-red light, this system is compatible with biosensors in these channels for microscopy-based quantification of the perturbation and cell response.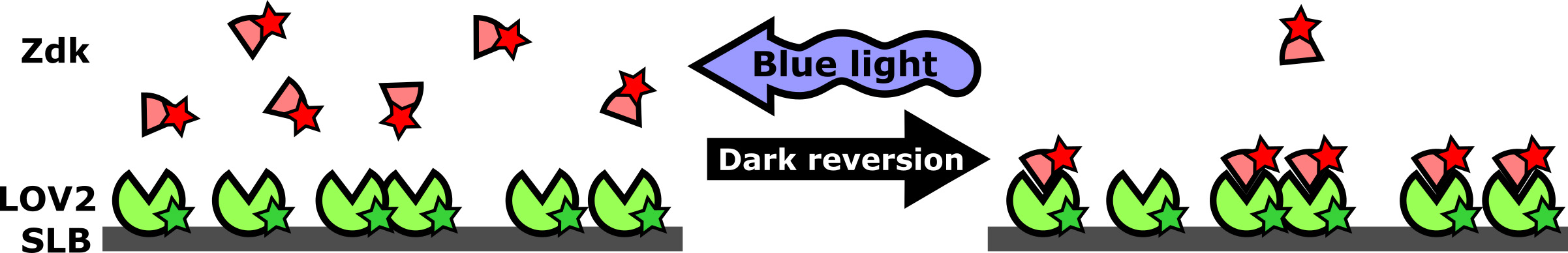
Figure 1. Diagram of the light-controlled protein-protein interaction system. The light-sensitive protein LOV2 is biochemically purified and attached to a supported lipid bilayer (SLB). Its binding partner, Zdk, binds to LOV2 in the dark and dissociates upon illumination with blue light. Zdk can be free in solution (as diagrammed here) or attached to a cell surface receptor to control cell signaling (as was done in Tischer and Weiner, 2019). In biochemical reconstitution, Zdk can be fused to a protein whose activity depends on membrane proximity, such as a lipid kinase or GTPase regulator.
We previously leveraged our optogenetic approach to stimulate the T cell signaling with precise temporal control, revealing that T cells measure the dynamics of ligand binding in their decision to activate (Tischer and Weiner, 2019). Ligands with longer binding half-lives signal disproportionately better than ligands with short binding half-lives, even when controlling for receptor occupancy. Such a conclusion was only possible because the optogenetic system allowed us to directly manipulate the variable of interest: protein-protein interaction half-life. This direct manipulation was not possible with existing experimental techniques. Because of the general nature of protein-protein interactions in initiating cell signaling, we detail our procedure here in the hopes that our approach will enable a powerful interrogation of other signaling systems.
Materials and Reagents
- Deep well 96-well block or alternative method for collecting fractions from an FPLC
- 0.22 μm filter
For large volumes: Millipore, catalog number: SE1M179M6
For small volumes: Millipore, catalog number: SLGV033RS - Glass coverslips for flow chamber (Ibidi, catalog number: 10812 )
- Tin foil
- Aluminum foil
- 1.5 ml Eppendorf tubes
- PCR tubes (Sigma-Aldrich, catalog number: CLS3744 )
- Plastic Pasteur pipettes or plastic tubing (Celltreat Scientific, catalog number: 229276 )
- Plastic tubing
- Superdex 200 Increase 10/300 GL column (Sigma-Aldrich, catalog number: GE28-9909-44 )
- HiPrep 26/10 desalting column (GE Healthcare, catalog number: 45-000-266 )
- Glass vials (Thermo Fisher Scientific, catalog number: B7800-2 )
- Tongs or sturdy wire to transfer Coplin jars from a hot water bath.
- Pasteur pipettes (Thermo Fisher Scientific, catalog number: 13-678-20A )
- Plasmid for expressing LOV2 (pDT552; From Tischer and Wiener, 2019)
This plasmid expresses the fusion protein 10xHis-TEV-AviTag-KCK-LOV2(V529N). Here, V529N denotes a mutation from the full length, wt phototropin 1 protein from Avena sativa. LOV2 refers to the second LOV domain of this protein. TEV denotes a cleavage site for the tobacco etch virus protease. AviTag is a 15 amino acid tag that can be biotinylated by the E. coli biotin ligase BirA. KCK is a tripeptide tag that makes the cysteine a stronger nucleophile; this helps to preferentially label this site over the LOV2 active site cysteine with maleimide-conjugated dyes. This construct is cloned into a modified pETM11-SUMO3 plasmid backbone. - Plasmid for expressing Zdk, (pDT482; From Tischer and Wiener, 2019)
This plasmid expresses the fusion protein 10xHis-SUMO3-KCK-SpyCatcher-Zdk1. SUMO3 is the human SUMO3 domain recognized by the SenP2 protease, which cleaves at the C-terminus of the domain. KCK is a tripeptide tag that makes the cysteine a stronger nucleophile; this makes the cysteine more reactive to maleimide-conjugated dyes. The SpyCatcher domain is not used for the current application but permits post-translational fusion to Spy-tag containing targets where desired. Zdk1 is the engineered binding partner to LOV2. This construct is cloned into a modified pETM11-SUMO3 plasmid backbone. - Liquid nitrogen
- LB broth (Sigma-Aldrich, catalog number: L3522 )
- Kanamycin (Goldbio, catalog number: K-120-10 )
- Isopropyl-β-D-thiogalactoside (IPTG; Sigma-Aldrich, catalog number: I6758 )
- Biotin (Sigma-Aldrich, catalog number: B4639 )
- Flavin mononucleotide (FMN; Bio-Rad, catalog number: 161-0501 )
