- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measuring Intracellular Vesicle Density and Dispersion Using Fluorescence Microscopy and ImageJ/FIJI



Published: Vol 10, Iss 15, Aug 5, 2020 DOI: 10.21769/BioProtoc.3703 Views: 8076
Reviewed by: Alexandros AlexandratosEvangelia XingiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cell signalling, cell secretion, and plasma membrane repair are processes that critically rely on intracellular vesicles, important components of the endocytic and secretory pathways. More specifically, the strategic distribution of intracellular vesicles is important for diverse cellular processes. The method presented here is a simple, affordable, and efficient tool to analyze the distribution of intracellular vesicles such as lysosomes, endosomes, Golgi vesicles or secretory granules under different experimental conditions. The method is an accessible way to analyze the density and dispersion of intracellular vesicles by combining immunofluorescence with pixel-based quantification software (e.g., ImageJ/FIJI). This protocol can be used widely within the scientific community because it utilizes ImageJ/FIJI, an open source software that is free. By tracking fluorescent vesicles based on their position relative to cell nuclei we are able to quantify and analyze their distribution throughout the cell.
Background
Intracellular vesicles are important components of the endocytic and secretory pathways, which are responsible for maintaining several critical cellular functions. For this reason, many studies have focused on the pathways that regulate the distribution of vesicles in both physiological and pathological conditions, while others have examined the consequences of aberrant vesicle distribution in many important cellular processes.
In 1988, Aunis and Bader showed that two pools of secretory vesicles exist in secretory cells (Aunis and Bader, 1988). The first pool was located just below the plasma membrane and the secretion of this pool was not regulated by the cytoskeleton, but attached to the plasma membrane as a result of being bound to elements of the cytoskeleton; the second pool was attached to actin filaments, slightly away from the membrane, which could be mobilized to the inner leaflet of the plasma membrane after the depletion of the first pool. This was achieved by depolarization of the membrane, rearrangement of the actin filaments and dissolution of the cytoskeleton barrier, leading to its detachment from the cytoskeleton to reach the exocytic sites.
Koseoglu et al. (2011) also provided evidence for the existence of different pools of secretory vesicles and that membrane cholesterol content is important for exocytosis of these pools. The authors observed that membrane cholesterol sequestration from chromaffin cells of the adrenal medulla did not alter either the kinetics or the amount of release of the first pool of vesicles, which was already pre-anchored to the plasma membrane. However, the subsequent release of the remaining vesicles, belonging to the second slow-releasing pool, also called the reserve pool, was compromised by changes in membrane cholesterol levels. The authors further suggested that the change in the polymerization of actin filaments induced by cholesterol depletion could be compromising the mobilization of the slow releasing or reserve vesicles.
Mobilization of lysosome pools is described in several situations (Brito et al., 2019; Lawrence and Zoncu, 2019). One example is plasma membrane repair (PMR), a mechanism widely used by nucleated cells to maintain plasma membrane integrity. Briefly, when a cell suffers a micro-injury, its membrane must be sealed to avoid depolarization, cytoplasm leakage and cell death. Upon injury, extracellular Ca2+ flows through the lesions into the cytoplasm, increasing cytoplasmic Ca2+ concentration, which in turn induces lysosomal exocytosis. Lysosomal exocytosis is then followed by a massive endocytosis, which reseals the plasma membrane by carrying wounded portions in endosomes into the cell. The whole process involves the participation of lysosomal hydrolases, which act on the extracellular leaflet of the plasma membrane and facilitates the endocytic process by inducing plasma membrane invagination and its inward budding (Idone et al., 2008; Tam et al., 2010; Castro-Gomes et al., 2016).
Some parasites, such as Trypanosoma cruzi and Leishmania amazonensis, can subvert PMR to invade different host cells (Horta et al., 2020). Tardieux et al. (1992) verified that the mobilization of lysosomes is a key event for effective cell infection by T. cruzi, which subverts PMR mechanism to invade cells. Hissa et al. (2012), studying how the protozoan parasite T. cruzi enters cardiomyocytes, showed that depletion of membrane cholesterol led to a decrease in host cell invasion by interfering with lysosome recruitment and fusion during parasite-host cell interaction. More recently, Cavalcante-Costa et al. (2019) showed that lysosomal positioning is also crucial for L. amazonensis invasion to non-phagocytic cells, using basically the same mechanism previously described for T. cruzi.
