- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification of Socially-activated Neurons
Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3744 Views: 7515
Reviewed by: Xi FengMohammed Mostafizur RahmanJuan Mauricio Garré
Original research article
The authors used this protocol in:

May 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Determining the neuronal circuitry responsible for specific behaviors is a major focus in the field of neurobiology. Activity-dependent immediate early genes (IEGs), transcribed and translated shortly after neurons discharge action potentials, have been used extensively to either identify or gain genetic access to neurons and brain regions involved in such behaviors. By using immunohistochemistry for the protein product of the IEG c-Fos combined with retrograde labeling of specific neuronal populations, precise experimental timing, and identical data acquisition and processing, we present a method to quantitatively identify specific neuronal subpopulations that were active during social encounters. We have previously used this method to show a stronger recruitment of ventral hippocampal neurons that project to the medial prefrontal cortex, compared to those that project to the lateral hypothalamus, following social interactions. After optimization of surgeries for the injection of retrograde tracers, this method will be useful for the identification and mapping of neuronal populations engaged in many different behaviors.
Keywords: c-FosBackground
A cornerstone goal of the neurobiology field is to determine how the brain processes sensory inputs and produces commands for behavioral outputs. In the past two decades, certain brain regions have become canonical examples in their control of specific behaviors, such as the hippocampus for location memory (Bird and Burgess, 2008). Recent work is beginning to parse out what these regions specifically do in a way that can be generalized to multiple behaviors (Aronov et al., 2017), and to determine the roles of multiple brain regions in a given behavior connected by long-range neuronal projections (Kohl et al., 2018). The first pass in determining which neuronal projections are involved in a particular behavior is to be able to measure the relative activity of neurons sending these projections during the behavior itself, with the concept that the most important neuronal projections will be selectively activated compared to neighboring neurons projecting to different brain regions.
The IEG c-Fos encodes for a transcriptional regulator that has become popular for the identification and mapping of recently active neurons due to its rapid transcription and translation following Ca2+ influx caused by the discharge of Na+-dependent action potentials. In neurons, one of the most influential molecular pathways leading to transcription and translation of c-Fos is the mitogen-activated protein kinase (MAPK) pathway, which is activated by transient increases of intracellular Ca2+ concentration brought up by activation of voltage-gated Ca2+ channels following depolarization by glutamate activation of AMPA-type receptors, as well as by Ca2+ influx through NMDA-type glutamate receptors (Herrera and Robertson, 1996; Hudson, 2018). It is, however, important to note that increases in intracellular Ca2+ concentration also lead to c-Fos expression in non-neuronal cells, including astrocytes and microglia (Eun et al., 2004; Groves et al., 2018). Accumulation of c-Fos protein begins approximately 45 min after action potential discharge, reaching peak levels after 1-2 h (Barros et al., 2015). While organic Ca2+-sensitive dyes and genetically encoded Ca2+ indicators (such as GCaMP) provide a more precise temporal estimation of neural activity in the millisecond-to-second timescale, dyes and sensors require invasive surgical procedures, and current attempts to co-register activity to specific cell subtypes is cumbersome and time-consuming.
Several genetic c-Fos-based strategies have been developed to tag active neurons during specific temporal windows using the rapid transcription and translation of IEGs, including the Fos-tTa and FosTRAP approaches. Fos-tTa uses the c-Fos promoter to drive the expression of the doxycycline-repressible tetracycline transactivator. In the absence of doxycycline (Dox), a reporter tags the cells with LacZ, which can then be visualized and quantified. However, the slow metabolism of Dox results in a long labeling window in the time frame of several days, which leads to low signal-to-noise (DeNardo and Luo, 2017). An improvement on temporal resolution was achieved with FosTRAP, which reduced the labeling window to ~12 h by using the Cre recombinase system, and a benefit of this system is that it allows genetic access to the labeled neurons (DeNardo and Luo, 2017). However, both of these genetic techniques reduce the temporal specificity of c-Fos itself. Due to their low temporal resolution and signal-to-noise ratio, quantification of relative neuronal activity is difficult and unreliable. An alternative to genetic approaches for labeling IEG-determined neuronal subsets is to perform immunohistochemistry for c-Fos protein after sacrificing the animal. This technique is commonly used to identify neurons and classifying them as either c-Fos positive or negative cells based on user-defined thresholds. However, immunohistochemical labeling of c-Fos has the potential to be used in a more quantitative manner by ensuring a precise timing interval between behavioral interactions and animal sacrifice, as well as ensuring identical tissue processing and imaging techniques. Quantification of c-Fos levels provides a more accurate depiction of neuronal activity, revealing relative activity of neurons based on subtype, projection, and/or experience, as well as reducing bias introduced by user-defined arbitrary cutoffs to score a neuron as a c-Fos positive cell.
Here, we describe a method that utilizes a precise timing between behavioral interactions and immunohistochemical detection of c-Fos that allows for reliable quantification of relative neuronal activity in different neuron subsets defined by their projection target regions. Fluorescent retrograde tracers are intracranially injected into projection targets of our primary brain region of interest. Retrograde tracers are taken up by axon terminals (Riddle et al., 1995) and transported back to the cell bodies, marking these neurons based on their projection targets. In our study, we injected green fluorescent microbeads (RetrobeadsTM) in the medial prefrontal cortex (mPFC) and red RetrobeadsTM in the lateral hypothalamus (lHYP) for the identification of projection neurons in the CA1 region of the ventral hippocampus (vCA1) (Phillips et al., 2019). This technique can be widely applied to other brain regions or by using alternative techniques to label specific neuronal subtypes. After allowing sufficient time for the retrograde transfer of the RetrobeadsTM and recovery from surgery (approximately 2 weeks), mice are subject to sequential interactions with either two conspecifics (social condition) or two fake toy mice (object condition). Mice are anesthetized and fixed by transcardiac perfusion at precisely 45 min after the conclusion of these interactions, and c-Fos levels, revealed by immunohistochemistry on sections from the ventral hippocampus, are used as a surrogate for neuronal activity during the interactions. By using a precise timing, we ensure the highest possible resolution and selectivity for neuronal activity driven by behavioral interactions. By comparing c-Fos levels within neurons with different projection targets, and between neurons from mice in either the social or object conditions, we determine the specificity of neuronal activity by projection region, as well as the selectivity for social encounters (Phillips et al., 2019). While this protocol has previously been used to gauge neuronal activation following social versus object interaction, the test mouse can be exposed to different stimuli during the behavioral paradigm to assess neural activation to other social cues such as male/female odor or social novelty.
