- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation and in Planta Functional Analysis of Potato Virus Y mutants
Published: Vol 10, Iss 14, Jul 20, 2020 DOI: 10.21769/BioProtoc.3692 Views: 4814
Reviewed by: Kristin L. ShinglerMatevz Papp-RuparPooja SaxenaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Potato virus Y (PVY), the type member of the genus Potyvirus (family Potyviridae), is the most widespread virus affecting potato and is included in the top five most economically detrimental plant viruses. Recently, the structure of the PVY virion has been determined by cryo-electron microscopy, which has opened the doors to functional studies that explore the involvement of selected amino acids in different stages of the viral cycle. The only way to functionally challenge in planta the role of particular amino acids in the coat protein of PVY, or in other viral proteins, is by using cDNA clones. The use and manipulation of PVY cDNA clones, unlike those of other potyviruses, has been traditionally impaired by the toxicity that certain sequences within the PVY genome pose to Escherichia coli. Here, we describe the use of a published PVY cDNA clone, which harbours introns that overcome the aforementioned toxicity, to explore the effects of different coat protein modifications on viral infection. The protocol includes manipulation of the cDNA clone in E. coli, biolistic inoculation of plants with the constructed clones, observation of the biological effects on plants, quantification of cDNA clones by reverse transcription quantitative PCR, and confirmation of virion formation by transmission electron microscopy. Future possibilities involve the use of PVY cDNA clones tagged with fluorescent protein reporters to allow further insights into the effects of coat protein mutations on the cell-to-cell movement of PVY virions.
Keywords: Potato virus YBackground
Potato virus Y (PVY) is the most economically important virus that infects cultivated potato and causes major crop losses worldwide (Valkonen, 2007). Besides potato, PVY can infect a broad range of other plant species, including mainly other cultivated Solanacea species such as tomato, pepper, or tobacco, as well as weeds and ornamentals (Kerlan, 2006). PVY is naturally transmitted by aphids in a non-persistent manner. Up to 65 aphid species have been reported to be potential vectors for PVY, which makes it difficult to implement efficient viral control and management measures (Lacomme and Jacquot, 2017). Typical for the Potyviridae family, the PVY virion is structured as a flexuous filamentous particle that encapsidates a 9.7 kb positive-sense single-stranded RNA genome. It encodes 10 proteins synthesised from a single open reading frame and one protein, P3N-PIPO, synthesised from an overlapping reading frame (Chung et al., 2008).
Understanding the complex interaction network established between PVY and its host plant is indispensable for designing more efficient and long-lasting virus control measures, such as targeted crop-breeding strategies (Lacomme and Jacquot, 2017). During the last decade, technological advancements have improved our knowledge of potato-PVY interactions (Križnik et al., 2017; Lukan et al., 2018; Stare et al., 2019) and viral mechanisms of the infection cycle (Mäkinen and Hafrén, 2014; Mäkinen et al., 2017). The potential of such technical developments has been demonstrated in our recent study (Kežar et al., 2019). By combining knowledge of the PVY virion structure (based on high-resolution cryo-electron microscopy) with in planta coat protein (CP) functional analysis (using a PVY cDNA clone), we provided insights into the crucial role of CP and its N- and C- terminal regions in PVY infectivity.
The study reveals the value of an easy-to-manipulate PVY cDNA infectious clone for in planta functional analysis of the virus. The production and use of PVY infectious clones has been more challenging than that for other potyviruses due to the expression of cryptic prokaryotic promoters in the PVY genome, which results in products that are cytotoxic to E. coli (Fakhfakh et al., 1996; Jakab et al., 1997). Thus, only a few PVY cDNA clones have been used to date to investigate the viral determinants involved in the vein necrosis symptom (Tribodet et al., 2005), the rate of PVY cell-to-cell movement via inclusion of a GFP reporter gene into the polyprotein (Rupar et al., 2015), and the role of temperature in Rysto-mediated extreme resistance (Grech-Baran et al., 2020).
However, the use of PVY infectious clones as a reverse genetics tool for in planta functional analysis of PVY is still scarce because of the low inoculation yields and complex manipulation of some PVY clones. Therefore, we here describe a protocol to functionally characterise the role of the viral CP by using the PVY-N605(123) cDNA clone (Bukovinszki et al., 2007), a full-length infectious clone that was previously developed to avoid the cytotoxic effects to E. coli through intron insertion. We present a detailed description of the cloning strategy to introduce desired CP deletions into the PVY-N605(123) cDNA clone followed by gene gun bombardment of Nicotiana clevelandii plants. The protocol includes characterisation of the resulting virus amplification and spread using reverse transcription quantitative PCR to quantify viral genome and transmission electron microscopy to confirm virion formation. We recently used this protocol to study the role of viral CP N- and C- terminal regions in virus infectivity (Kežar et al., 2019). The same procedure, with an appropriate modification of the mutagenesis strategy, can also be used to introduce any mutation into PVY cDNA or alternatively to insert fluorescent protein into cDNA and tag specific PVY proteins for purification or imaging purposes. Therefore, the protocol described here offers an indispensable tool for in planta analysis of the viral cycle and functional studies of viral proteins.