Therefore, lysosomal positioning is crucial for PMR and cell invasion by some pathogens, as well as many other cellular mechanisms. These vesicles can be found around cell nuclei or spread throughout the cell, moving fast along cell microtubules towards the cell periphery in a stop-and-go manner (Cabukusta and Neefjes, 2018), depending on cell type or special conditions, such as the presence of certain drugs or plasma membrane injury by cytotoxins. Since lysosomes function as Ca2+ sensitive vesicles (Rodriguez et al., 1997), their positioning and exocytosis are finely regulated by the presence of this ion in the cytosol.
The examples above show that the distribution of intracellular vesicles can be disturbed by several conditions and that their correct positioning is pivotal for physiological cellular functions. Thus, it is critical to have a reliable method to measure the distribution and movement of intracellular vesicles. Here we describe a method to quantify lysosomes in cells that takes advantage of immunofluorescence, a commonly-used technique, and open source software ImageJ/FIJI (Schneider et al., 2012; Schindelin et al., 2012). This protocol provides a way for researchers to compare changes in the distribution of lysosomes in diverse processes and it can be adapted to studies involving different types of intracellular vesicles such as endosomes, Golgi vesicles or secretory granules.
Materials and Reagents
- 13 mm round coverslips (Perfecta, catalog number: 10210013CE )
- 24-well plates
- Primary antibody: rat anti-LAMP1 IgG (1:50, 1D4B) or rat anti-LAMP2 IgG (1:50, ABL-93) (obtained from Developmental Studies Hybridoma Bank–DSHB–University of Iowa) or any marker of the desired vesicle
- Alexa-Fluor-488-conjugated equivalent anti-rat secondary antibodies (Life Technologies, catalog number: A11006 )
- Texas Red Dextran (Thermo Fisher Scientific, catalog number: D1863 )
- Phosphate Buffered Saline (PBS) (Sigma-Aldrich, catalog number: P3813 )
- Bovine Serum Albumin (BSA) (Sigma-Aldrich, catalog number: A7906 )
- Saponin (Sigma-Aldrich, catalog number: S4521 )
- 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) (Thermo Fisher Scientific, catalog number: D1306 )
- Paraformaldehyde (Vetec, catalog number: 694 )
Equipment
- Epifluorescence or confocal microscope
Software
- ImageJ (NIH/https://imagej.nih.gov/ij/download.html) or "FIJI Is Just ImageJ" FIJI (NIH/http://fiji.sc/Fiji)
Procedure
Cell labeling:
- Labeling of lysosomes
- For this assay, we start with subconfluent cell monolayers. We use mouse embryonic fibroblasts (MEF) in this assay. To get subconfluent cell monolayers 24 h before the assay, we plate cells on 13 mm round coverslips at a density of 4 x 104 cells/well in 24-well plates. Treat each cell-containing well as follows.
- Fix with 4% paraformaldehyde for 15 min.
- Rinse 3 times with 500 µl of PBS.
- Block and permeabilize cells by incubating them in 500 µl of PBS containing 2% BSA and 0.5% saponin (PBS/BSA/Saponin) for 20 min.
- Incubate for 45 min with any of the following primary antibodies: rat anti-LAMP1 IgG (1:50, 1D4B); rat anti-LAMP2 IgG (1:50, ABL-93) (obtained from Developmental Studies Hybridoma Bank) or any desired marker.
Note: Antibodies must be diluted in PBS/BSA/Saponin solution. - Rinse 3 times with 500 µl of PBS/BSA/Saponin to remove wash primary antibody.
- Incubate for 30 min with Alexa-Fluor-488-conjugated equivalent secondary antibodies (Life Technologies), in a dilution of 1:250 in PBS/BSA/Saponin, or any appropriate labeled secondary antibody.
- Rinse 3 times with 500 µl of PBS/BSA/Saponin to wash secondary antibody.
- Stain with DAPI (10 µg/ml) to visualize cell nuclei.
- Rinse 3 times with 500 µl of PBS.
- Mount coverslips on microscope slides using anti-fading mounting medium. Let the microscope slides dry overnight.
- Analyze slides by fluorescence microscopy, acquiring images of representative cells.
Note: Use 60x and/or 100x objectives to acquire images of the entire cell. Prefer individual cells, rather than groups of cells (an alternative method for analyzing a group of cells is explained below).
- Labeling of endocytic vesicles
- Obtain subconfluent monolayers by plating cells on 13 mm round coverslips at a density of 4 x 104 cells/well in 24-well plates 24 h before assay.
- At selected time points, incubate cells with Texas Red-Dextran (0.25 mg/ml) (or any other labeled dextran), a membrane impermeable fluorescent dye.
Note: If you intend to study whether a treatment or another condition can stimulate or compromise endocytosis, perform the stimuli in the presence of Texas Red-Dextran. - At the researcher-determined time points of treatment, rinse at least 3 times with 500 µl of PBS to eliminate extracellular Texas-Red Dextran. This is a crucial step to avoid high background staining. Treat each cell-containing well as follows.