Materials and Reagents
- Surgery
- 32 gauge, small hub RN NDL; custom specs: point style 4 12 beveled, length 1 inch (Hamilton Company, catalog number: 7803-04 )
- 2.5 μl Model 62 RN syringe (Hamilton Company, catalog number: 7632-01 )
- Alcohol wipes (Covidien)
- Test mice, aged 28-80 postnatal days
- Red RetrobeadsTM (Lumafluor Inc.)
- Green RetrobeadsTM (Lumafluor Inc.)
- Isoflurane (Vedco, IsoSol)
- Nutra-Gel diet (Bio-Serve)
- Paralube opthalamic ointment (Dechra)
- Vetbond (3M)
- Sterile 0.9% NaCl solution (Hospira)
- Local analgesics: Lidocaine (Astra, Xylocaine)
- Triple antibiotic ointment (Actavis)
- Antibiotics: Baytril (Bayer)
- Analgesics and nonsteroidal anti-inflammatory:
Carprofen (Zoetis, Rimadyl)
Ketoprofen (Zoetis, Ketofen)
Ibuprofen (Children’s Motrin)
- 32 gauge, small hub RN NDL; custom specs: point style 4 12 beveled, length 1 inch (Hamilton Company, catalog number: 7803-04 )
- Behavior
- Testing box, sized (60 x 60 cm)
- Plastic toy mice, 2 different styles (Amazon)
- Group-housed familiar sentinel mice, age-matched and co-housed with test mice
For disease models, it is best practice to use wildtype mice that are of the same genetic strain as the experimental subjects. - Group-housed novel sentinel mice, age-matched to test mice
- Standard mouse bedding
- Isopropyl alcohol (Fischer Scientific)
- Transcardiac perfusion
- Butterfly needles (Fine Science Tools)
- BD general use and precision glide hypoderminc needles, 25 Gauge, 1.5 inch (Fisher Scientific, catalog number: 14-826 )
- BD slip-tip sterile syringes (Fisher Scientific, catalog number: 14-823-434 )
- Parafilm
- Ketamine (Vet ONE, Zetamine)
- Xylazine (Lloyd, AnaSed)
- Na2HPO4·7H2O (Sigma-Aldrich, catalog number: S7907 )
- NaCl (Sigma-Aldrich, catalog number: S7653 )
- KCl (Sigma-Aldrich, catalog number: P9333 )
- KH2PO4 (Sigma-Aldrich, catalog number: 74092 )
- Paraformaldehyde 32% aqueous solution (Electron Microscopy Sciences, Fisher Scientific, catalog number: 50-980-494 )
- Agar
- Anesthetic mixture for intraperitoneal injection (see Recipes)
- 1x phosphate buffered saline (see Recipes)
- 4% paraformaldehyde in 1x PBS (see Recipes)
- Agar blocks (see Recipes)
- Tissue preparation
- Immunohistochemistry
- Aluminum foil
- Silicone glue (DAP)
- Anti-c-Fos antibodies (Synaptic Systems, catalog number: 226004 )
- Anti-NeuN antibodies (Synaptic Systems, catalog number: 266006 )
- Biotinylated goat anti-guinea pig antibodies (Vector Laboratories, catalog number: BA-7000 )
- Streptavidin conjugated to Alexa Fluor 405 (Thermo Fisher Scientific, catalog number: S-32351 )
- AffiniPure goat anti-Chicken IgY (H+L) conjugated to Alexa Fluor 647 (Jackson Immuno Research, catalog number: 103-605-155 )
- GibcoTM goat serum, New Zealand origin, standard (sterile-filtered) (Fisher Scientific, catalog number: 16-210-064 )
- Bovine serum albumin (Sigma-Aldrich, catalog number: A7030 )
- Triton X-100 (Sigma-Aldrich, catalog number: X100-500ML )
- Fisherbrand Colorfrost plus microscope slides (Fisher Scientific, catalog number: 12-550-17 )
- Vectasheild anti-fade mounting medium (Vector Laboratories, catalog number: H-1000 )
- Clear nail polish (Electron Microscopy Sciences, catalog number: 72180 )
- Immunohistochemistry permeabilization solution (see Recipes)
- Immunohistochemistry block solution (see Recipes)
- Immunohistochemistry antibody diluent solution (see Recipes)
- Well inserts (see Recipes)
Equipment
- Surgery
- Small animal stereotactic frame with digital display console (Kopf, catalog number: 942 )
- Isoflurane vaporizer machine (Ohmed)
- Hair clippers (Norelco, Phillips)
- Medical oxygen tank (AirGas)
- Heating pad (Watlow)
- DC temperature controller (FHC)
- Drill (Foredom)
- Drill bits (Fine Science Tools)
- Micro injector (WPI, Micro4)
- Surgical tools (Fine Science Tools)
- Behavior
- Headlamp with red light (Amazon), or, red overhead lights (Amazon)
- Optional: Recording camera with long-pass infrared filter
- Optional: Infrared light
- Perfusion
- Fisherbrand straight micro scissors (Fisher Scientific, catalog number: 08-953-1B )
- Peristaltic pump (Ismatec)
- Surgical tools (Fine Science Tools)
- Brain sectioning
- General Data Healthcare SHUR/MarkTM Camel Hair Brush Set
- Vibratome (Ted Pella, model: Pelco100 )
- General Data Healthcare SHUR/MarkTM Camel Hair Brush Set
- Fluorescence microscopy
- Confocal microscope (Nikon A1) with 405, 488, 594, and 647 nm wavelength lasers
- 20x objective, NA 0.8 or higher (Nikon)
Software
- ImageJ (NIH), or FIJI
- Matlab (Mathworks) or Excel (Microsoft)
- Prism (GraphPad)
Procedure
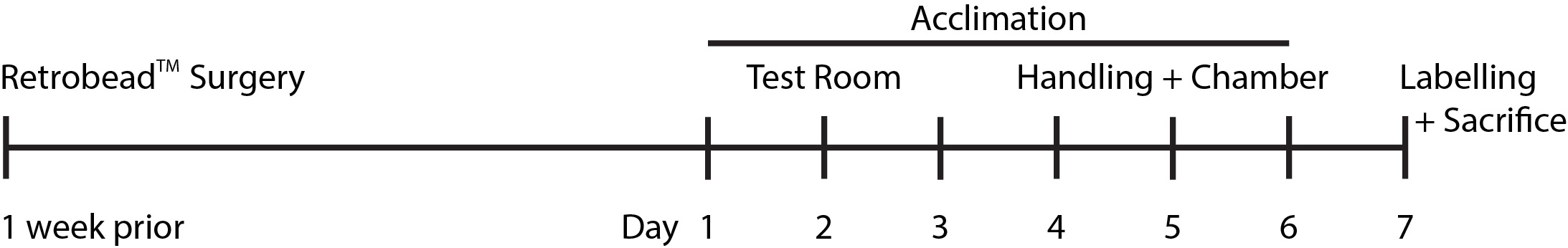
Figure 1. Timeline of experimental procedures. Mice are injected with RetrobeadsTM one week prior to behavioral acclimation. On days 1-3, mice are acclimated to the testing room. On days 4-6, mice are handled (to reduce their anxiety) and the test mice are acclimated to the testing chamber. On day 7, mice are exposed to the behavioral paradigm for labeling and sacrificed.