Materials and Reagents
- 384-well plates (Thermo Fisher Scientific, Applied Biosystems, catalog number: 4309849 )
- 15 ml polypropylene centrifuge tubes (Corning, catalog number: 430791 )
- Filter paper for laboratory use (Sartorius)
- 1.5 ml microfuge tube (Brand, catalog number: 780502 )
- 10 ml syringe (BD, catalog number: 309110 )
- 0.22 μm disposable filter (Sartorius, catalog number: 16532-Q )
- N. clevelandii plants grown from seeds at 24 °C and 60%-70% humidity
Note: Seeds were from an in-house collection. - PVY cDNA clone PVY-N605(123) (Bukovinszki et al., 2007)
Note: The plasmid can be provided by the National Institute of Biology, Slovenia, upon request. - QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, catalog number: 200522 )
- Bacto tryptone (BD, catalog number: 211705 )
- Bacto yeast extract (BD, catalog number: 212750 )
- Bacto agar (BD, catalog number: 214010 )
- NaCl (Merck, Supelco, catalog number: 1.06404.1000 )
- KCl (Merck, Supelco, catalog number: 1.04936.0500 )
- MgCl2 (Merck, Sigma, catalog number: M8266-100G )
- Glucose, anhydrous (Kemika, catalog number: 0 705007 )
- Ampicillin (Merck, Sigma-Aldrich, catalog number: A9393-5G )
- KAPA2G Robust HotStart Polymerase with dNTP (Kapa Biosystems, catalog number: KK5516 )
- GenEluteTM Plasmid Miniprep Kit (Merck, Sigma-Aldrich, catalog number: PLN350-1KT )
- Monarch Plasmid Miniprep Kit (NEB, catalog number: T1010 )
- Agarose (Merck, Sigma-Aldrich, catalog number: A9539 )
- Ethidium bromide solution 10 mg/ml (Merck, Sigma-Aldrich, catalog number: E1510-10ML )
- Trizma base (Tris base) (Merck, Sigma-Aldrich, catalog number: T1503 )
- Acetic acid (glacial) 100% (Merck, Supelco, catalog number: 1.00063 )
- EDTA (disodium salt, dihydrate) (Merck, Sigma-Aldrich, catalog number: E5134 )
- Fresh 100% ethanol (Ethanol for spectroscopy Uvasol®, Merck, Sigma-Aldrich, Supelco, catalog number: 1009800500 )
- 0.05 M spermidine (Merck, Sigma-Aldrich, catalog number: S0266-1G )
- 1 M CaCl2 (Merck, Sigma-Aldrich, catalog number: C1016-100G )
- MagMAXTM-96 Total RNA Isolation Kit (Invitrogen, catalog number: AM1830 )
- Plant RNA Isolation Aid (Invitrogen, catalog number: AM9690 )
- Lysing Matrix A: tubes containing garnet and one 1/4" ceramic sphere (MP Biomedicals, catalog number: 116910050-CF )
- OneStep RT-PCR Kit (Qiagen, catalog number: 210210 )
- AgPath-ID One-Step RT-PCR Reagents (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: AM1005 )
- NaH2PO4·2H2O (Merck, SAFC, catalog number: 1.06345.1000 )
- Na2HPO4 (Merck, Supelco, catalog number: 1.06586.0500 )
- 1% (w/v) uranyl acetate in ddH2O (SPI Supplies, catalog number: 6159-44-0 )
- S.O.C. media (see Recipes)
- LB medium liquid (low salt) (see Recipes)
- Antibiotic (see Recipes)
- 0.1 M phosphate buffer (see Recipes)
- 50x TAE buffer (see Recipes)
- 0.5 M EDTA (see Recipes)
Equipment
- Tweezers
- Pipettes
- T100TM Thermal Cycler (Bio-Rad, catalog number: 1861096 )
- Water bath WB-13 (Kambič, catalog number: 1201 )
- Incubator, orbital shaker SI500 (Stuart)
- Agarose gel electrophoresis (Bio-Rad)
- UVP ChemStudio PLUS (Analytik Jena, catalog number: 849-97-0847-04 )
- NanoDropTM 1000 Spectrophotometer (Thermo Fisher Scientific, out of production)
- Sanger sequencing (Eurofins Scientific)
- Helios Gene Gun System (Bio-Rad, catalog number: 165-2432 ), includes:
- Helium hose assembly with regulator and a tubing prep station (Bio-Rad, catalog number: 165-2420 )
- Syringe kit, Tefzel tubing, tubing cutter, optimisation kit (Bio-Rad, catalog number: 165-2424 , includes 0.6 μm, 1.0 μm, and 1.6 μm gold microcarriers, PVP (Polyvinylpyrrolidone), desiccant, five cartridge storage vials, 50 feet of Tefzel gold-coat tubing, for 1,000 samples)
- Helium tank (Messer, grade 6.0)
- Nitrogen tank (Messer, grade 5.0)