- Fix with 4% paraformaldehyde (500 µl) for 15 min.
- Rinse 3 times with 500 µl of PBS.
- Follow Steps A9-A12.
Data analysis
- Density of lysosomes and dispersion of fluorescence intensity in perinuclear and pericellular regions
By labeling the lysosomes as described, it is possible to distinguish two regions in the cytoplasm and measure the fluorescence intensity of labeled vesicles in these regions. The first region surrounds the nucleus, and will be referred to as the perinuclear region, which can be defined as having 0.5 of the nuclei radius. Since it is common for the nucleus to be an irregular size, or oval, a short (r) and a long (R) radius have to be defined to define the perinuclear region accordingly (adapted from Nabavi et al., 2008 and as illustrated in Figure 1). Measurements will be made throughout the whole area defined by the perimeter traced around the nucleus considering the starting point from the center of the nuclei to r + 0.5r or R + 0.5R, removing the DAPI channel. The second region (pericellular) is determined as the cell periphery ranging from the end of the perinuclear region to the border of the cell. These measurements can be made using ImageJ/FIJI software or similar. Just remember to calibrate the software to provide measurements in μm instead of pixels. To calibrate, open an image that has been taken using the same microscope (with the same objective) of an element of a well-known size, for instance a scale bar or a ruler with a micrometric scale. Using the line selection tool, draw a line with the same measures of the known element, press analyze > set scale. ImageJ/FIJI will provide the distance in pixels. Complete blank fields with the known distance and the unit (μm). The calibration can be applied to the open image or it can be applied to other images by checking the "Global" option.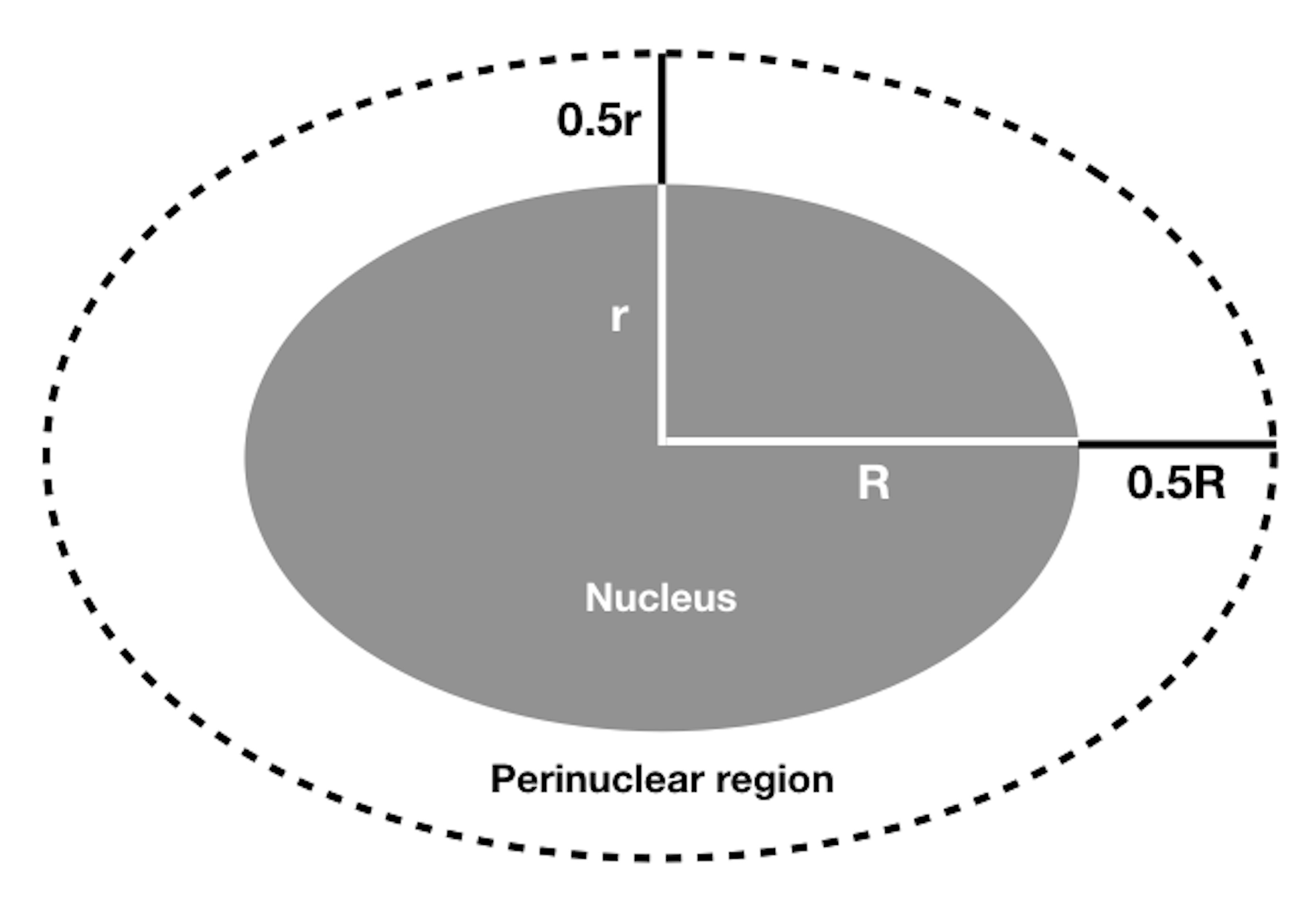
Figure 1. Definition of the perinuclear region used to quantify fluorescence-labeled vesicles distribution around an oval-shaped nucleus. r is the nucleus short radius, R is the nucleus long radius. Perinuclear region radii are calculated starting from the nuclei center as r + 0.5r and R + 0.5R for short and large radius, respectively, to define the area inside the perimeter obtained.