- Methanol (Sigma-Aldrich, catalog number: 179337 )
- Flow chamber sticky tops (Ibidi, catalog number: 80608 )
- POPC (Avanti Polar Lipids, catalog number: 850457C )
- PEG-PE (Avanti Polar Lipids, catalog number: 880230C )
- Biotinyl CAP PE (Avanti Polar Lipids, catalog number: 870277X )
- Alexa Fluor 488 C5 Maleimide (Thermo Fisher Scientific, catalog number: A10254 )
- Sulfo-Cyanine3 maleimide (Lumiprobe, catalog number: 11380 )
- Flavin mononucleotide (Bio-Rad, catalog number: 161-0501 )
- Anhydrous DMSO (Thermo Fisher Scientific, catalog number: D12345 )
- Chloroform (Electron Microscopy Sciences, catalog number: 12550 )
- DPBS (Thermo Fisher Scientific, catalog number: 14190144 )
- Hellmanex III (Sigma-Aldrich, catalog number: Z805939 )
- Acetone (Sigma-Aldrich, catalog number: 534064-4L )
- 190 proof ethanol (Koptec, catalog number: V1101 )
- KOH (Fisher, catalog number: P251 )
- Hydrogen peroxide (Fisher Scientific, catalog number: H325-500 )
- HCl (Acros, catalog number: 42379-5000 )
- Streptavidin (Rockland, catalog number: S000-01 )
- ProLong Live Antifade Reagent (Thermo Fisher Scientific, catalog number: P36975 )
- Roche cOmplete Mini, EDTA-free protease inhibitor tablet (Sigma-Aldrich, catalog number: 11836170001 )
- NaOH (Fisher, catalog number: 19232 )
- KH2PO4 (Sigma-Aldrich, catalog number: P5379 )
- K2HPO4
- NaCl (Sigma-Aldrich, catalog number: 793566 )
- Beta-mercaptoethanol (βME; Fisher, catalog number: 34461 )
- Imidazole (Fisher, catalog number: O3196-500 )
- N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES; Sigma-Aldrich, catalog number: H3375-250G )
- KCl (Fisher, catalog number: S77375-1 )
- Tris(2-carboxyethyl)phosphine (TCEP; Thermo, catalog number: 20491 )
- Tryptone (BD Biosciences, catalog number: 211705 )
- Yeast extract (BD Biosciences, catalog number: 288620 )
- Glycerol (Fisher, catalog number: BP229 )
- Beta-casein (Fisher, catalog number: 11461422 )
- Phenylmethylsulfonyl fluoride (PMSF; Pierce, catalog number: 36978 )
- 6xHis-TEV protease (Prepared recombinantly from E. coli in house. Commercial substitutes are easy to come by e.g., Sigma-Aldrich, catalog number: T4455-1KU )
- Protein concentrator (Vivaspin 20 MWCO 10,000; GE Life Sciences, catalog number: 28932360 )
- Dithiothreitol (DTT; Sigma-Aldrich, catalog number: D9779 )
- 6xHis-SenP2 protease (Prepared recombinantly from E. coli in house. Commercial substitutes are available e.g., Novus Biologicals, catalog number: E-710-050 )
- Compressed inert gas (Compressed nitrogen; Airgas, catalog number: NI 200 )
- Ethylenediamine tetraacetic acid (EDTA; Fisher Scientific, catalog number: S80007-1 )
- Cobalt(II) chloride hexahydrate (CoCl2·6H2O; Sigma-Aldrich, catalog number: 255599 )
- Clear nail polish
- IMAC binding buffer (see Recipes)
- IMAC elution buffer (see Recipes)
- HEPES buffered saline (HBS; see Recipes)
- Terrific Broth (TB; see Recipes)
- DPBS-BB (see Recipes)
Equipment
- 500 ml flask
- 2.8 L Baffled flask
- Heat block
- 1 L glass beaker
- Temperature controlled shaker, capable of 37 °C to 18 °C
- EmulsiFlex-C3 (or some other method of cell lysis)
- AKTA pure 25 (GE Healthcare, or similar FPLC system)
- 5 ml HiTrap Chelating column (GE Healthcare, catalog number: 17040801 )
- Milli-Q water purification system (or similar)
- Hamilton syringes (Hamilton Company, Gastight 1700 series, catalog numbers: 80265 and 81165 )
- Glass Coplin jar (Sigma-Aldrich, catalog number: BR472800 )
- Peristaltic pump and tubing
- Vacuum desiccator
- Dounce homogenizer
- Bath sonicator
- Tabletop ultracentrifuge
- Floor ultracentrifuge (Beckman Coulter, model: Optima L-90K ) with Ti-45 rotor
- Swinging bucket floor centrifuge
- 470 nm LED (Lightspeed Technologies Inc., model: HPLS-36 )
- Inverted epifluorescence microscope, ideally capable of Total Internal Reflection (TIRF) microscopy (Nikon Eclipse Ti inverted microscope)
Software
- Microsoft Excel (Microsoft)
- Fiji (Schindelin et al., 2012)
- Micromanager (Edelstein et al., 2014)
Procedure
- LOV2 purification
- In a 500 ml flask, inoculate 200 ml of LB Kan (30 μg/ml) with E. coli BL21(DE3) transformed with a bicistronic plasmid expressing LOV2 and BirA (plasmid pDT552). Grow overnight (> 8 h) at 37 °C shaking at 225 rpm.
