- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

BRIDGE: An Open Platform for Reproducible Protein-Ligand Simulations and Free Energy of Binding Calculations


(*contributed equally to this work) Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3731 Views: 8709
Reviewed by: Prashanth N SuravajhalaTianjiao HuangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Protein-ligand binding prediction is central to the drug-discovery process. This often follows an analysis of genomics data for protein targets and then protein structure discovery. However, the complexity of performing reproducible protein conformational analysis and ligand binding calculations, using vetted methods and protocols can be a challenge. Here we show how Biomolecular Reaction and Interaction Dynamics Global Environment (BRIDGE), an open-source web-based compute and analytics platform for computational chemistry developed based on the Galaxy bioinformatics platform, makes protocol sharing seamless following genomics and proteomics. BRIDGE makes available tools and workflows to carry out protein molecular dynamics simulations and accurate free energy computations of protein-ligand binding. We illustrate the dynamics and simulation protocols for predicting protein-ligand binding affinities in silico on the T4 lysozyme system. This protocol is suitable for both novice and experienced practitioners. We show that with BRIDGE, protocols can be shared with collaborators or made publicly available, thus making simulation results and computations independently verifiable and reproducible.
Keywords: Protein DynamicsBackground
The Googleplex of protein ligand binding possibilities is best reduced to a focussed set of drug hits, that can be interrogated in vitro and in vivo to produce a candidate drug for clinical studies, using computational chemistry methods. As a consequence, correct use of accurate compute methods that provide estimates of the energy of binding between a potential drug molecule, and a protein is central to the drug discovery process (Cournia et al., 2017). The idea is that the ligand should preferentially bind to the protein and inhibit its function. In this protocol, you will learn how to calculate relative protein-ligand binding free energies using open-source free energy tools and the Biomolecular Reaction and Interaction Dynamics Global Environment (BRIDGE) platform (Senapathi et al., 2019) that is built on the Galaxy platform (Afgan et al., 2018). The free energy differences between two ligands are calculated through an in silico perturbation from one ligand to another as they are i) bound to a protein (e.g., T4 lysozyme) and ii) solvated in water. Setting up free energy molecular simulations to run on High-Performance Computing (HPC) hardware resources, often done using scripts and the Linux command line, is challenging and a barrier to obtaining reliable results. In addition to the computational challenges, not all steps are repeatable as when run using varied software and hardware resources available through different laboratories, universities, public facilities, and cloud platforms. However, the objective of BRIDGE development is to provide a web-based platform that includes reliable methods for simulation, analysis, and analytics. A similar approach to BRIDGE is employed by the BioExcel Building Blocks software (Andrio et al., 2019). The BRIDGE-Galaxy platform makes the transition from genetics to proteomics to chemistry a seamless one and enables repeatable computer simulations and analysis using curated workflows.
We study T4 lysozyme as an illustration of a protocol to compute the relative binding of ligands, and we use benzene vs. p-xylene bound to T4 lysozyme as an example. The following tools are used: ProtoCaller for setup (Suruzhon et al., 2020), GROMACS for molecular dynamics (MD) simulations (Abraham et al., 2015), and alchemical analysis for simulation analysis (Klimovich et al., 2015). At least two simulations are required to compute the relative free energy of binding. First, the transformation between ligands in water and then the transformation between ligands bound to the solvated protein. Ligand 2 can be transformed into Ligand 1 or vice versa; the choice of direction is important to understand the meaning of the free energy calculated. Calculating in both directions is not required but can be used as an additional metric to confirm the computational convergence of the free energy result.
The identification of a protein target may be made through genomics experiments and bioinformatics analysis of gene expressions or arrived at through a literature search that may point to one or many protein targets. The protein structures are sourced from the RCSB Protein Data Bank. The identification of ligand hits is often guided by molecules that share a similarity to the known substrate or inhibitors found in the PDB, or derived from existing pre-screened molecule libraries, using similarity measure and docking calculations. Public molecular databases are good resources for finding possible ligands, for example, ChEMBL (Gaulton et al., 2012), ZINC (Irwin and Shoichet, 2005) and PubChem (Kim et al., 2016).
Preparing the protein from a PDB file into a state ready for simulation, posing the ligand in the binding site, and selecting appropriate parameters for accurate simulation are some of the immediate barriers to an MD simulation, even more so for free energy simulation. There are numerous alternative methods to prepare the protein, e.g., PDBFixer (Eastman et al., 2013). However, the preparation often requires specialist training to treat challenges such as missing loops from PDBs properly, repairing or installing mutated residues, disulfide bonds creation and assigning experimentally consistent protonation states. In the case of ligands, accurate charges require parameterization and assignment. In the protocol presented here, the automated setup of protein and ligand is done using the Alchemical Setup tool that links several specialized tools to perform protein setup and parameterization.