- Using ImageJ/FIJI, open an image of the DAPI-labeled nuclei. Determine the diameters of the nucleus (2R and 2r). To do so, use the DAPI channel image, choose the Line Selection Tool to draw a line from one border to the opposite border of the DAPI staining, and measure the length of the line (make sure the parameter "Length" is set in the "Set Measurements" dialog box, then press Analyze > Measure).
- Calculate the radii (r and R) of the nucleus.
- Open an image of Alexa488 or Texas Red-labeled vesicles.
- Determine the perinuclear region, as described and indicated in Figure 1.
Note: Use the composite image of DAPI and Alexa488 or Texas Red-labeled vesicle to determine the radius of the perinuclear region. - Once the perinuclear region sizes are established, measure fluorescence intensity inside this gated area, exclude the DAPI channel. Consider these values as Integrated Density (ID).
Note: Performed measurements can be specified in the "Set Measurements" dialog box (Analyze > Set measurements). Check "Area", "Mean Gray Value" and "Integrated Density” boxes. To create the statistics according to the defined parameters, press Analyze > Measure. - Measure fluorescence intensity of the whole cell image (ID).
- Measure fluorescence intensity in at least 4 areas of the background (use Area Selection Tools) and calculate the mean gray value (mean background) of these areas.
- Correct the Fluorescence Intensity (IDcor) of the Perinuclear region and of the whole image subtracting the mean gray value of the background (mean background) from the ID using the equation below:IDcor = ID – (area x mean background)
- After correcting the fluorescence intensity, determine the pericellular region using the equation below.Pericellular region = IDcorwhole image – IDcorperinuclear region
- If the image contains more than one cell, use the addition of all IDcorperinuclear region for the equation above. Then, normalize the result by the number of cells.
- Plot the values as preferred.
- When a particular effect in the distribution of the vesicles has been observed and quantified as explained above, a typical representation of the effect can be shown choosing a typical cell and obtaining the mean fluorescence intensity along a line drawn from the middle of the nuclei to cell periphery (Figure 2).