- Back-dilute 100 ml of the overnight culture into 1 L of TB Kan (30 μg/ml) in a 2.8 L baffled flask. Shake at 180 rpm at 37 °C until the OD600 (optical density at 600 nm) is between 0.5-0.6.
- Reduce the temperature to 18 °C, wait 30 min for the media to cool near to 18 °C, and induce expression with IPTG (250 μM, final concentration). From stock solutions, bring biotin to a final concentration of 50 μM and flavin mononucleotide (FMN) to 1 mM. Grow overnight.
Note: Carry out all future steps on ice or at 4 °C.- Pellet cells in a swinging bucket centrifuge (or similar) pre-chilled to 4 °C. Discard the supernatant. A typical spin is 800 RCF for approximately 10 min. The pellet usually appears noticeably yellow due to the high expression of LOV2 and the FMN cofactor.
- Use a scale and an empty centrifuge container to estimate the mass of the wet cell pellet. Add 2 ml of IMAC binding buffer for every 1 g wet cell pellet, and vigorously pipette up and down to crudely suspend the majority of the pellet.
- Transfer the cell suspension (some chunks are okay) to a dounce homogenizer that is surrounded by ice and add one Roche cOmplete Mini, EDTA-free protease inhibitor tablet. Thoroughly resuspend the cell pellet until it is completely homogenized (typically about around 10 plunges up and down).
- Immediately before lysing the cell on an EmulsiFlex-C3, add PMSF (phenylmethylsulfonyl fluoride) to a final concentration of 1 mM. PMSF has a half-life in aqueous environments of around 30 min, so it is important to add PMSF near the time of cell lysis. Make sure your PMSF stock is prepared in anhydrous methanol. If you think water might have gotten into your stock, make fresh PMSF. It is optional, but not required, to run the lysate through the EmulsiFlex a second time to reduce its viscosity. This can make the subsequent loading onto the IMAC column faster.
- Spin the lysate in an ultracentrifuge in a Ti-45 rotor at 40k rpm for one hour at 4 °C. Decant and retain the supernatant.
- Use a peristaltic pump to recirculate the supernatant over a 5 ml HiTrap Chelating column charged with Co2+ and equilibrated with the IMAC binding buffer. In the recirculation setup, the eluate from the column should gently drip back into the reservoir of supernatant that feeds the IMAC column. Make sure the inlet tube of the peristaltic pump is at the bottom of the reservoir so that all the supernatant will flow over the column. Estimate the flow rate by capturing some liquid in a 20-30 s interval. 1.5-2.0 ml/min is typical. Load the column for a duration that enables the entire supernatant to pass over the column twice. The column will turn visibly yellow from the top down as LOV2 binds the column.
- Transfer the loaded column to an AKTA and wash with IMAC binding buffer until the A280 of the effluent is less than 60 mAu. Elute the protein with a 10-column volume gradient (going from IMAC binding buffer to IMAC elution buffer) and collect in 1.5 ml fractions in a deep-well 96-well plate. After elution, equilibrate the column in IMAC binding buffer.
- Pool visibly yellow fractions in the 96-well block and then buffer exchange into IMAC binding buffer using a HiPrep 26/10 desalting column.
- Add purified 6xHis-TEV protease 1:10 w/w (weight to weight) TEV:LOV2 and incubate overnight at 4 °C covered in tin foil.
Note: TEV protease was purified in house, but can be obtained commercially. - Recirculate the digested LOV2 mixture over a HiTrap Chelating column (this can be the one used previously to elute LOV2 after equilibrating with IMAC binding buffer) with a peristaltic pump as before to remove the TEV protease, free His-tags, and any uncut LOV2. Recirculating at least twice over the column.
- Collect the flow-through and exchange into HEPES buffered saline (HBS) with the desalting column.
- Concentrate to approximately 4 mg/ml with a Vivaspin protein concentrator.
- Make a fresh 10 mM stock of Alexa Fluor 488 C5 Maleimide in anhydrous DMSO.
- On ice, add the dye to the protein at a final molar ratio of 2:1 dye:LOV2 and let the reaction proceed for 30 s.
- Quench with DTT at a final concentration of 10 mM. This procedure preferentially labels the N-terminal KCK tag (65) over the active site cysteine of LOV2. Restricting the final volume after quenching to under 500 μl is convenient, as you can proceed to gel filtration without concentrating the protein further.
- On an AKTA, run the quenched dye/LOV2 mixture over a Superdex 200 Increase 10/300 GL column equilibrated with HBS. Collect 1.5 ml fractions in a deep-well 96-well block.
- Pool fractions that have a high (> 100 mAu) A280 and are visibly yellow and concentrate to approximately 1.5 mg/ml using a Vivaspin protein concentrator.
- Add glycerol to a final volume of 10%.
- Make 3 μl aliquots in PCR tubes and snap freeze in liquid nitrogen.
- Store at -80 °C. The aliquots have shown no signs of reduced activity after > 2 years stored at -80 °C.
- Zdk purification
Purify and label Zdk in an identical manner to LOV2, except for the following changes.- Do not supplement the bacterial growth media with biotin or flavin mononucleotide.
- Add the SUMO protease 6xHis-SenP2 to the IMAC eluted protein at 1:1,000 w/w instead of TEV protease (6xHis-SenP2 was purified in house, but can be obtained commercially).
- Label the protein with Sulfo-Cyanine3 maleimide instead of Alexa Fluor 488 C5 Maleimide.