The BRIDGE interface (Figure 1) is straightforward and follows the Galaxy design. Tools are in the left panel, and a history of progress is in the right panel and information about the current tool or dataset selected is in the central panel. Data can be uploaded using the green upload icon at the top of the Tools menu (left panel), while the top menu bar includes your user account and other information. To search for tools, type a keyword into the search bar and tools with matching words will be displayed. Changes to the tools are versioned and the available versions can be selected via the central panel interface. Example histories of this process that include the outputs can be found by browsing to shared data and clicking on published histories (https://galaxy-compchem.ilifu.ac.za/histories/list_published). Choose the example history and click on the ‘+’ icon to add to your histories. Browse the example outputs using the eye icon. The free energy workflow illustrated in Figure 2 is also available (Reference 15).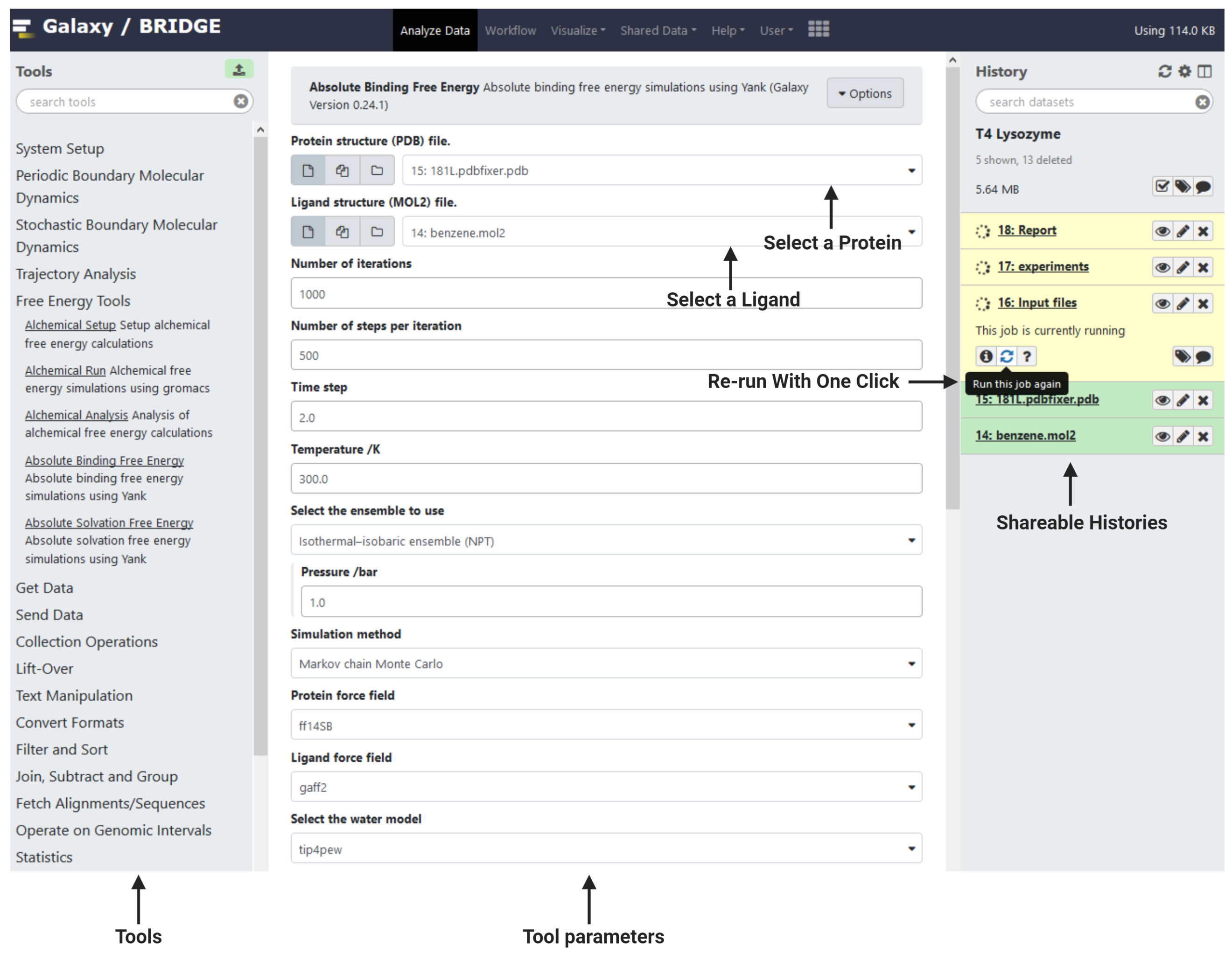
Figure 1. An overview of the Galaxy interface. Tools are in the left panel. Tool information or data are shown in the central panel and a history is displayed in the right panel.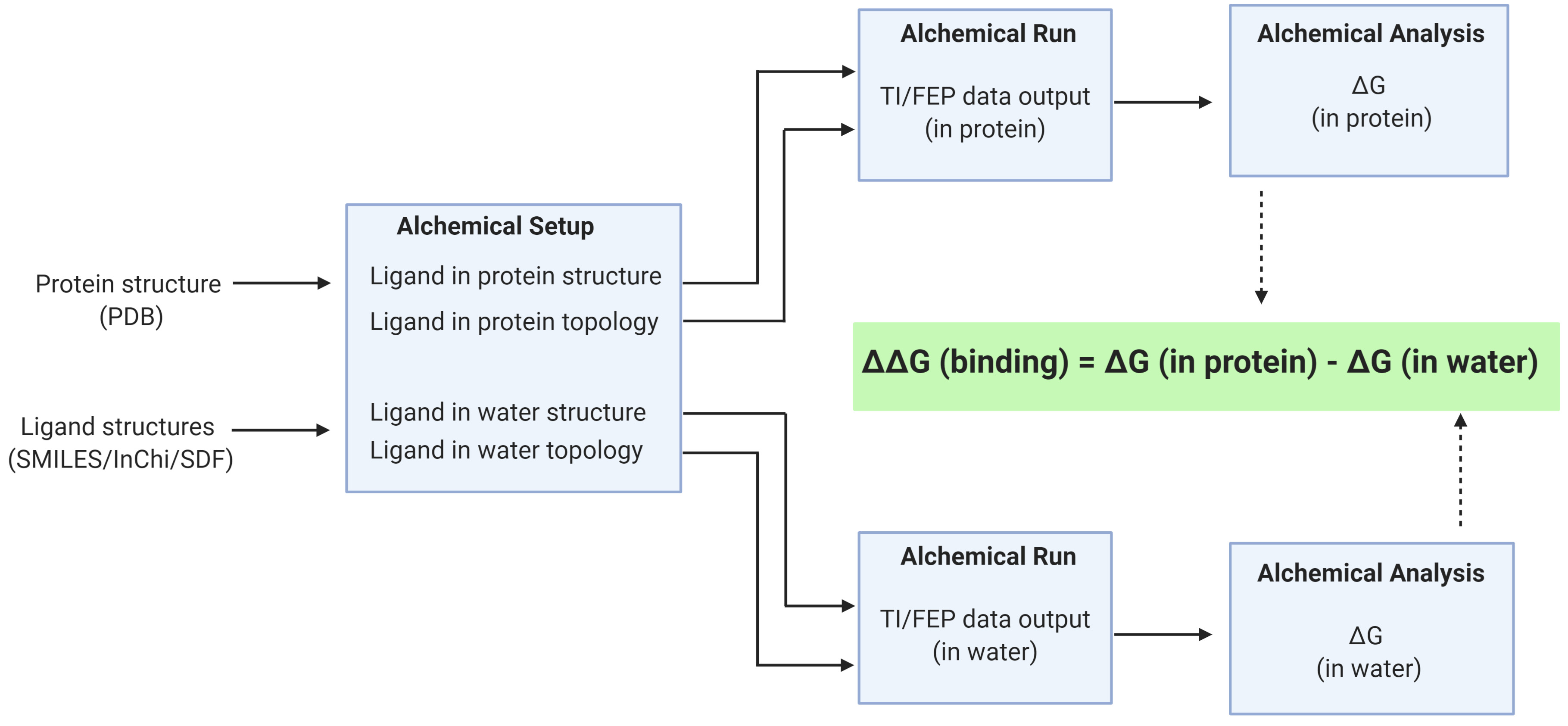
Figure 2. An overview of the Relative Binding Free Energy Workflow. The receptor in PDB format and ligand in SMILES are inputs to the Alchemical Setup. The multiple simulations are run using the Alchemical Run and the Alchemical Analysis tool is used to check convergence.
As part of the procedures below, a brief analysis is provided that is used to confirm that the free energy simulations are converged. An analysis of the molecular trajectories of the lead inhibitor can be conducted to identify the molecular interactions responsible for ligand binding and consider the conformation of ligand and protein. The aim of this further analysis would be to understand the rationale for binding. Further analysis might, for example, include hydrogen bond analysis and Ramachandran analysis, Root Mean Square Deviation (RMSD) and Principal Component Analysis (PCA). An example workflow is provided (Reference 14). PCA will reveal the flexibility of the catalytic domain and principal protein motions affecting inhibitor binding, while hydrogen bonding analysis would indicate key hydrogen bonding interactions between the ligand and the protein binding site.
Software
- Local computer
To run this on your local compute resources, obtain the BRIDGE Docker (https://github.com/scientificomputing/bridge-docker) and install the Docker image on your local machine. All libraries, compilers and links are packaged within the Docker image, so installation is seamless. A quad-core processor with 8GB RAM and 80GB hard drive space is sufficient for testing with Docker on any operating system. - Public servers