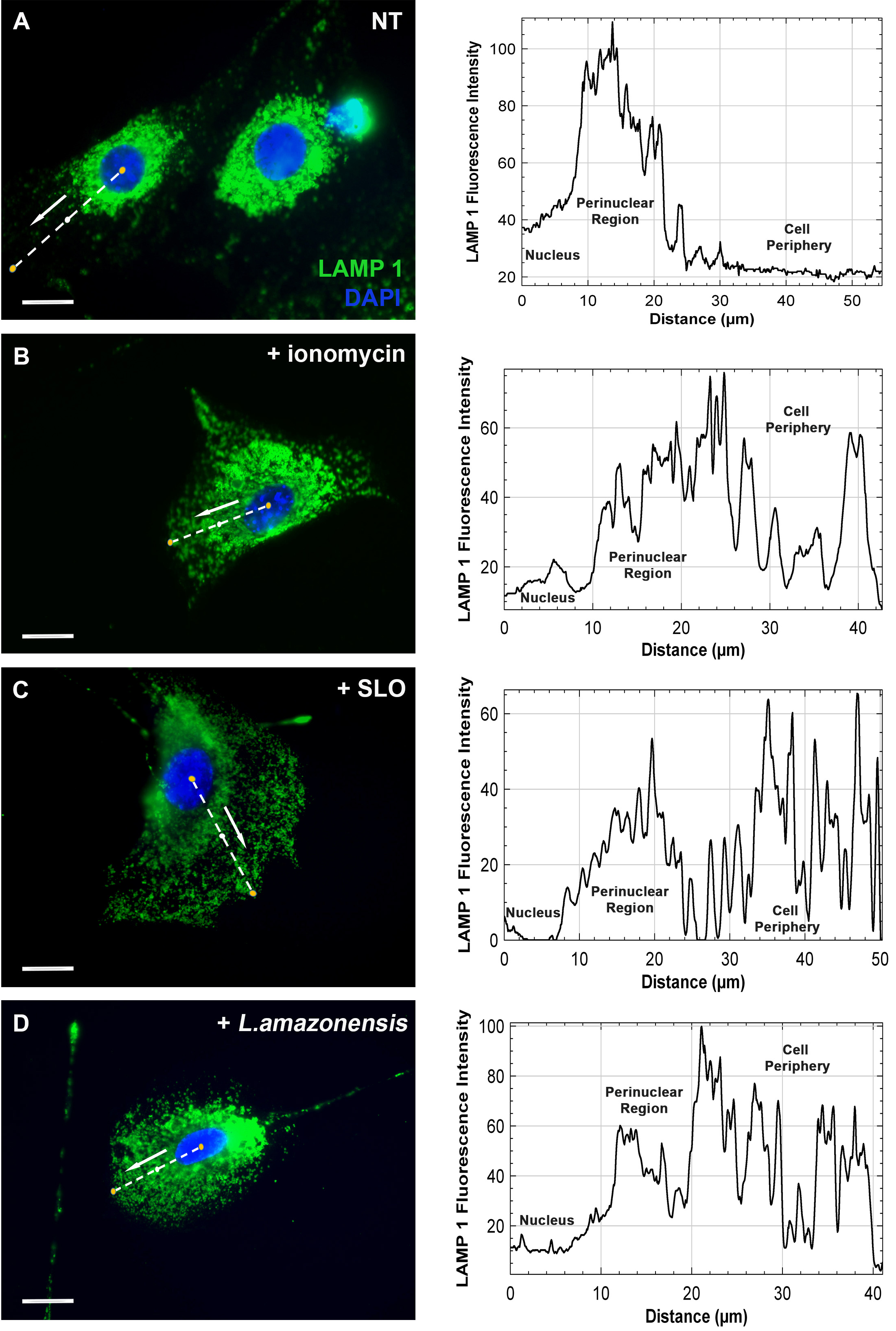
Figure 2. Lysosomes distribution in fibroblasts under different treatments. MEF cells were cultivated, treated as indicated in each image, fixed and labeled as described. Green, lysosomes (LAMP1), blue, nuclei (DAPI). A. Non-treated cells with its typical perinuclear cloud of lysosomes. Cells pre-treated with B 10 μM ionomycin or C 100 ng/ml streptolysin-O (SLO), to induce Ca2+ influx and lysosome spread, or D parasite Leishmania amazonensis (10 per MEF) which induces Ca2+-dependent lysosome spread towards the plasma membrane. For each indicated typical cell, a line was drawn from the middle of the nuclei to the edge of the cell and the fluorescence along this line is shown at the side of each image to visualize the re-distribution of the vesicles from the nuclear cloud located at the perinuclear region to cell periphery. Bars = 20 mm.
- Dispersion of labeled endosomal vesicles along the cell–calculating the number of vesicles from the perinuclear region to cell periphery
With this approach, it is possible to assess the distance between labeled vesicles and the center of the cell nuclei. Consequently, it generates an overview of the cell and the areas where labeled vesicles accumulate. This method is an interesting alternative to follow the occurrence of new endocytosis over time, for instance.- On ImageJ/FIJI, open image of the DAPI-labeled nuclei.
- Measure mean nucleus’ radius (R), or radii (r and R) when the shape of the nucleus is irregular, using the same approach described previously in steps A1-A2 of Data analysis.
- Determine approximate center of the nucleus.
- Open an image of the Alexa488 or Texas Red-labeled vesicles.
- Determine the regions of interest (ROIs) around the nucleus using the area selection tools (rings) with known mean distances arising from the approximate nucleus center (D), as shown in the figure (Figure 3).