- Cobalt column charging procedure
This charging protocol assumes the column begins stored in 20% ethanol. Throughout the procedure, be sure to capture any effluent that could contain cobalt(II) for proper waste disposal.- Attach plastic tubing to the peristaltic pump and flush the tubing with ddH2O.
- Attach the plastic tubing to the IMAC column, taking care not to introduce any air bubbles.
- Remove the storage solution by flushing the column with 5-10 column volumes of ddH2O. Typical flow rates are between 1.5-2.0 ml/min.
- Pause the pump and switch the tubing intake to a 100 mM CoCl2 solution that has been passed through a 0.22 μm filter. Take care not to introduce bubbles into the line. Turn the pump back on.
- Flow the cobalt solution over the column until the column and the effluent are visibly pink.
- Pause the pump and let the column incubate in the cobalt solution for 10 min.
- Switch the tubing intake back to ddH2O and turn on the pump. Wash the column with at least 10 column volumes of ddH2O. It is important to completely remove any traces of free cobalt(II), as cobalt(II) is easily reduced in buffers with reducing agents, causing it to precipitate and clog the column.
- Switch the tubing intake to the desired buffer (that has been passed through a 0.22 μm filter) and wash the column with 5 column volumes.
- The column is now ready to use.
- Column stripping, cleaning and storage
Throughout the procedure, be sure to capture any effluent that could contain cobalt(II) for proper waste disposal.- Attach the column to a peristaltic pump and tubing that has been flushed with ddH2O. Wash the column for 10 column volumes of ddH2O.
- Switch the tubing intake to a 1 M EDTA solution (that has been passed through a 0.22 μm filter) and flow over the column until it is visibly white again.
- Switch the tubing intake into ddH2O and wash for 5 column volumes.
- Switch the tubing intake into a 20% ethanol solution (that has been passed through a 0.22 μm filter) and wash for 5 column volumes.
- Cap both ends of the column and store at 4 °C.
- Glassware cleaning
SUV preparation and glassware cleaning protocols are modified from those previously published (Taylor et al., 2017).- Add 8 glass vials (without the caps) and 8 Pasteur pipettes to a 1 L glass beaker and cover with 3 M NaOH. Place the beaker in a bath sonicator for 30 min.
- Decant the NaOH (it can be saved to future glass cleaning) and wash the glassware 5 times with ddH2O by filling the beaker and gently agitating.
- Cover the glassware with a 5% (volume/volume) Hellmanex III solution and incubate overnight (> 8 h).
- Decant the Hellmanex III solution and extensively wash the glassware with ddH2O. It is critical to remove all traces of the Hellmanex detergent from glassware. To thoroughly wash with ddH2O, attach a plastic pasteur pipette (or some small tubing that can fit inside the glass vials) to the ddH2O source with plastic tubing. Continually rinse the inside of the vial by holding it up-side-down and placing the plastic Pasteur pipette inside as far from the neck as possible. Rinse for approximately 30 s or until well after the water no longer makes bubbles. Also wash the outside of the vials extensively. Similarly, aggressively wash the inside and outside of each glass Pasteur pipette. The inside of the glass Pasteur pipettes can be effectively rinsed by threading the end of the glass Pasteur pipette just inside of the end of the plastic Pasteur pipette (you may have to trim the end of the plastic Pasteur pipette with scissors a bit) to ensure a continuous rinsing with water. For both the vials and the glass Pasteur pipettes, you may have to alternate between rinsing the inside and outside of the vial to ensure there is no cross contamination of the Hellmanex solution.
- Remove excess water by blow-drying with compressed nitrogen, air, or other compressed inert gas.
- Gently wrap the glassware in aluminum foil (to keep out dust) and set on an 80 °C heat block (or use a glass drying oven) for several hours until completely dry.
- Store glassware in a clean beaker covered with aluminum foil to protect from dust.
- To clean the plastic vial caps, sonicate in a beaker of ddH2O for 30 min, then dry and store in the same manner as the vials and glass Pasteur pipettes.
- Preparation of small unilamellar vesicles (SUVs)
- Rinse an NaOH-cleaned 4 ml glass vial with chloroform by adding approximately 500 μl chloroform with an NaOH-cleaned Pasteur pipette. Swirl several times to coat the inside of the vial and discard the chloroform. Repeat once more.
- Add approximately 500 μl of chloroform to the rinsed vial.
- Using Hamilton syringes, add 4 μmoles total lipids in a molar ratio of 97.5% POPC, 0.5% PEG-PE, and 2% biotinyl CAP PE to the vial. It is useful to dedicate a single Hamilton syringe to handle each lipid type, to reduce the change of cross contamination.
- Remove the chloroform by rotating the vial at an angle while slowly flowing nitrogen gas (or other inert gas) into the top. Once all visible solvent is evaporated (there should be white rings of residue on the inside of the vial), loosely cover the vial with a pre-washed cap and place in a vacuum desiccator overnight.
- The next day, rehydrate the lipids by adding 1.5 ml of 0.22 μm filtered DPBS, firmly screw on the cap, and gently vortex for 10 min. The vial can be secured to a benchtop vortexer by placing it vertically in the middle and running a piece of tape from under the vortex head, over the vial cap, and back down to the underside on the opposite side of the vortex head. Repeat with another piece of tape at 90 degrees to the first one. The vortexing speed is important. It should be no faster than is necessary to make the DPBS reach the neck of the vial so that all of the lipids can rehydrate. The vortexing is too fast if there are bubbles after 10 min of vortexing; this risks oxidizing the lipids.