Access is obtained through a web browser and software installation is not required. The BRIDGE platform can be accessed at https://galaxy-compchem.ilifu.ac.za/. The tools are available on Galaxy Europe (http://cheminformatics.usegalaxy.eu).
Procedure
- System Preparation with Alchemical Setup
- Login to the BRIDGE server.
- To start preparing the simulation of lysozyme (protein, or receptor) and the benzene ligand, click on the Alchemical Setup tool to set up the simulation (Figure 3). Nine outputs will be generated–one report, four structures and four topologies. A structure and topology are created for the Ligand 1 to alchemically transform to Ligand 2 and vice versa in water and the protein.
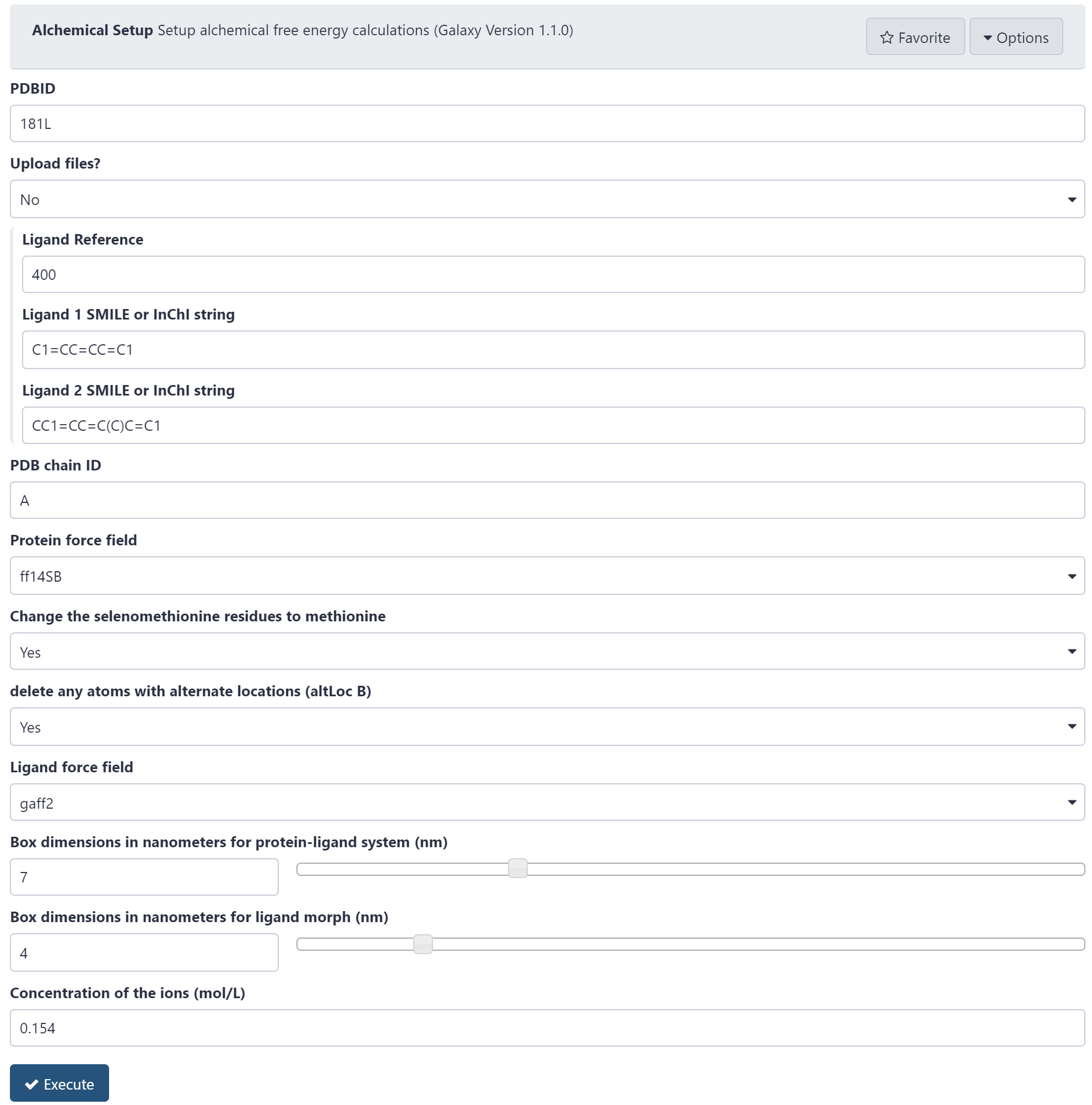
Figure 3. The Alchemical Setup tool. A protein and two ligands are specified and GROMACS compatible outputs will be generated. - Type in the PDB ID for T4 lysozyme: “181L” (https://www.rcsb.org/structure/181L).
- Choose “Upload files” and change to “No”.
- Choose the “Ligand Reference” as “400”. This is a benzene molecule in the active site (residue ID is 400). We are going to map the two ligands (benzene and p-xylene) onto this benzene. Therefore, Ligand Reference should be given as “400”.
- ProtoCaller can build ligands using the SMILES notation of a molecule. Insert a SMILES string for Ligand 1–use benzene “C1=CC=CC=C1”. This string can be generated using molecular software or an online database.
- Insert a SMILES string for Ligand 2 -use p-xylene "CC1=CC=C(C=C1)C.
- Choose the “PDB chain ID” as chain “A”.
- Choose the “Protein force field” as “ff14SB”. This is the AMBER Force Field that has been developed for proteins, and alternative force fields can be selected.
- Change any mutated selenomethionines back to methionine Set this option to “Yes".
- Set “delete any atoms with alternate locations (altLoc B)” to “Yes”. Only the primary coordinates will be used.
- Select “gaff2”, the General Amber Force Field 2, for “Ligand force field”.
- The protein-ligand complex will need to be solvated in a box of water molecules. Choose the box dimension for the protein-ligand system to be “7” nm. The longest side of lysozyme is approximately 5 nm and we use nonbonded cutoffs of 1.2 nm. We need to leave a buffer region for the protein to move freely in the water box and ensure there is sufficient space so that it does not interact with its periodic image. Adjust this value depending on the system under consideration.
- The ligand will also be solvated in a water box. Choose the size of the simulation box. “4” nm will be sufficient for most ligands, but this can be increased if the ligand is longer than 2.5 nm. Simulations of larger water boxes will take more time to complete.