Note: To hold multiple ROIs, and manipulate them separately, select the first ROI, press Analyze > Tools > ROI Manager and press “Add”. Select a second ROI and add to the ROI Manager. Repeat these steps to continue adding ROIs to the image.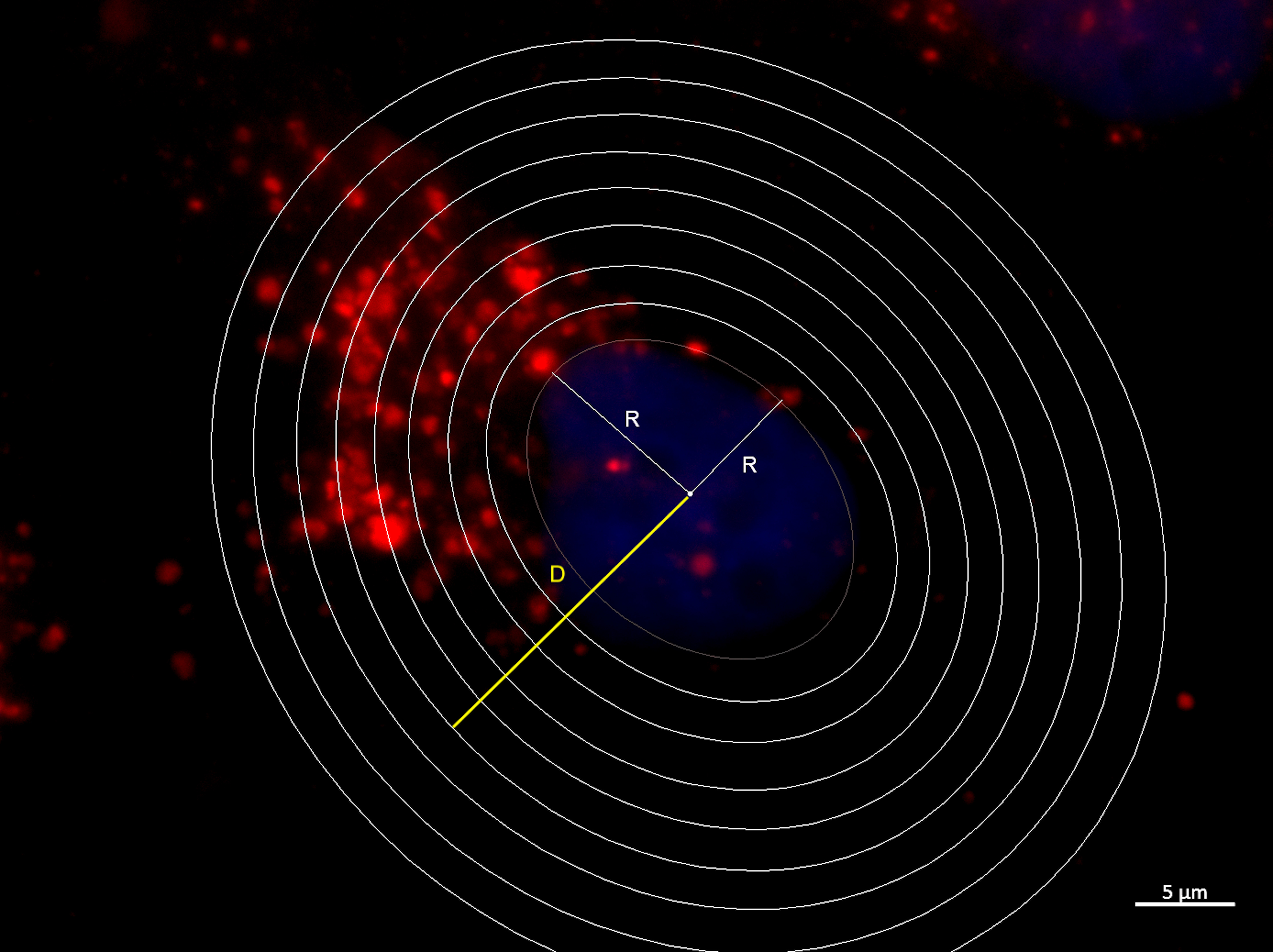
Figure 3. Definition of the areas around the nuclei used to quantify fluorescence-labeled vesicles distribution. Measurement of mean nucleus radius (R) and areas around the nuclei with a known distance (D) starting from the center of cell nucleus. The nucleus is stained with DAPI (blue) and endocytic vesicles are labeled with Texas-Red Dextran (red). - Determine the D/R ratio. Values close to one indicate vesicles located closer to the perinuclear area, whereas values higher than one indicate locations further from the cell center and nearer the cell border.
- Count the number of labeled vesicles (if countable) or measure the fluorescence intensity inside of each ring.