- Once rehydrated, split the mixture equally between two 1.5 ml Eppendorf tubes and close under streaming nitrogen or other inert gas.
- Carry out 20 freeze-thaw cycles by moving the tubes between a dewar of liquid nitrogen and a 42 °C water bath or heat block. The mixture should turn from cloudy to noticeably clearer. If the solution doesn’t increase in clarity, it likely means the large lipid aggregates and multi-lamellar vesicles are not being broken up into SUVs. Since the PEG-PE helps the lipid aggregates to break up into SUVs, a cloudy solution could indicate either too much or too little PEG-PE was used or that the lipid has gone bad. Consider ordering new PEG-PE and/or double checking the correct molar amount of PEG-PE was used.
- Spin the lipid mixture at 60k RCF for 40 min at 4 °C in a tabletop ultracentrifuge. There will likely be a small, glassy pellet at the bottom of the tubes.
- Remove the supernatant to a 1.5 ml Eppendorf tube and store in liquid nitrogen until ready for use.
- RCA cleaning of microscopy coverslips
- Place one glass coverslip into each slot of a glass Coplin jar, cover with acetone and place in a bath sonicator for 10 min.
- Decant the acetone and repeat with 190 proof ethanol.
- Decant the ethanol and repeat with ddH2O. Fill and decant the Coplin jar 5 times with ddH2O before and after the ddH2O sonication step to remove excess organic solvents.
- Heat 1 L of ddH2O in a microwave (or hotplate) to approximately 70 °C. The warmed water will help prevent the Coplin jar from cracking from temperature stress during the upcoming washes.
- Separately, prepare a 70-80 °C water bath with enough water to submerge most of the Coplin jar without overrunning the lip.
- Dissolve 3.75 g KOH into 45 ml of the warmed ddH2O and add to the coverslips. Then add 15 ml 30% hydrogen peroxide.
- Use tongs or wire securely wrapped around the neck of the Coplin jar to transfer it to the water bath and let it react for 12 min: approximately 2 min for the solution to get up to temperature and 10 min for the reaction to proceed. The solution will constantly produce many small bubbles once it warms up.
- Remove the Coplin jar from the water bath. Carefully decant the base solution and wash 5 times with the warmed ddH2O.
- Add the following components in order to the Coplin jar: 38 ml warmed ddH2O, 9.5 ml 37% HCl and 12.6 ml 30% hydrogen peroxide.
- Incubate in the water bath for 12 min as before. The solution will bubble similarly to the base solution, but slightly less strongly.
- Remove the Coplin jar from the water bath, decant the acid solution, and wash 5 times with the warmed ddH2O. Cover the coverslips with warmed ddH2O, add the Coplin jar lid, and store at room temperature for up to one week.
- Forming SLBs
While drying and preparing the glass coverslips, take care to only handle the edges and avoid touching any part near the center.- Use forceps to remove a glass coverslip from the ddH2O in the Coplin jar. Hold it in one gloved hand by pinching between the two long sides and immediately blow dry with compressed nitrogen (or compressed air or other inert gas). The reflection off the coverslip should be spotless.
- Stack a sheet of microscopy lens paper on top of two paper towels and place the coverslip on top.
- Firmly press a six-well Ibidi sticky chamber onto the coverslip. Use very firm pressure to ensure contact, especially between the channels and between the ends of the channels and the sides of the coverslip.
- Seal the interface along the long edges of the coverslip with clear nail polish. The seal helps prevent any solution in the channels from wicking to the sides and evaporating during long microscopy time lapse experiments.
- Dilute a 30 μl aliquot of SUVs with 800 μl 0.22 μm filtered DPBS and evenly distribute between the six wells.
- Incubate at 37 °C for one hour. Once the SUVs are added, take care to never let the channel dry out during the subsequent washing and functionalization steps.
- Proceed the SLB functionalization
- Functionalizing SLBs
- To functionalize a well, flush out excess lipids with 500 μl of 0.22 μm filtered DPBS. It is useful to push the liquid in one end with a pipette and carefully aspirate from the other end (Figure 2).