- The last option is to define the concentration of ions to place inside the water box. By default, “0.154” M of sodium (Na+) and chloride (Cl-) ions are included, which is the physiological concentration.
- Click the “Execute” button to run this tool.
- The simulation will have started and will appear in the history panel. The files created are the GROMACS topology (.top) and structure (.gro) files required for the Alchemical Run tool. This will generate files to perturb Ligand 2 into Ligand 1 inside the protein and in a water box in both forward and backward directions.
- Check the progress of the simulations by clicking the eye icon in the report. This will take some time. The time taken will depend on available compute resources and may also be due to tool dependency checking and installation. This tool is parameterizing the ligand force field, which should take less than 20 minutes. If the history items remain grey for more than 1 day, contact a Galaxy Admin. e.g., via Gitter (https://gitter.im/galaxycomputationalchemistry/Lobby).
- The simulation is finished when the history items turn green.
- Relative free energy simulations in protein (Ligand 2 to Ligand 1) with Alchemical Run
Once complete with setup, the alchemical free energy simulations for each ligand are started and these are needed to calculate the ligand perturbation in water and the active site. The alchemical simulation is carried out in four steps for each free energy window, energy minimization, MD simulations in the Canonical ensemble (NVT) ensemble followed by Isothermal-Isobaric ensemble (NpT) ensemble. This pre-equilibration is vital to remove any Hamiltonian lags. Finally, the production simulation is carried out in an NpT ensemble. These steps are all carried out by the Alchemical Run tool in Galaxy (see Figure 4). It is from these simulations that the relative binding free energies can be calculated. Here, a simple perturbation of p-xylene (Ligand 2) to benzene (Ligand 1) will be carried out, and we choose a short simulation length for the purposes of illustrating the protocol. In reality, lengthier simulations are required. Often production simulations are at least 1 ns for free energy runs. For our converged lysozyme simulations we used the following parameters:
Minimization steps:10000, NVT steps: 500000 (500 ps), NpT steps: 500000 (50 ps), MD (production) steps: 1000000 (1000 ps), time step: 0.001 ps. Whichever parameters are chosen the analysis tools must be used to check convergence.- Select the Alchemical Run tool. This tool requires the topology and structure outputs from the alchemical setup tool.