- Nitrogen regulator (e.g., Bio-Rad, catalog number: 165-2425 )
- Ultrasonic cleaning bath Sonis 3 GT (Iskra Pio)
- FastPrep-24TM Classic Instrument (MP Biomedicals, catalog number: 116004500 )
- KingFisher Instrument (Thermo Scientific, out of production)
- QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4485701 )
- Automated liquid handling system (Microlab STARlet, Hamilton)
- Transmission electron microscope (Philips, CM 100) equipped with couple-charged device camera ORIUS SC 200 (Gatan Inc., Pleasanton, USA)
Software
- SnapGene (https://www.snapgene.com/snapgene-viewer/)
- QuantGenius software (Baebler et al., 2017, http://quantgenius.nib.si/user/login)
- Digital Micrograph, version 2.11.1404.0 (Gatan Inc., Pleasanton, USA)
Procedure
- Gene cloning to prepare the mutated PVY cDNA clone
In Procedure A, the mutation is introduced into the desired location of the PVY plasmid, followed by transformation of the mutant plasmid into E. coli, and confirmation of the introduced mutation by colony PCR. In the end, mutated and WT plasmids are multiplied and purified before they are introduced into the plants.- Introduce mutation into the plasmid (PVY cDNA clone PVY-N605(123), Bukovinszki et al., 2007) by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent technologies).
- Design primers for mutation (e.g., deletion).
- The desired mutation should be in the middle of the primer.
- Minimum GC content should be 40%.
- Terminate in one or more C or G bases.
- The primer should be 40-50 bases long.
- The melting temperature (Tm) of the primer is ≥ 78 °C (Tm calculation: 81.5 + 0.41(%GC) – (675/N); N is the primer length in bases and does not include the bases that are being inserted or deleted.
- Purify with HPLC.
- An example of designing a forward primer to introduce a deletion of 60 amino acid residues at the C terminus of PVY CP is provided (Figure 1):
The forward primer (excluding the nucleotides to be deleted) is annealed to the parental plasmid, forming a hairpin in the parental plasmid’s region to be deleted. This results in an amplified fragment that includes the desired deletion after PCR.
Figure 1. An example of designing a forward primer using SnapGene viewer - Set up the PCR reaction with the reagents specified in Table 1 and the cycling program shown below.
Table 1. Reaction components for mutagenic PCR
Thermal cycler conditions92 °C 2 min 92 °C 50 s 65 °C to 55 °C 50 s, touch down 10 cycles 68 °C 2 min/kb 92 °C 50 s 55 °C 50 s 8 cycles 68 °C 2 min/kb 68 °C 60 min
- Design primers for mutation (e.g., deletion).
- Digest unmodified plasmid with DpnI enzyme from the QuikChange II XL Site-Directed Mutagenesis Kit. Use 4 µl of enzyme per 25 µl of reaction, incubate 2 h at 37 °C.
- Transform E. coli XL10-Gold ultracompetent cells (included in the QuikChange II XL Site-Directed Mutagenesis Kit):
- Gently thaw the XL10-Gold ultracompetent cells on ice. Aliquot 45 μl of the ultracompetent cells to a 2 ml tube for each sample reaction.
- Add 2 μl of the β-ME mix (provided with the kit) to the 45 μl of cells.
- Mix gently. Incubate the cells on ice for 10 min, mixing gently every 2 min.
- Transfer 2 μl of DpnI-treated DNA to each aliquot of the ultracompetent cells.
- Gently mix the transformation reactions and incubate on ice for 30 min.
- Heat shock tubes in a 42 °C water bath for 30 s. The duration of the heat shock is critical for obtaining the highest efficiencies.
- Incubate the tubes on ice for 2 min.
- Add 0.5 ml of preheated (42 °C) S.O.C. media to each tube.
- Incubate at 37 °C for 1 h with shaking at 225-250 rpm.
- Plate the entire volume of each transformation reaction (250 µl x 2 plates) on LB agar plates containing the appropriate antibiotic for the plasmid (e.g., ampicillin).