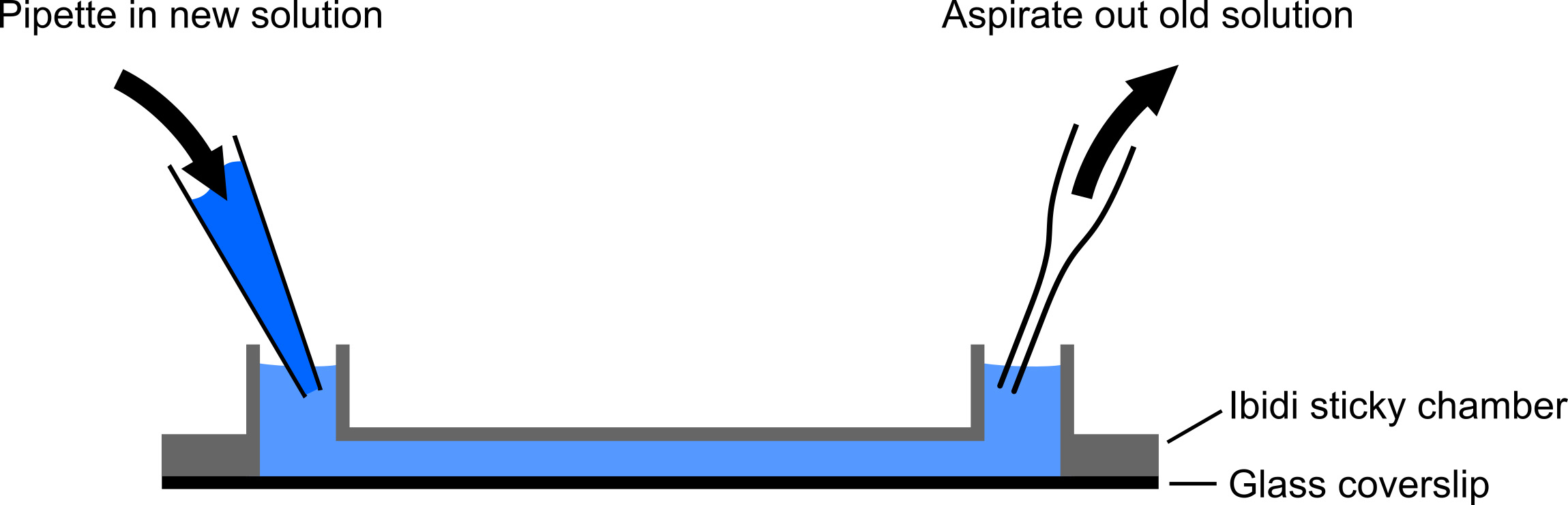
Figure 2. Exchanging solutions during SLB preparation and functionalization. Diagram of how to exchange solutions in the channels of the Ibidi chamber. In order to have an efficient exchange of solutions in the channels of the Ibidi chamber while keeping the SLB hydrated, it is useful to simultaneously push new solutions in one end and aspirate out the other. Keeping the tip of the aspirator high in the exit, above the top of the intervening channel, helps ensure liquid is only removed as a new solution is being pushed in by the pipette. Adding a small diameter pipette tip to the end of the aspirator can help moderate the suction to avoid over-aspirating the channel. - Flush the well with 200 μl of streptavidin diluted into DPBS-BB (2 μg/ml final) and incubate at room temperature for 5 min.
- Flush the well with 500 μl DPBS-BB.
- Flush the well with 200 μl of LOV2 diluted into DPBS-BB to the desired concentration (typically between 20-200 nM) and incubate at room temp for 5 min.
- Finally, flush the channel with 500 μl of DPBS-BB. The SLB is now functionalized with LOV2.
Regardless of the intended use of the LOV2-functionalized SLBs, it is important that the solution the experiment takes place in has been pretreated with an oxygen scavenger. This helps prevent blue light from bleaching the LOV2 chromophore and dramatically increases the possible rounds of recruitment and release of Zdk to LOV2. One method that works well is to dilute ProLong Live Antifade Reagent 1:100 into the solution in which microscopy will be performed and let it incubate at room temp for at least 90 min for the dissolved oxygen to be depleted. It is useful to begin the oxygen scavenger treatment before functionalizing the SLBs so that the solution is ready by the time the SLBs are fully functionalized. - To functionalize a well, flush out excess lipids with 500 μl of 0.22 μm filtered DPBS. It is useful to push the liquid in one end with a pipette and carefully aspirate from the other end (Figure 2).
- Microscope
A microscope capable of TIRF (total internal reflection fluorescence) microscopy is necessary for quantifying Zdk recruitment and release from a LOV2-functionalized bilayer and is a natural choice for imaging a resulting change in cell signaling or of a biochemical reaction.
We imaged using an Eclipse Ti inverted microscope (Nikon) with two tiers of dichroic turrets to allow simultaneous fluorescence imaging and optogenetic stimulation. The microscope was also equipped with a motorized laser TIRF illumination unit, a 60x Apochromat TIRF 1.49 NA objective (Nikon), an iXon Ultra EMCCD camera, and a laser merge module (LMM5; Spectral Applied Research) equipped with 405-, 440-, 488-, 514-, and 561-nm laser lines. The microscope and associated hardware were controlled with MicroManager (Edelstein et al., 2014) in combination with custom-built Arduino controllers (Advanced Research Consulting Corporation). Blue light for optogenetic stimulation was from a 470 nm LED independently controlled with MATLAB. - Checking bilayer fluidity
Before performing experiments, it is important to confirm that your bilayers are functionalized with LOV2 and fluid. This is easily done with a fluorescence recovery after photobleaching (FRAP) experiment. One can use almost any fluorescent imaging modality (e.g., TIRF, confocal, wide field) to check that the bilayer is functionalized with LOV2.- Load an SLB functionalized with a saturating amount of fluorescently labeled LOV2 onto the microscope and use a 60x or 100x objective to focus at the glass-water interface. It may be useful to dope in a small concentration of fluorescent beads to make locating this interface easier.
- Close down the episcopic field diaphragm so that only part of the field of view is illuminated. There should be an obvious increase in fluorescence inside the illuminated region if the bilayer is functionalized with fluorescently labeled LOV2.
- A qualitative FRAP measurement is usually all that is needed to assess bilayer fluidity. While it is useful to have a proper FRAP system or high power TIRF laser, it is sometimes possible to use a lower-powered light source (such as a wide field epifluorescence light source) if neither is available. In such a case, it is useful to label the LOV2 with a fluorophore like FITC (fluorescein isothiocyanate) that is easy to bleach. Not treating the imaging buffer with an oxygen scavenger also makes fluorophores easier to bleach.