- Select the “Structure file” to be “Ligand 2 to Ligand 1 in Protein Structure”.
- Select the “Topology file” to be “Ligand 2 to Ligand 1 in Protein Topology”.
- Set the number of “Minimization steps” to “10000”.
- Set the number of “NVT equilibration steps” to “500” (1 ps).
- Set the number of “NpT equilibration steps” to “500” (1 ps).
- Set the number of “MD (production) steps” to “1000” (2 ps).
- The “seed” is set to 19880924 by default and it must be changed. This is the seed to start the random number generator for molecular dynamics, change this to the current date or any random number, for example, “20200130”.
- Set the “time step” to “0.002” ps.
- Create hydrogen bond constraints. Choose “Yes” to apply constraints to the ligands and choose “H-bond constraints”. Choose “LINCS order” “4”, “LINCS iterations” “1”, “LINCS maximum angle” “30”.
- Select the free energy perturbation (FEP) path and choose to go from a larger ligand (p-xylene) to a smaller ligand (benzene).
- Type in “39” for the “number of free energy windows”. The more windows, the more likely the simulations will overlap and converge. The number of windows is zero-indexed, typing in 39 means there are 40 windows. First the charges are perturbed, then the Van der Waals and Bonding terms are perturbed. Here we are alchemically perturbing from Ligand 2 (0.00) to Ligand 1 (1.00) on a scale from 0 to 1. Any number between 0 and 1 represents how the charges, Van der Waals, Bonding interactions are scaled to form a hybrid system.
- Scale the charges of molecules in the simulations for the first 10 windows from 0.00 to 1.00 in increments of 0.11. The rest of the free energy windows are not perturbed. The exact lambda values must be described, by typing in the following 40 numbers: “0.00 0.11 0.22 0.33 0.44 0.56 0.67 0.78 0.89 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00”.
- Scale the Van der Waals interaction in the simulations for the final 29 windows after the charge has been perturbed. These are scaled from 0.00 to 1.00 in increments of 0.04 and then the change tapers to 0.02 and 0.01 at the end point to prevent end point catastrophes. The exact lambda values must be described, type in the following 40 numbers: “0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.04 0.08 0.11 0.15 0.19 0.23 0.27 0.30 0.34 0.38 0.42 0.46 0.49 0.53 0.57 0.61 0.65 0.68 0.72 0.76 0.80 0.84 0.87 0.91 0.95 0.97 0.98 0.99 0.99 1.00”.
- Scale the Bonding terms in the simulations in tandem with the Van der Waals interaction scaling. The exact lambda values must be described, type in the following 40 numbers: “0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.04 0.08 0.11 0.15 0.19 0.23 0.27 0.30 0.34 0.38 0.42 0.46 0.49 0.53 0.57 0.61 0.65 0.68 0.72 0.76 0.80 0.84 0.87 0.91 0.95 0.97 0.98 0.99 0.99 1.00”.
- Use the default free energy options.