- Surgery
Notes:- For the behavioral paradigm, you will need one non-surgical mouse per cage acting as a known sentinel. In group housed males, it is recommended that the groups including the sentinel are littermates and that the housing is kept to no more than 5 mice per cage. Additional non-surgical novel sentinel mice (age-, sex-, and size- matched, also group housed) should be made available for the behavioral testing.
- Test mice will be injected with RetrobeadsTM 2 weeks prior to final behavioral exposure and sacrifice, which should be considered in the case of age- or disease- related studies (Figure 1).
- To increase labeling with RetrobeadsTM, 2 injections of small volumes (approximately 250 nl) per region are suggested. This is easily done by adding an additional site in the dorsoventral axis, determined by choosing sites equidistance from region boundaries per a standard mouse brain atlas.
- RetrobeadsTM can easily clog the injection needle. Be sure not to let the RetrobeadsTM sit in the needle for any longer than strictly necessary. To reduce this time, only draw up enough RetrobeadsTM per dorso-ventral axis and give test pulses regularly (where a small amount ~1 nl is dispensed). Between dorso-ventral axes, wipe the tip of the needle with alcohol wipes. Ensure the needle is not clogged before and after insertion into the brain by giving a test pulse and observing the ejection of RetrobeadsTM. If RetrobeadsTM are not ejected, the needle is clogged and will not (and may not have) deliver the necessary amount of RetrobeadsTM. The needle can be unclogged by pipetting PBS, insertion of cleaning pipes in the needle, or soaking the tip in ethanol. Allowing the needle to clog often will decrease the lifetime of the needle.
- Injection coordinates used in this paper are the prelimbic (PL) region of the mPFC and the lHYP.
- To determine injection sites, coordinates were found using the Paxinos & Franklin mouse atlas (Paxinos and Franklin, 2008), and verified with pilot studies. To pilot coordinates, black food-coloring can be injected in place of RetrobeadsTM and the brain immediately removed and cut with a razor blade. Sites can be visualized using a standard bright-field microscope.
- Because RetrobeadsTM injections can be difficult to master, be sure to test the injection protocol on a pilot dataset before proceeding to the remainder of the protocol.
- All procedures on experimental animals should be reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
- Aliquot RetrobeadsTM into small PCR tubes, adding one volume more than will be used to account for hub loss (250 nl x number of injection sites per mouse x # of mice + 250 nl).
- Insert Hamilton needle into syringe, priming with sterile saline, ensuring no bubbles are in the needle.
- Anesthetize mice in induction chamber using 4-5% vaporized isoflurane diluted with 100% oxygen at a constant flow rate of 2.5 L/min.
- Give mouse subcutaneous injection of analgesics (e.g., 2 mg/kg Ketaprofen).
- Shave the head of the mouse using standard clippers, sterilizing the scalp with alcohol wipes.
- Position the mouse in the stereotactic frame. Reduce isoflurane to ~2%, adjusting throughout the surgery as necessary.
- Apply ophthalmic ointment to the eyes.
- Subcutaneously inject mouse with analgesics and antibiotics.
- Using micro-scissors, make an incision in the scalp that is long enough to see both bregma and lambda.
- Ensure the head is leveled and straight using the arms of the stereotactic frame. Tighten ear bars to secure mouse, being careful to avoid changing the orientation of the head.
- Zero medio-lateral and anterio-posterior positions of the drill (attached to one arm of the stereotactic frame) at bregma.
- Drill holes in the skull above the injection site, being careful not to puncture brain. Injection site coordinates are generated using the Paxinos & Franklin mouse brain atlas (Paxinos and Franklin, 2008) and verified by pilot studies.
- Draw up enough RetrobeadsTM for the number of injections per needle insertion + 250 nl. Give a small test pulse to ensure the needle is not clogged.
- Zero needle at bregma, move needle to injection site medio-lateral and anterio-posterior coordinates. Lower needle into brain, expanding burr hole if necessary. Zero dorso-ventral position when the needle just enters the brain.
- Slowly lower the needle to 0.002 mm below the deepest injection site, wait 2 min. Give small test pulse to clear needle.
- Retract the needle to the first injection site, give small test pulse to keep needle clear, wait 2 min. Inject RetrobeadsTM at a speed of 250 nl per min. Wait 2 min after injection completion.
- Retract the needle to the most superficial injection site, give small test pulse, wait 2 min and inject RetrobeadsTM.
- Slowly retract the needle, over the course of 5-10 min depending on depth of injection. Give test pulse to ensure needle is not clogged. Wipe needle with ethanol wipes.
- Repeat injection procedure for the other hemisphere and any additional regions.
- After all injections are completed, drop approximately 1 ml of sterile saline on the mouse skull. Dry with sterile tissue paper.