- Bleach a small region of the field of view with the method of your choice. If you are not using a proper FRAP system (e.g., using a TIRF laser or wide field epifluorescence light source instead), close down the episcopic field diaphragm so only a small portion of the field of view is illuminated. If using a TIRF laser, point it straight up. For non-FRAP systems, the power will likely need to be increased to its maximum. The time it takes to bleach the area of interest will vary depending on the intensity of the light source, the small molecule fluorophore attached to LOV2, and the imaging media, among other factors. Bleaching times longer than 30 seconds are generally not effective, as diffusion in the bilayer (if it is fluid) will equilibrate LOV2 between the illuminated and non-illuminated regions.
- Image the entire field of view every 5-10 s for the next 2-3 min. (Open the episcopic field diaphragm if it was closed down.) A bleached hole with a diameter of ~5-10 µm should largely recover in ~30-60 s if the bilayer is fluid (Figure 3 and Video 1). No border from the original bleached hole should remain visible.

Figure 3. Assessing bilayer fluidity with FRAP. A small hole was bleached into a bilayer functionalized with fluorescent LOV2 using a TIRF laser. Because the bilayer is fluid, new molecules of LOV2 diffused into the bleached area, largely returning to its initial state in 60 s. Non-fluid bilayers fail to recover over this time course. Scale bar indicates 1 μm. Video 1. Time lapse of a FRAP experiment showing the SLB is fluid and functionalized with LOV2. A small hole was photobleached into an SLB functionalized with fluorescent LOV2. Over the course of about 60 s, the fluorescence signal recovers, owing to new molecules of LOV2 diffusing in from outside the bleached region. A bilayer that was not functionalized with LOV2 would not show a "hole" in the beginning. A bilayer that was functionalized but not fluid would not recover in about 60 s. The exact recovery rate depends on the diameter of the bleached region. Scale bar indicates 1 μm.
Video 1. Time lapse of a FRAP experiment showing the SLB is fluid and functionalized with LOV2. A small hole was photobleached into an SLB functionalized with fluorescent LOV2. Over the course of about 60 s, the fluorescence signal recovers, owing to new molecules of LOV2 diffusing in from outside the bleached region. A bilayer that was not functionalized with LOV2 would not show a "hole" in the beginning. A bilayer that was functionalized but not fluid would not recover in about 60 s. The exact recovery rate depends on the diameter of the bleached region. Scale bar indicates 1 μm.
- Testing LOV2 functionality
The most direct way to test LOV2 functionality on the bilayer is to use TIRF to measure the recruitment and release of fluorescently labeled Zdk. This test should be performed after you have confirmed the bilayers are functionalized with LOV2 and fluid.- Functionalize an SLB with saturating amounts of LOV2 and flow in imaging media containing 250 nM Zdk that has been treated with the oxygen scavenger. It is best to use the same media you intend to perform your experiments in, but DPBS can serve as a good starting point. Make sure the Zdk is labeled with a dye that is excited by light with a wavelength longer than 560 nm so that you can continuously image it without activating LOV2.
- Set up a 10 min time course where Zdk is imaged with TIRF every 5 s. Every 2 min, expose the LOV2 to blue light of around 470 nm for 1 s. Any sort of GFP imaging setting usually works well. Depending on the intensity of blue light, Zdk should be rapidly released from the bilayer in ~1-10 s and then slowly recover over the following 2 min. Similar cycles of release and recruitment should be observed over the next 10 min (Figure 4). If more than a 50% reduction in Zdk recruitment occurs over 10 min, the most likely reason is that the oxygen scavenger treatment was not effective.