- Set the temperature to “300” K and the pressure to “1” bar.
- Choose “Perform the simulation”. The input files can be generated and simulated outside of Galaxy if required.
- Click “Execute”.
- Check the report in your history for the simulation progress. Output will also include ‘.tar’ files (archives) of the FEP/TI (Thermodynamic Integration) data and trajectories. View these trajectories to ensure no unphysical events have occurred during the FEP calculations.
- Optional: Repeat this for Ligand 1 to Ligand 2 in protein.
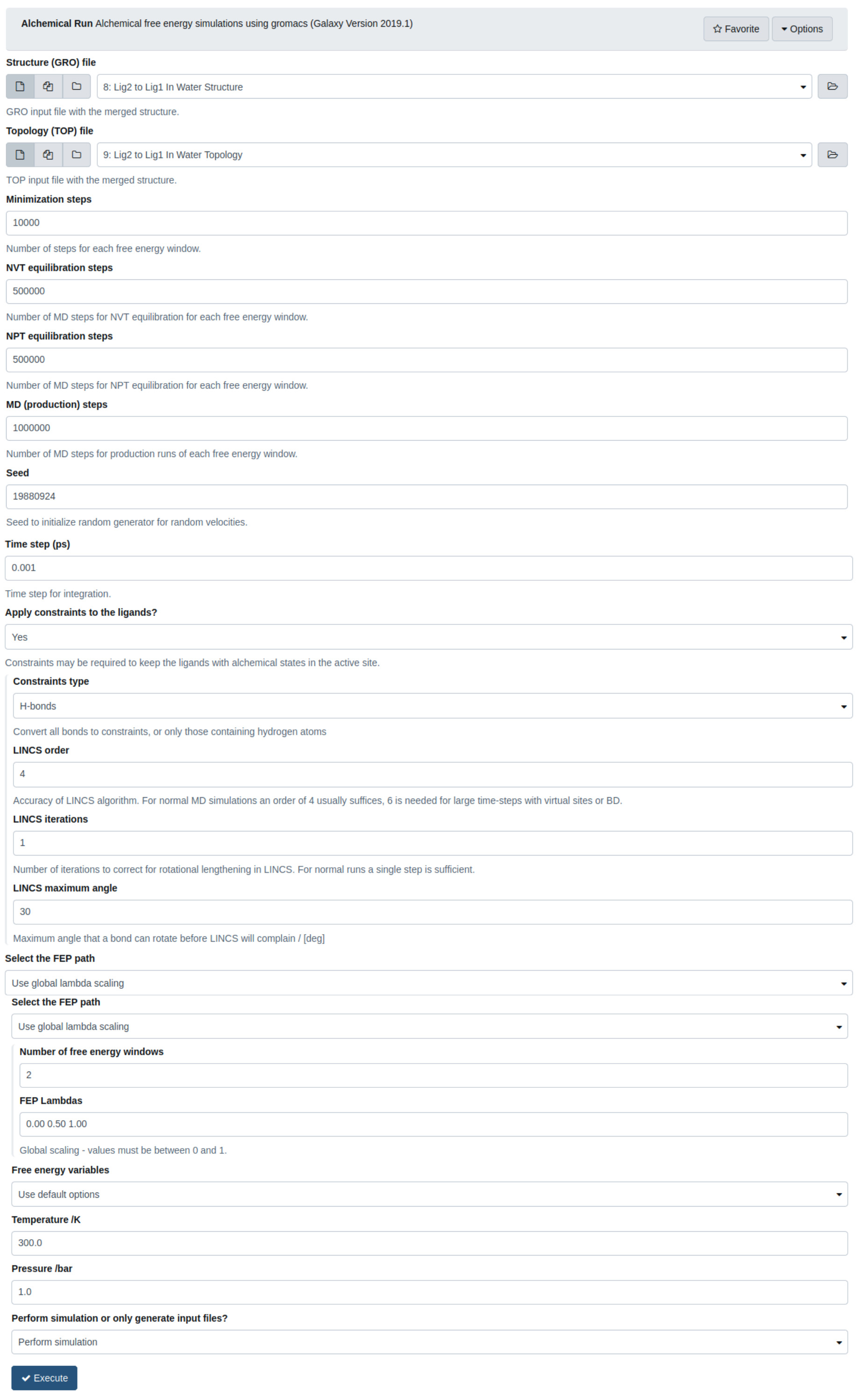
Figure 4. The Alchemical Run tool. This tool is used to run the minimization, equilibration and production simulations in GROMACS for the alchemical transformation between the ligands in a particular environment.
- Relative free energy simulations in water (Ligand 2 to Ligand 1) with Alchemical Run
- As for B, but choose the “Structure file” as “Ligand 2 to Ligand 1 in Water Structure” and the “Topology File” as “Ligand 2 to Ligand 1 in Water Topology”.
- Optional: Repeat this for Ligand 1 to Ligand 2 in water.
- Analysis and convergence testing of Ligand 2 to Ligand 1 in the protein with Alchemical Analysis
The Alchemical Analysis tool (Figure 5) provides a comprehensive analysis (although the GROMACS inbuilt bar module can be used). The data.tar file from the Alchemical Run tool is a required input. The expected tool outputs are as follows:
•Report and free energy outputs.
•Overlap matrix of free energy windows (as PNG).
•Convergence plot (as PNG).
•Curve Fitting Method (CFM) based consistency inspector (as PNG).
•Free energy change breakdown (as PNG).
•Thermodynamic Integration plot (as PNG).
For more details about Alchemical Analysis, read the article (Klimovich et al., 2015) and the code repository (https://github.com/MobleyLab/alchemical-analysis).- Open the Alchemical Analysis tool.
- Select the Protein data output; this will be in the “tar” format.
- Set the prefix to “md”, the alchemical run uses the prefix md to save the simulation data by default.
- Set the temperature to “300” K.
- The “Equilibration time” is set as “0” ps. This can be changed to discard some simulation data. It would be useful in a scenario where the simulations are found not to have converged and the first few ps need to be removed.
- Report energies in “kcal”.
- If required, include lambda states to skip. By default, none are skipped.
- Click “Execute”.
- Optional: Repeat for Ligand 1 to Ligand 2 in protein.
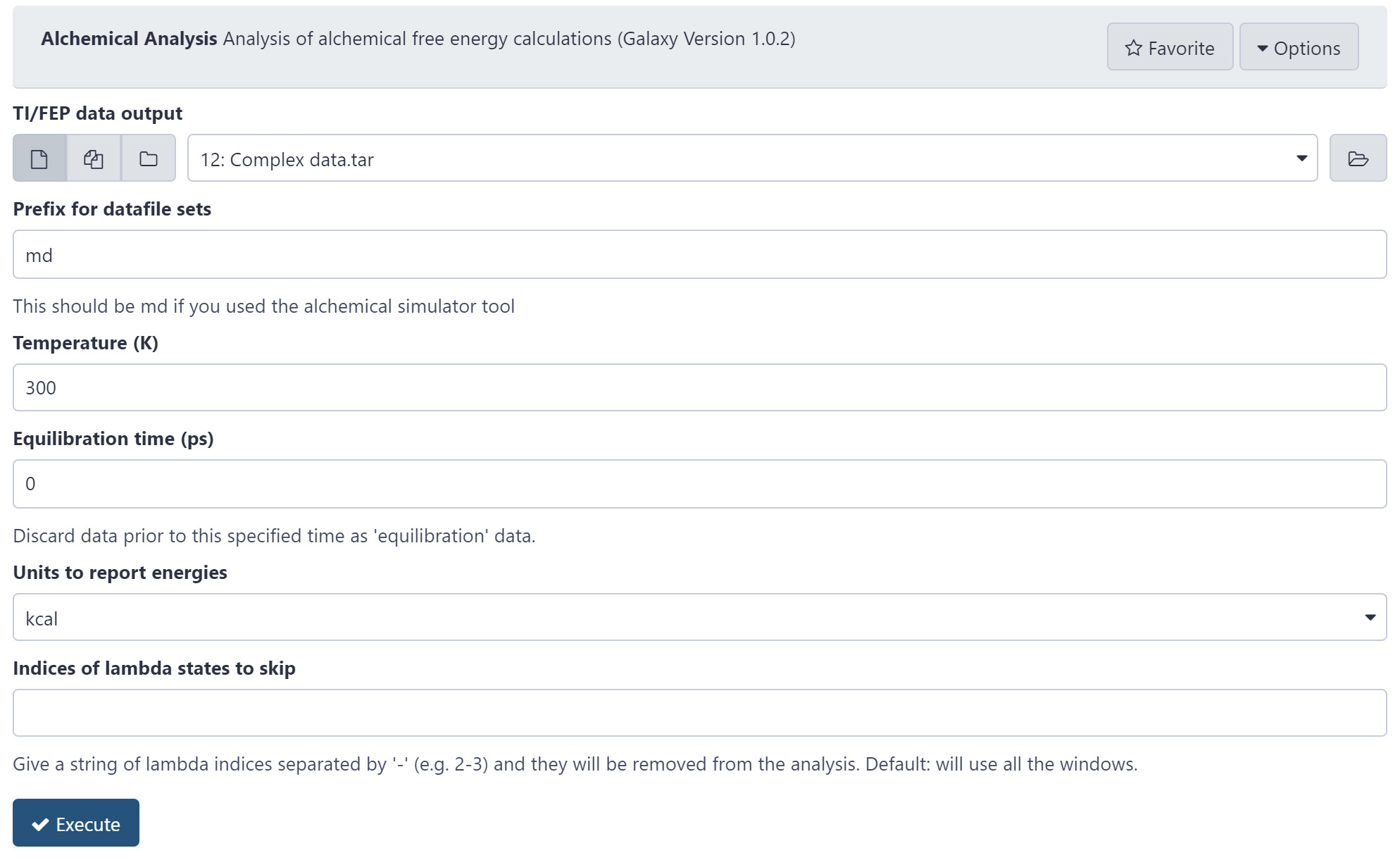
Figure 5. The Alchemical Analysis tool. This tool is used to analyze free energy simulations. The user provided simulation data from GROMACS in tar format along with further simulations details such as the temperature. The outputs provided assist the user in evaluating if the calculations have converged. - Analysis and convergence testing of Ligand 2 to Ligand 1 in water with Alchemical Analysis