Note: Counting vesicles can be done manually or automatically by choosing the desired image and channel (type RGB or 8-bit), adjusting the threshold (Image > Adjust > Threshold) to define a better adjustment for a B&W image. Then press Process > Binary > Convert to mask (in case of touching particles, separate them by pressing Process > Binary > Watershed) and, finally, proceed to counting: Analyze > Analyze particles. In cases where you are measuring the fluorescence intensity, consider values of ID, correcting for background, as shown previously in items 5 to 8 of the alternative method described above. - Plot values as number of vesicles (% of total) or fluorescence intensity versus D/R, as shown in figure (Figure 4).
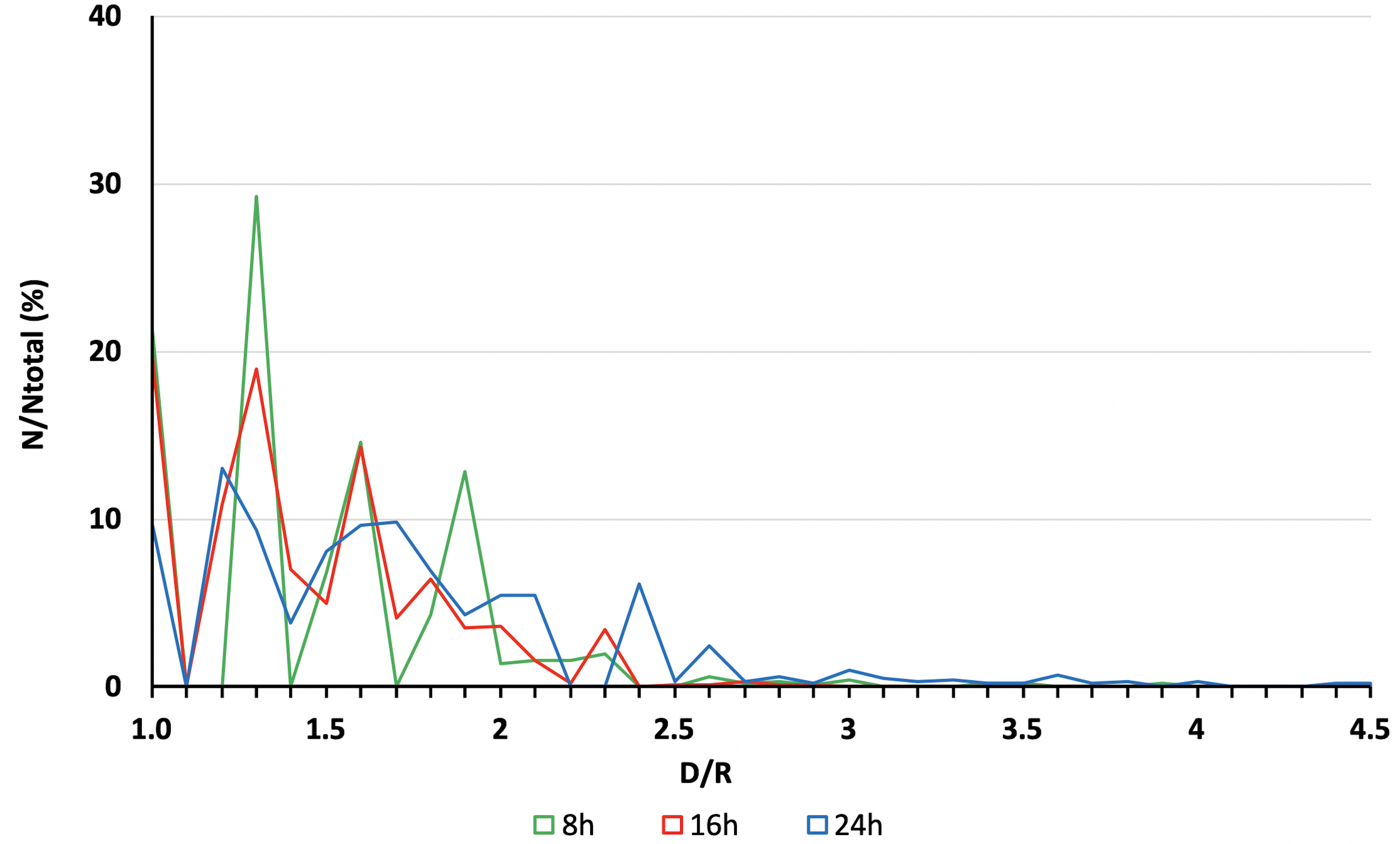
Figure 4. Example of plot. Quantitative analysis of Texas-Red Dextran labeled vesicle distribution, relative to cell nuclei, in endothelial cells (Eahy.926) over 24 h. The mean distance between a vesicle and its respective nuclei center is represented by the letter D and the mean vesicle distance relative to the mean nucleus’ radius (R) was defined as the ratio of D/R. Data are expressed as the percentage of vesicles from total (N/N total) located at a specific D/R.