- Pull the skin on sides of incision together, being careful not to fold or overlap them. Apply vet bond avoiding the eyes, nose/whiskers, and ears.
- Remove mouse from isoflurane, place the mouse on a paper towel in the recovery cage that is positioned so that half the cage is on a heating pad. The mouse should be placed on the heated section. One recovery cage per mouse is required, mice can be placed back in their previous social groups once they are up and walking.
- Check mice daily for incision opening and lethargy. Follow approved IACUC protocols for humane endpoints, if necessary.
- Figure 2 shows representative examples of successful retrogradely labeled neurons in ventral CA1 with projections to the mPFC and the lHYP, and immunostained for the neuronal marker NeuN and the IEG c-Fos.
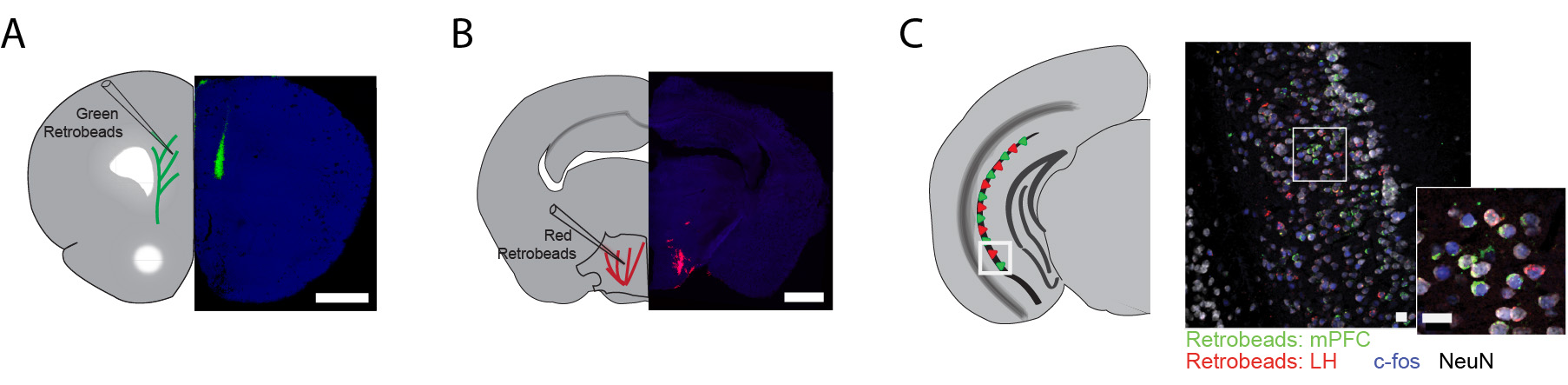
Figure 2. Example of successful retrobead injection. Green RetrobeadsTM were injected into the mPFC (A) and red RetrobeadsTM were injected into the lHYP (B). Minimal spread of RetrobeadsTM is observed. Two weeks following injection, neuronal cell bodies containing RetrobeadsTM can be observed in the target region, vHIP, determined by their colocalization with NeuN (C). - Table 1 shows the stereotactic coordinates used to target the mPFC and the lHYP during injections of RetrobeadsTM in adult C57/Bl6 mice.
Table 1. RetrobeadsTM injection coordinates. Coordinates for stereotactic injection targeting the mPFC and lHYP for labeling vHIP projection neurons (from Phillips et al., 2019).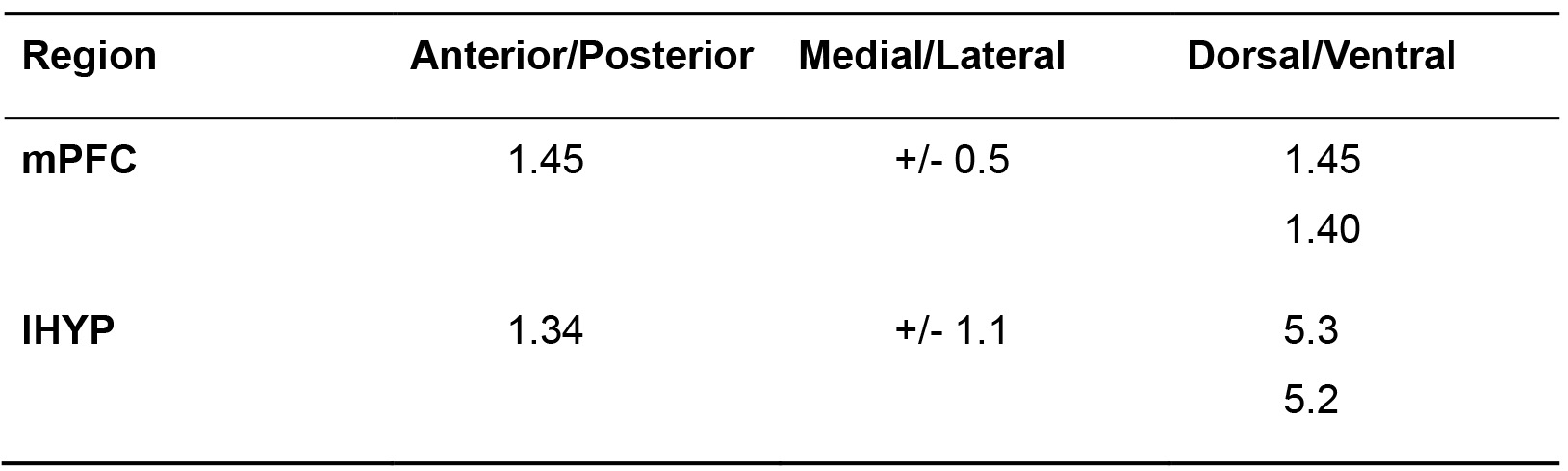
- Behavior
Notes:- Mice should be acclimated, handled, and tested during their most active phase (i.e., dark cycle).
- Timing of interactions is critical because c-Fos expression is temporally linked to behavior.
- A red headlamp should be worn so that the experimenter can see but without disrupting the circadian rhythm of the mice.
- Note excessive fighting in any of the mouse-mouse interactions, removing mice from the test group if injuries occur. Using sequential interactions, as opposed to combined, reduces aggression and enhances time spent with each mouse.