- As for D but choose the Ligand 2 to Ligand 1 in Water data output.
- Optional: Repeat for Ligand 1 to Ligand 2 in Water.
- Interpret the analysis for Ligand 2 to Ligand 1 in the protein
- Check the results of the calculated free energy values with different estimators and methods (Thermodynamic integration [TI], Deletion exponential averaging [DEXP], Insertion exponential averaging [IEXP], Bennett acceptance ratio [BAR], Multistate Bennett acceptance ratio [MBAR]); see Figures S1 and S2.
- Check the free energy change dF(t) output which is a text file containing the calculated free energies as a function of time in both forward and backward directions. The backward direction simulation was not run but this tool can extract that data from the forward direction simulation.
- Check the Convergence Plot dG(t) to see if the simulation has converged. The forward and reverse ΔG should be the same within tolerance, preferably within the purple shaded band (Figure 6).
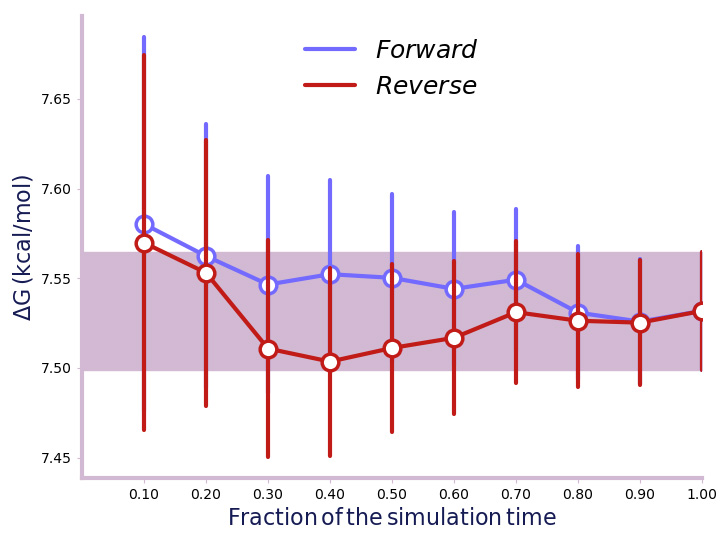
Figure 6. Free Energy Convergence plot. This plot is used to confirm the forward and reverse ΔG values fall within tolerance. - Check the Overlap Matrix which shows the overlapping of each free energy window in the simulation. There should be a good overlap between neighbouring free energy windows. If so, this indicates the stratification (number of free energy windows) is sufficient for the simulation.
- Consider the Free energy change breakdown. This gives the visualization of the calculated free energy for each window using each free energy estimator used. This plot shows that the IEXP exhibits a huge deviation from the other methods.
- Consider the Thermodynamic Integration Plot. This visualization of the data is useful for calculating free energy using TI.
- Consider the Curve Fitting Method based consistency inspector. This will indicate if all simulations have converged. If not, the simulations can be rerun using a longer simulation time (repeat protocol B with a longer simulation time) or the unconverged fraction of data of the simulation can be removed using alchemical analysis (rerun protocol D and set the equilibration time to be an appropriate number of ps.
- We have completed the analysis of the perturbation from Ligand 2 to Ligand 1 in the protein.
- Optional: Repeat for Ligand 1 to Ligand 2 in protein.
- Interpret the analysis for Ligand 2 to Ligand 1 in water
- As for F but choose the Ligand 2 to Ligand 1 in Water analysis results.
- Optional: Repeat for Ligand 1 to Ligand 2 in water.
- Calculate the relative free energy of Ligand 2 to Ligand 1
- Find the free energy estimated with MBAR for Ligand 2 to Ligand 1 in protein (10.212 ± 0.278 kcal/mol).