Acknowledgments
We acknowledge Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Instituto Nacional de Ciência e Tecnologia de Fluidos Complexos (INCT/FCx) for supporting this work and Centro de Aquisição e Processamento de Imagens (CAPI / ICB) for the fluorescence equipments. We also acknowledge the previous work of Hissa et al. (2012 and 2013) from which this protocol was adapted.
Competing interests
The manuscript has no conflict of interest. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Aunis, D. and Bader, M. F. (1988). The cytoskeleton as a barrier to exocytosis in secretory cells. J Exp Biol 139: 253-266.
- Brito, C., Cabanes, D., Sarmento Mesquita, F. and Sousa, S. (2019). Mechanisms protecting host cells against bacterial pore-forming toxins. Cell Mol Life Sci 76(7): 1319-1339.
- Cabukusta, B. and Neefjes, J. (2018). Mechanisms of lysosomal positioning and movement. Wiley Traffic 19(10): 761-769.
- Castro-Gomes, T., Corrotte, M., Tam, C. and Andrews, N. W. (2016). Plasma Membrane Repair Is Regulated Extracellularly by Proteases Released from Lysosomes. PLoS ONE 11(3): e0152583.
- Cavalcante-Costa, V. S., Costa-Reginaldo, M., Queiroz-Oliveira, T., Oliveira, A. C. S., Couto, N. F., Dos Anjos, D. O., Lima-Santos, J., Andrade, L. O., Horta, M. F. and Castro-Gomes, T. (2019). Leishmania amazonensis hijacks host cell lysosomes involved in plasma membrane repair to induce invasion in fibroblasts. J Cell Sci 132(6).
- Hissa, B., Duarte, J. G., Kelles, L. F., Santos, F. P., del Puerto, H. L., Gazzinelli-Guimaraes, P. H., de Paula, A. M., Agero, U., Mesquita, O. N., Guatimosim, C., Chiari, E. and Andrade, L. O. (2012). Membrane cholesterol regulates lysosome-plasma membrane fusion events and modulates Trypanosoma cruzi invasion of host cells. PLoS Negl Trop Dis 6(3): e1583.
- Hissa, B., Pontes, B., Roma, P. M., Alves, A. P., Rocha, C. D., Valverde, T. M., Aguiar, P. H., Almeida, F. P., Guimaraes, A. J., Guatimosim, C., Silva, A. M., Fernandes, M. C., Andrews, N. W., Viana, N. B., Mesquita, O. N., Agero, U. and Andrade, L. O. (2013). Membrane cholesterol removal changes mechanical properties of cells and induces secretion of a specific pool of lysosomes. PLoS One 8(12): e82988.
- Horta M. F. , Andrade L. O., Martins-Duarte É. S., Castro-Gomes T. (2020). Cell invasion by intracellular parasites - the many roads to infection. J Cell Sci 133(4): jcs232488.
- Idone, V., Tam, C., Goss, J. W., Toomre, D., Pypaert, M. and Andrews, N. W. (2008). Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol 180(5): 905-914.
- Koseoglu, S., Love, S. A. and Haynes, C. L. (2011). Cholesterol effects on vesicle pools in chromaffin cells revealed by carbon-fiber microelectrode amperometry. Anal Bioanal Chem 400(9): 2963-2971.
- Lawrence, R. E. and Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21(2): 133-142.
- Nabavi, N., Urukova, Y., Cardelli, M., Aubin, J. E. and Harrison, R. E. (2008). Lysosome dispersion in osteoblasts accommodates enhanced collagen production during differentiation. J Biol Chem 283(28): 19678-19690.
- Rodriguez, A., Webster, P., Ortego, J., Andrews, N (1997). Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. The Journal of Cell Biology 137 (1): 93-104.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
- Tam, C., Idone, V., Devlin, C., Fernandes, M. C., Flannery, A., He, X., Schuchman, E., Tabas, I. and Andrews, N. W. (2010). Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol 189(6): 1027-1038.
- Tardieux, I., Webster, P., Ravesloot, J., Boron, W., Lunn, J. A., Heuser, J. E. and Andrews, N. W. (1992). Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71(7): 1117-1130.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
do Couto, N. F., Queiroz-Oliveira, T., Horta, M. F., Castro-Gomes, T. and Andrade, L. O. (2020). Measuring Intracellular Vesicle Density and Dispersion Using Fluorescence Microscopy and ImageJ/FIJI. Bio-protocol 10(15): e3703. DOI: 10.21769/BioProtoc.3703.
Category
Cell Biology > Organelle isolation > Extracellular vesicle
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.