- One week after the RetrobeadsTM injection surgery, mice should start being acclimated to the behavior room. For the first 3 days, move the mouse cages (including any sentinel cages) to the room and let them acclimate for 2-4 h before returning them to the animal facility (Figure 1).
- On days 4-6, mice should be acclimated to handing and the interaction chamber. Put clean mouse bedding in the bottom of the acclimation chamber. Handle each mouse for approximately 3 min by picking them up and allowing them to sit in your hands. After 3 min, or when the mice have calmed down (whichever is longest), transfer the mice to the interaction chamber. The sentinel mice should be handled, but not moved to the interaction chamber.
- After 10 min of acclimation, remove the mice from the interaction chamber and return to the home cage. Remove the used bedding and replace with clean. At the end of each day, clean the interaction chamber with 70% isopropyl alcohol.
- On labeling day (day 7), remove sentinels from home cages and place singly in clean cages for the duration of the testing. Be sure to mark the cages well for ease in determining which mice will be the familiar and novel sentinel.
- Use a random number generator to separate surgical mice into 2 groups: those that will interact with other mice, and those that will interact with inanimate objects (e.g., toy mice). Be sure to fill the bottom of the interaction chamber with clean bedding before each interaction session.
- For test mice that will interact with other mice, place the test mouse in the interaction chamber and quickly add the familiar sentinel (cage mate). Allow to interact for 10 min before returning to respective cages (Figure 3).
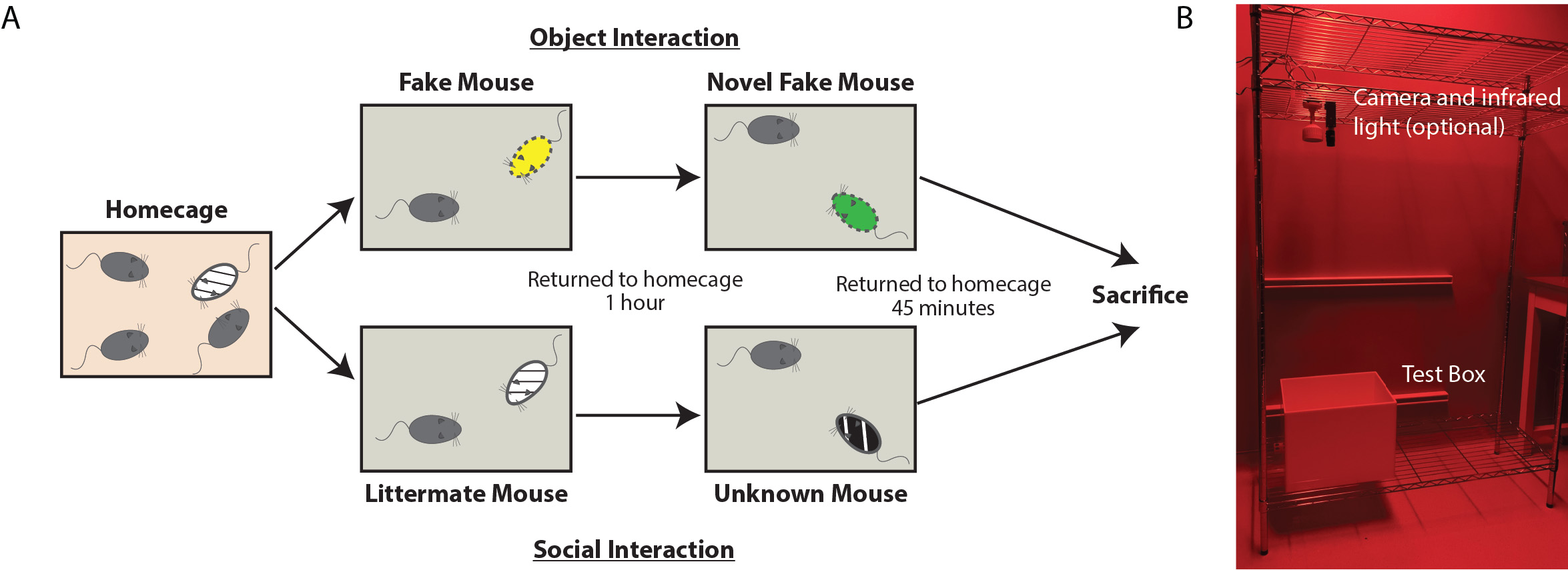
Figure 3. Schematic and Image of Labeling Day. A. Schematic of behavioral interactions on labeling day. Test mice reside in their homecage until immediately prior to interactions. One group of randomly selected mice will interact with toy mice (Object condition) while the other will interact with live mice (Social condition). Each test mouse is moved to the testing arena, immediately after which either the toy mouse or live littermate mouse is added. After 10 min of unrestricted interaction, the test mouse is returned to the home cage for 1 h. The test mouse is placed in the same, cleaned testing arena immediately joined by either a second toy mouse or live novel mouse. After 10 min of unrestricted interaction, the test mouse is returned to their home cage. The test mouse is anesthetized and perfused 45 min following the cessation of the last interaction. B. Image of behavioral set up, consisting of optional video camera (housing a long-pass filter) with infrared light positioned above the test box. In this set up, the room is equipped with red lights, but a red head lamp is also acceptable. - One hour following the initial exposure, return the test mouse to the interaction chamber. Quickly add the novel sentinel (non-cage mate) and allow them to interact for 10 min.
- For test mice interacting with objects, place one of the fake toy mice in the corner of the interaction chamber prior to adding the test mouse. Allow to interact for 10 min before returning the test mouse to the home cage.
- After 1 h, place the second fake toy mouse in an alternative corner. Add the test mouse and allow to interact for 10 min before returning the test mouse to the home cage.
- Forty-five minutes after the last interaction, anesthetize mice using an intraperitoneal injection of anesthetic mixture and proceed to the transcardiac perfusion fixation.
- Perfusion
- Prepare 1x PBS and 4% paraformaledehyde solutions, placing both in ice baths.
- Assemble perfusion system and prime tubing, removing air bubbles. Make sure that the line up to the needle is clear of paraformaldehyde before beginning because perfusion should start with 1x PBS to clear the blood from all vessels. The flow rate should be approximately 4 ml/min.