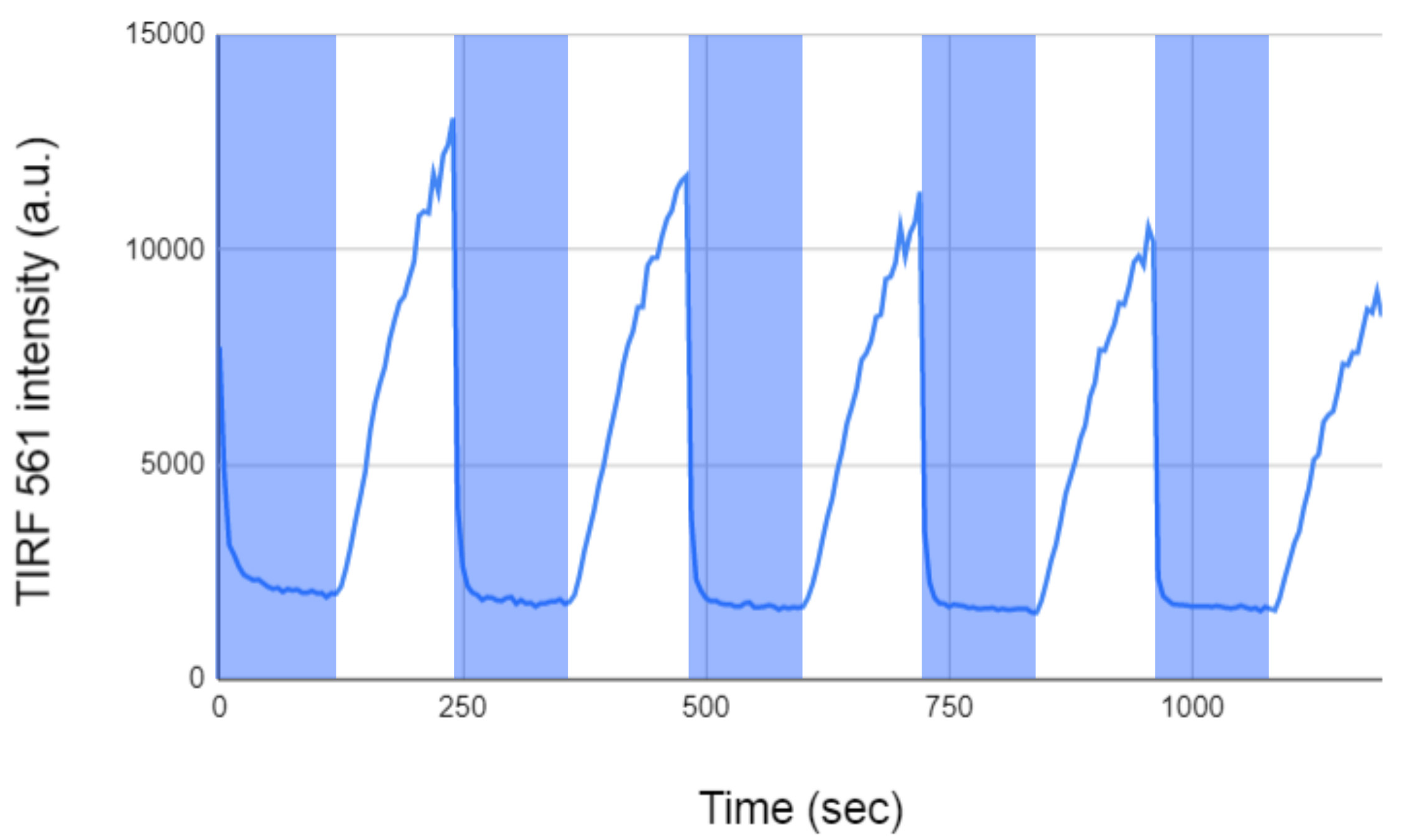
Figure 4. Time course of Zdk recruitment and release from lipid bilayers. Fluorescently labeled Zdk was recruited and released from a LOV2 functionalized bilayer for five cycles. LOV2 quickly releases Zdk after being excited with blue light (470 nm light from an LED) and slowly rebinds after the blue light is removed. (Vertical blue bars indicate the 470 nm LED is on.) Even if the microscope is only capable of periodic blue light illumination (instead of continuous illumination as ours is), the recruitment and release kinetics of Zdk should be very similar.
Recipes
Media and buffers:
- IMAC binding buffer
50 mM KH2PO4
400 mM NaCl
0.5 mM βME
pH 7.5 - IMAC elution buffer
50 mM KH2PO4
400mM NaCl
500 mM imidazole
0.5 mM βME
pH 7.5 - HBS
20 mM HEPES
100 mM KCl
0.5 mM TCEP
pH 7.5 - Terrific Broth (TB)
Nutrient Base
12 g tryptone
24 g yeast extract
4 ml glycerol
900 ml ddH2O
10x TB Salts
170 mM KH2PO4
720 mM K2HPO4- Autoclave the nutrient base and 10x TB salts separately
- Then add 100 ml of 10x TB salts to 900 ml of the nutrient base to make 1 L of TB
Buffers for SLB functionalization and imaging:
- DPBS-BB
DPBS (Thermo Fisher Scientific)
1 mg/ml beta-casein
0.5 mM βME
Filter with a 0.22 μm filter immediately prior to use
Acknowledgments
This work was supported by a Genentech Fellowship (D.T.), NIH grant GM118167 and the Novo Nordisk Foundation (O.D.W.) and the Center for Cellular Construction (DBI-1548297), an NSF Science and Technology Center. This protocol was adapted from Tischer and Weiner (2019).
Competing interests
The authors declare no financial or non-financial competing interests.
References
- Edelstein, A. D., Tsuchida, M. A., Amodaj, N., Pinkard, H., Vale, R. D. and Stuurman, N. (2014). Advanced methods of microscope control using μManager software. J Biol Methods 1(2): e11.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Taylor, M. J., Husain, K., Gartner, Z. J., Mayor, S. and Vale, R. D. (2017). A DNA-based T cell receptor reveals a role for receptor clustering in ligand discrimination. Cell 169(1): 108-119 e120.
- Tischer, D. K. and Weiner, O. D. (2019). Light-based tuning of ligand half-life supports kinetic proofreading model of T cell signaling. Elife 8: 42498.
- Toettcher, J. E., Gong, D., Lim, W. A. and Weiner, O. D. (2011). Light-based feedback for controlling intracellular signaling dynamics. Nat Methods 8(10): 837-839.
- Toettcher, J. E., Weiner, O. D. and Lim, W. A. (2013). Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155(6): 1422-1434.
- Wang, H., Vilela, M., Winkler, A., Tarnawski, M., Schlichting, I., Yumerefendi, H., Kuhlman, B., Liu, R., Danuser, G. and Hahn, K. M. (2016). LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat Methods 13(9): 755-758.
Article Information
Copyright
![]() Tischer and Weiner. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Tischer and Weiner. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Tischer, D. and Weiner, O. D. (2020). Optogenetic Tuning of Protein-protein Binding in Bilayers Using LOVTRAP. Bio-protocol 10(17): e3745. DOI: 10.21769/BioProtoc.3745.
- Tischer, D. K. and Weiner, O. D. (2019). Light-based tuning of ligand half-life supports kinetic proofreading model of T cell signaling. Elife 8: 42498.
Category
Biophysics > Microscopy
Immunology > Immune cell function > Lymphocyte
Biochemistry > Protein > Interaction > Protein-lipid interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.