- Find the free energy estimated with MBAR for Ligand 2 to Ligand 1 in water (10.600 ± 0.258 kcal/mol).
- Calculate the relative free energy of binding (the difference), -0.388 ± 0.379 kcal/mol. This value is negative, which means that Ligand 1 (benzene) binds more favorably than Ligand (2) p-xylene. This compares well to the experimental difference, which is -0.52 ± 0.17 kcal/mol (Morton et al., 1995).
- Optional: Carry out a trajectory analysis of the first and last free energy windows.
Notes
- Cautionary points on free energy calculations should be noted (Reference 13).
- “These rules are not the end-all set and you should be familiar with why each one is suggested before just accepting them”.
- “More states are better than fewer. Variance shrinks rapidly with the number of states. You want the difference between intermediaries to be between 2-3 kBT”.
- These simulations are repeatable using workflows and histories (Reference 15).
- There will be some variability in the molecular ensembles if a new random seed is chosen, and it is recommended to choose a new random seed for each simulation.
Acknowledgments
We would like to acknowledge the Galaxy community, the Galaxy EU team and the Galaxy computational chemistry team on GitHub for tool and code review.
An original paper and the workshop from which this protocol has been derived is unpublished at the time of this writing. However, this may be found when searching for i) “BRIDGE an open platform for reproducible high throughput free energy” Tharindu Senapathi, Miroslav Suruzhon, Christopher B. Barnett, Jonathan Essex and Kevin J. Naidoo.
This work is based on research supported by the South African Research Chairs Initiative (SARChI) of the Department of Science and Technology (DST) and National Research Foundation (NRF) grant 449130 and supported by the South African Medical Research Council under a Self-Initiated Research Grant and the Medical Research Council grant (KJN). Even though the work is supported by the MRC, the views and opinions expressed are not those of the MRC but of the authors of the material produced or publicized. TS thanks SARChI for a doctoral award. We thank the University of Cape Town, the National Research Foundation of South Africa (NRF) for support in hosting BRIDGE on the ilifu data centre and the Centre for High Performance Computing for the use of their clusters when developing BRIDGE.
Competing interests
There are no competing interests.
References
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B. and Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1: 19-25.
- Afgan, E., Baker, D., Batut, B., Van Den Beek, M., Bouvier, D., Čech, M., Chilton, J., Clements, D., Coraor, N. and Grüning, B. A. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46(W1): W537-W544.
- Andrio, P., Hospital, A., Conejero, J., Jordá, L., Del Pino, M., Codo, L., Soiland-Reyes, S., Goble, C., Lezzi, D., Badia, R. M., Orozco, M., Gelpi, J. L. (2019). BioExcel Building Blocks, a software library for interoperable biomolecular simulation workflows. Scientific Data 6(1): 169.
- Cournia, Z., Allen, B. and Sherman, W. (2017). Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J Chem Inf and Model 57(12): 2911-2937.
- Eastman, P., Friedrichs, M. S., Chodera, J. D., Radmer, R. J., Bruns, C. M., Ku, J. P., Beauchamp, K. A., Lane, T. J., Wang, L.-P., Shukla, D., Tye, T., Houston, M., Stich, T., Klein, C., Shirts, M. R. and Pande, V. S. (2013). OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J Chem Theory Comput 9(1): 461-469.
- Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., Light, Y., McGlinchey, S., Michalovich, D. and Al-Lazikani, B. (2012). ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40(D1): D1100-D1107.
- Irwin, J. J. and Shoichet, B. K. (2005). ZINC− a free database of commercially available compounds for virtual screening. J Chem Inf Model 45(1): 177-182.
- Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., Han, L., He, J., He, S. and Shoemaker, B. A. (2016). PubChem substance and compound databases. Nucleic Acids Res 44(D1): D1202-D1213.
- Klimovich, P. V., Shirts, M. R. and Mobley, D. L. (2015). Guidelines for the analysis of free energy calculations. J Comput Aided Mol Des 29(5): 397-411.
- Morton, A., Baase, W. A., & Matthews, B. W. (1995). Energetic origins of specificity of ligand binding in an interior nonpolar cavity of T4 lysozyme. Biochemistry 34(27): 8564-8575.
- Senapathi, T., Bray, S., Barnett, C. B., Grüning, B. and Naidoo, K. J. (2019). Biomolecular Reaction and Interaction Dynamics Global Environment (BRIDGE). Bioinformatics 35(18): 3508-3509.
- Suruzhon, M., Senapathi, T., Bodnarchuk, M. S., Viner, R., Wall, I. D., Barnett, C. B., Naidoo, K. J. and Essex, J. W. (2020). ProtoCaller: Robust Automation of Binding Free Energy Calculations. J Chem Inf Model 60(4): 1917-1921.
- Constructing a Pathway of Intermediate States - AlchemistryWiki. (2016, August 12). Retrieved May 19, 2020, from http://www.alchemistry.org/wiki/Constructing_a_Pathway_of_Intermediate_States.
- Galaxy Training: Analysis of molecular dynamics simulations. (2020, January 13). Retrieved May 19, 2020, from https://galaxyproject.github.io/training-material/topics/computational-chemistry/tutorials/analysis-md-simulations/tutorial.html.
- Scientificomputing/bioprotocol-paper-2020-sm: Data and workflows - Bioprotocol. (2020, May 19). Retrieved May 19, 2020, from https://zenodo.org/badge/latestdoi/264202784.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Senapathi, T., Barnett, C. B. and Naidoo, K. J. (2020). BRIDGE: An Open Platform for Reproducible Protein-Ligand Simulations and Free Energy of Binding Calculations. Bio-protocol 10(17): e3731. DOI: 10.21769/BioProtoc.3731.
Category
Molecular Biology > Protein > Protein-protein interaction
Systems Biology > Proteomics > Spatial Proteomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.