- Forty-five min after the conclusion of the last behavioral encounter, anesthetize mice with an intraperitoneal injection of anesthetic mixture of Ketamine (100 mg/kg) and Xylazine (15 mg/kg). Ensure mouse is properly anesthetized by the absence of toe-pinch reflexes before proceeding.
- (Optional) Secure mouse to perfusion tray. Open the chest cavity, using a hemocytometer hold the rib cage out of the way.
- Slide perfusion needle into the left ventricle of the heart and secure it in place. Begin perfusion with 1x PBS, and quickly make small incision in the right atrium to allow discharge of perfusion solutions.
- Perfuse with 1x PBS until the perfusate is clear from blood, approximately 2 min for an adult mouse. Switch to 4% paraformaldehyde solution. Perfuse for 20 min.
- Dissect the brain, being careful not to create nicks in the tissue. Immerse the brain in 4% paraformaldehyde, and store at 4 °C overnight.
- Replace 4% paraformaldehyde with brain storage solution (see Recipe 5).
- Be sure to clear final perfusion line of paraformaldehyde using 1x PBS before next perfusion.
- Sectioning
- Soak vibratome cutting blades in acetone for ~5 min prior to use (to remove shipping glue and oil).
- Cut brain with large blocking cuts using a razor blade; make sure to save injection sites for confirmation. Make straight cuts, leaving ample room for the brain to be glued to the cutting chuck.
- Prepare agar block in a way that the sides are cut straight and it is slightly wider and taller than the brain. Glue this side to the back of the cutting chuck using cyanoacrylate glue (e.g., Super GlueTM).
- Quickly remove excess brain storage solution using tissue paper, and glue brain directly in front of (closer to the blade) the agar block on the cutting chuck.
- Position the chuck in the vibratome, fill vibratome well with 1x PBS.
- Cut brain sections at 30 µm thickness for c-Fos immunohistochemistry, and 100 µm thickness for visualization of injection sites. Store serial sections in order within 12-well plates containing brain storage solution. Seal the well plates with ParafilmTM, and store at 4 °C and covered from light until staining.
- Immunohistochemistry
Notes:- Use only 4 well inserts in each 12-well plate to make media changes easier and reduce errors. When reusing wells, suction used media from the wells, rinse with 1x PBS and suction dry. To change media, fill dry wells with the upcoming solution, and move well inserts into fresh solution.
- Keep well plates covered with tin foil while not changing media to prevent bleaching of RetrobeadsTM with ambient light.
- For best results, stain all sections from an experiment at the same time to avoid unnecessary variability from tissue processing.
- Place one well insert per brain inside 12-well plates. Add 2-3 ml of 1x PBS.
- Using a paint brush, carefully transfer five 30 µm sections per brain to the wells.
- Fill 1 empty well per insert with 1 ml permeabilization solution. Move the inserts from the 1x PBS to the wells containing permeabilization solution. Place well plate on a shaker for 15 min, covered with tin foil.
- Fill 1 empty well per insert with 1 ml blocking solution, transfer wells to the block, replace plate on shaker, cover, leave for 1 h.
- Prepare primary antibody solution. Mix [1.25 ml antibody diluent + 2.5 µl anti c-Fos + 2.5 µl anti NeuN] x number of wells.
- Fill 1 empty well per insert with 1 ml primary antibody solution, transfer inserts to well, replace plate on shaker, cover with tin foil. Leave sections on shaker for ~36 h.
- Fill empty wells with 2 ml 1x PBS, transfer inserts and wash for 5 min. Repeat 3 times with fresh PBS each wash.
- Prepare amplification antibody solution. Mix [1.25 ml antibody diluent + 6.25 µl biotinylated goat anti-guinea pig] x number of wells.
- Place well inserts in 1 ml of amplification solution, let amplify for 2 h.
- Fill empty wells with 2 ml 1x PBS, transfer inserts and wash for 5 min. Repeat 3 times with fresh PBS each wash.
- Prepare secondary antibody solution. Mix [1.25 ml antibody diluent + 1.25 µl streptavidin conjugated to Alexa Fluor 405 + 6.25 µl goat anti-chicken conjugated to Alexa Fluor 647] x number of wells.
- Place well inserts in 1 ml of secondary antibody solution, let stain for 4 h.
- Fill empty wells with 2 ml 1x PBS, transfer inserts and wash for 5 min. Repeat 3 times with fresh PBS each wash.
- Put 1 ml of 1x PBS on a microscope slide. Using a paintbrush, transfer brain sections to the slide. Carefully position and flatten sections.
- Using a pipette, aspirate the excess 1x PBS while repositioning the sections. Dry remaining 1x PBS with tissue paper. Cover and leave until just dry, ~5 min.
- Place a small drop of Vectashield mounting media over each section. Carefully position coverslip avoiding bubbles.
- Using clear nail polish, seal the edges of the coverslip and slide.
- Store in the dark at 4 °C until imaging.
- Confocal microscopy and image analyses
Note: For quantitative immunohistochemistry, it is best practice to image all sections on the same day or immediately consecutive days to avoid unnecessary variability. It is of utmost importance that all images are taken using the exact same settings in the microscope, e.g., objective lens (especially NA), laser power, dwell time (scan rate), pinhole aperture, PMT gain and offset, distance between optical sections in z-stack, number of optical sections in each z-stack, pixel resolution, scanning area.- To begin, set laser power, and PMT gain and offset so that ~75% of the range of pixel intensities is used. Be sure to check a few brain sections from each condition to ensure these settings are suitable for the entire dataset. No pixels in any image should be brighter than the maximum in any of the experimental conditions (i.e., avoid image saturation).
- Recommended settings include:
- Pinhole set to 1 Airy unit, as determined by the image acquisition software of the confocal microscope.
- Lower gain can be compensated by slightly increasing laser power.
- Use only minimal PMT offset.
- Use consecutive excitation of all channels through line scan (i.e., do not use simultaneous excitation to avoid spectral “bleed-through” and shifting).
- Use line averaging (2 should be sufficient).
- Use 1,024 x 1,024 pixel image size, saved at 16 bit pixel depth resolution.
- Z-axis interval set to optimal as determined by confocal software.
- Depending on the number of neurons labeled per section, determine how many images you will take per section for the desired sample number (“n”). In Phillips et al. (2019), the entire region from each section was imaged, and every RetrobeadsTM-labeled neuron analyzed. This may not be feasible in other brain regions, depending on density of labeled neurons.
Data analysis
Note: See Figure 3 for the workflow of data analysis.
- Open images in ImageJ (or FIJI) (Figure 4A).
- Subtract the background from all channels, setting to 30 pixels (Figure 4B).
- Despeckle the c-Fos (blue) channels (Figure 4C).
- Merge the RetrobeadsTM and NeuN channels (Figure 4D).
- Set Measurement settings to record the mean gray value and redirect to c-Fos channel (Figure 4E).
- If using two RetrobeadsTM colors for different projection neurons, turn off one of RetrobeadsTM channels using the Channels tool. Adjust brightness and contrast in order to identify individual neurons that contain RetrobeadsTM.
- For each RetrobeadsTM and NeuN positive neuron, zoom in and scroll through the z-planes until you find the plane in which the neuron is most in focus (usually the largest soma size). Trace the neuron’s profile using the Freehand selection tool (Figure 4F).
- Change the z-plane on the c-Fos channel window to match the RetrobeadsTM/NeuN window. Measure the gray value (Figure 4F).
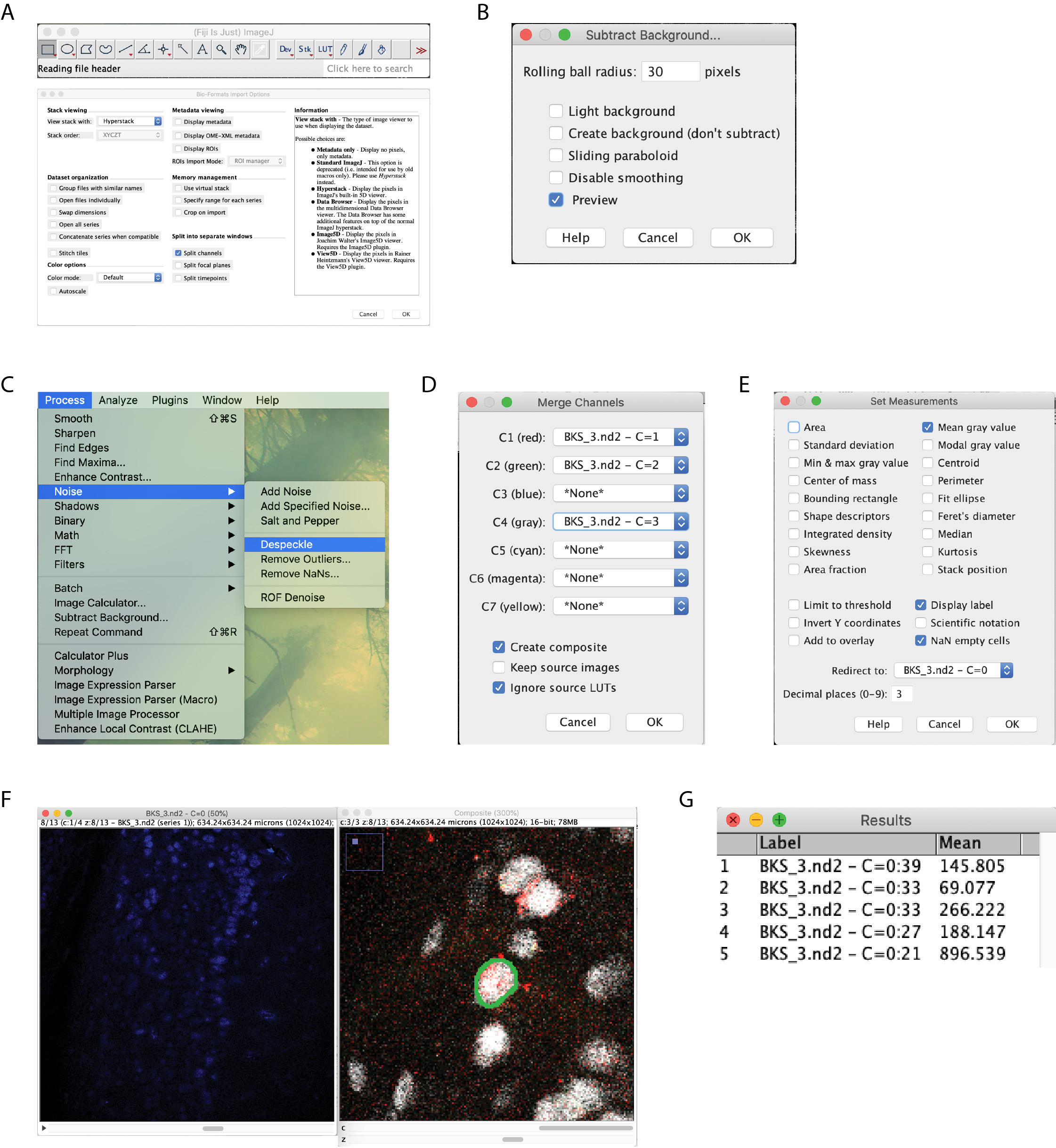
Figure 4. Image analysis workflow. Begin by opening images, check the split channels box so that options box matches that pictured (A). Subtract background with a 30 pixel rolling ball radius on all channels (B). Despeckle the c-Fos channel (C). Merge the RetrobeadsTM and NeuN channels (D). Set measurement settings to record the mean gray value, redirect to the c-Fos channel (E). Manually circle the NeuN label that contains RetrobeadsTM (F). Being sure to move the c-Fos channel to the same z-plane, measure the intensity (G). - Repeat steps 7 and 8 for all RetrobeadsTM-labeled neurons.
- Measure the c-Fos intensity of several neuron-sized regions of background and record for each section.
- When intensity collection is finalized, load the averaged background intensity per section into a statistical program (i.e., Prism) to determine outliers using Grubb’s test set at 1%. Remove neurons from sections whose background was determined to be an outlier.
- Data can be displayed either by using each neuron as an individual “n”, or, by averaging neurons from each animal. Phillips et al. (2019) chose to display data in both ways, with statistical testing being performed on the later.
- Be sure to use appropriate testing for the number of conditions in the experiment (e.g., 3- or 2-way ANOVA). Benjamani-Hochberg post-hoc testing can be used for comparisons between conditions.
Recipes
- 10x PBS
25.6 g Na2HPO4 7H2O
80 g NaCl
2 g KCl
2 g KH2PO4
Bring to 1 L double distilled water (ddH2O)
Store at room temperature for up to 1 month - 1x PBS
100 ml 10x PBS
900 ml ddH2O
Store at room temperature for up to 1 month - Anesthesia mixture for intraperitoneal injection: Ketamine (100 mg/kg) and Xylazine (15 mg/kg)
0.9 ml Ketamine
0.1 ml Xylazine
Store at room temperature for up to 1 month - 4% paraformaldehyde in 1x PBS
10 ml 37% PFA
70 ml 1x PBS
Make fresh each use - Brain section storage solution
10 mg Sodium azide
Add 100 ml 1x PBS
Store at room temperature for up to 1 month - Agar blocks
200 ml 1x PBS
800 mg Agar- Heat 1x PBS and agar until agar is fully dissolved
- Pour into 100 mm plate
- Allow to cool to room temperature, store wrapped in ParafilmTM at 4 °C
- Immunohistochemistry permeabilization solution
10 ml 1x PBS
250 µl Triton X-100
Store at 4° C for up to 1 month - Immunohistochemistry block solution
10 ml 1x PBS
10 µl Triton X-100
1.1 ml Serum from the host of secondary antibody (goat)
200 mg BSA
225 mg Glycine
Make fresh each staining protocol - Immunohistochemistry antibody diluent solution
10 ml Brain section storage solution
100 µl Triton X-100
505 µl Serum host of secondary antibody (Goat)
200 mg BSA
Make fresh each staining protocol, store at 4 °C while not in use - Well inserts
5 syringes cut to 1 inch in height
Mosquito netting
Silicone gel- Pull mosquito netting tight over the top of glassware, using rubber bands to hold in place
- Apply silicone gel to cut end of syringe
- Place syringe, silicone coated side down, on top of the netting
- Add weights (such as coins) to the top of the syringe to apply pressure
- Allow to dry overnight, carefully trim netting surrounding the insert and excess silicone
- Soak in 1x PBS overnight
Acknowledgments
This protocol was first reported in Phillips et al. (2019). This work was supported by NIH grant R56-NS103089-01A1 (LP-M), the Civitan International Foundation (MLP), NIH training grant T32-NS061788-04 (MLP), and Rettsyndrome.org (LP-M). We thank Ms. Karen Ayala-Baylon for mouse colony maintenance. We thank the Biostatistics Clinic of UAB’s Center for Clinical and Translational Science for assistance with statistical analyses.
Competing interests
The authors declare no competing financial interests.
Ethics
Mice were handled and housed according to the Committee on Laboratory Animal Resources of the National Institutes of Health (https://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf). All experimental protocols were reviewed and approved annually by the Institutional Animals Care and Use Committee of the University of Alabama at Birmingham (IACUC-21145; first approved on February 2, 2018).
References
- Aronov, D., Nevers, R. and Tank, D. W. (2017). Mapping of a non-spatial dimension by the hippocampal-entorhinal circuit. Nature 543(7647): 719-722.
- Barros, V. N., Mundim, M., Galindo, L. T., Bittencourt, S., Porcionatto, M. and Mello, L. E. (2015). The pattern of c-Fos expression and its refractory period in the brain of rats and monkeys. Front Cell Neurosci 9: 72.
- Bird, C. M. and Burgess, N. (2008). The hippocampus and memory: insights from spatial processing. Nat Rev Neurosci 9(3): 182-194.
- DeNardo, L. and Luo, L. (2017). Genetic strategies to access activated neurons. Curr Opin Neurobiol 45: 121-129.
- Eun, S. Y., Hong, Y. H., Kim, E. H., Jeon, H., Suh, Y. H., Lee, J. E., Jo, C., Jo, S. A. and Kim, J. (2004). Glutamate receptor-mediated regulation of c-fos expression in cultured microglia. Biochem Biophys Res Commun 325(1): 320-327.
- Groves, A., Kihara, Y., Jonnalagadda, D., Rivera, R., Kennedy, G., Mayford, M. and Chun, J. (2018). A Functionally Defined In Vivo Astrocyte Population Identified by c-Fos Activation in a Mouse Model of Multiple Sclerosis Modulated by S1P Signaling: Immediate-Early Astrocytes (ieAstrocytes). eNeuro 5(5). doi: 10.1523/ENEURO.0239-18.2018.
- Herrera, D. G. and Robertson, H. A. (1996). Activation of c-fos in the brain. Prog Neurobiol 50(2-3): 83-107.
- Hudson, A. E. (2018). Genetic Reporters of Neuronal Activity: c-Fos and G-CaMP6. Methods Enzymol 603: 197-220.
- Kohl, J., Babayan, B. M., Rubinstein, N. D., Autry, A. E., Marin-Rodriguez, B., Kapoor, V., Miyamishi, K., Zweifel, L. S., Luo, L., Uchida, N. and Dulac, C. (2018). Functional circuit architecture underlying parental behaviour. Nature 556(7701): 326-331.
- Paxinos, G. and Franklin K. B. J. (2008). The mouse atlas in stereotaxic coordinates (3rd Edition). Academic Press.
- Phillips, M. L., Robinson, H. A. and Pozzo-Miller, L. (2019). Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. Elife 8: 44182.
- Riddle, D. R., Lo, D. C. and Katz, L. C. (1995). NT-4-mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature 378(6553): 189-191.
Article Information
Copyright
![]() Phillips and Pozzo-Miller. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Phillips and Pozzo-Miller. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Phillips, M. L. and Pozzo-Miller, L. (2020). Identification of Socially-activated Neurons. Bio-protocol 10(17): e3744. DOI: 10.21769/BioProtoc.3744.
- Phillips, M. L., Robinson, H. A. and Pozzo-Miller, L. (2019). Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. Elife 8: 44182.
Category
Neuroscience > Neuroanatomy and circuitry > Brain nerve
Neuroscience > Neuroanatomy and circuitry > Immunofluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.