- Incubate plates overnight at 37 °C.
- Set up Colony PCR using the KAPA2G Robust HotStart PCR Kit (Kapa Biosystems).
- Resuspend five transformed colonies in 50 µl sterile ddH2O.
- Use 1 µl of colony suspension to prepare a colony PCR.
Note: Store the remaining colony suspension at 4 °C; it will be used to prepare overnight cultures of positive colonies. - Set up the PCR reaction with the reagents specified in Table 2 and the cycling program shown below.
Note: Amplify also non-mutated plasmid (10 ng/µl) as a control.
Table 2. Reaction components for colony PCR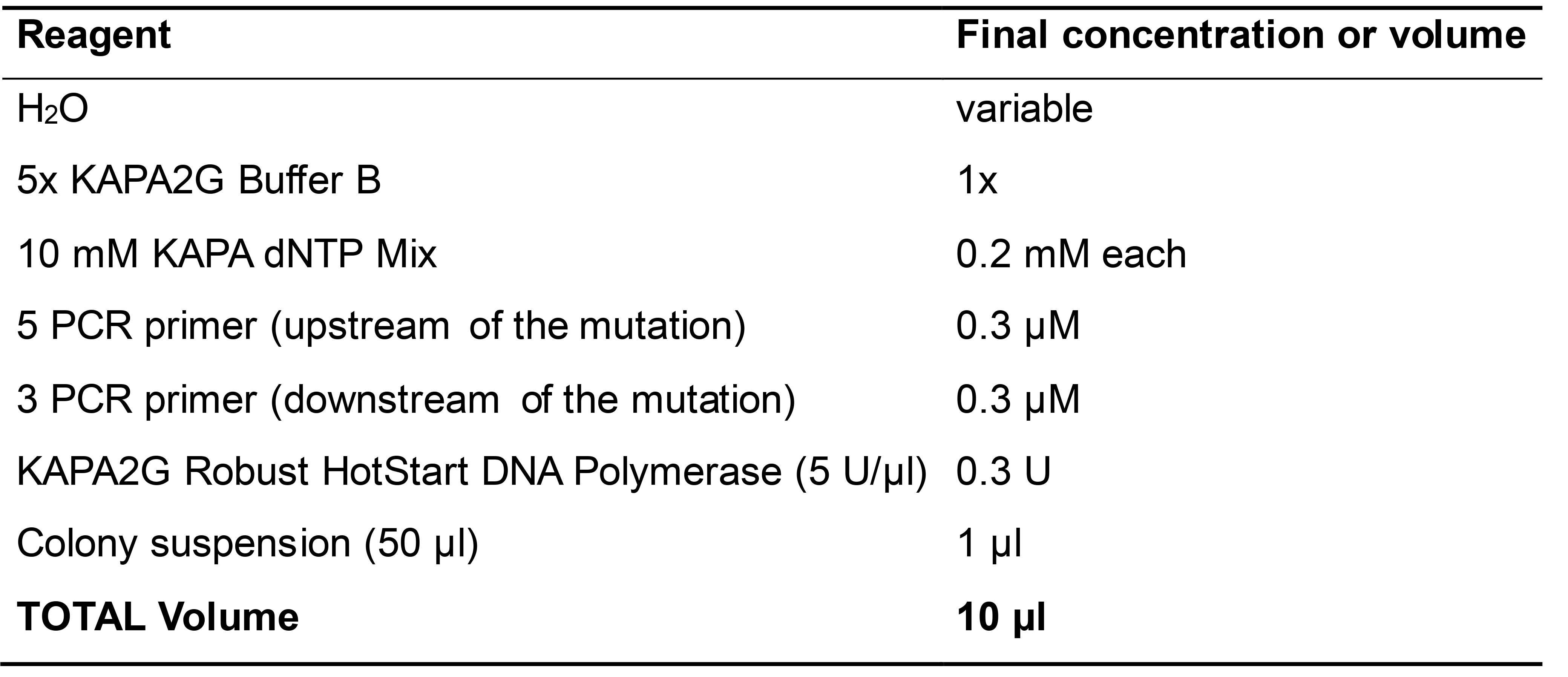
Thermal cycler conditions95 °C 5 min 95 °C 30 s Tm °C 15 s 30 cycles 72 °C 1 min/Kb 72 °C 5 min - Load 1 µl of each PCR product (also non-mutated plasmid) onto 1% agarose gel, made in 1x TAE buffer with ethidium bromide (final concentration 0.5 µg/ml).
- For colonies with mutations (observed as a shift on the gel, see Figure 2), add 5 µl of colony suspension to 5 ml of LB liquid media with antibiotic (e.g., ampicillin).
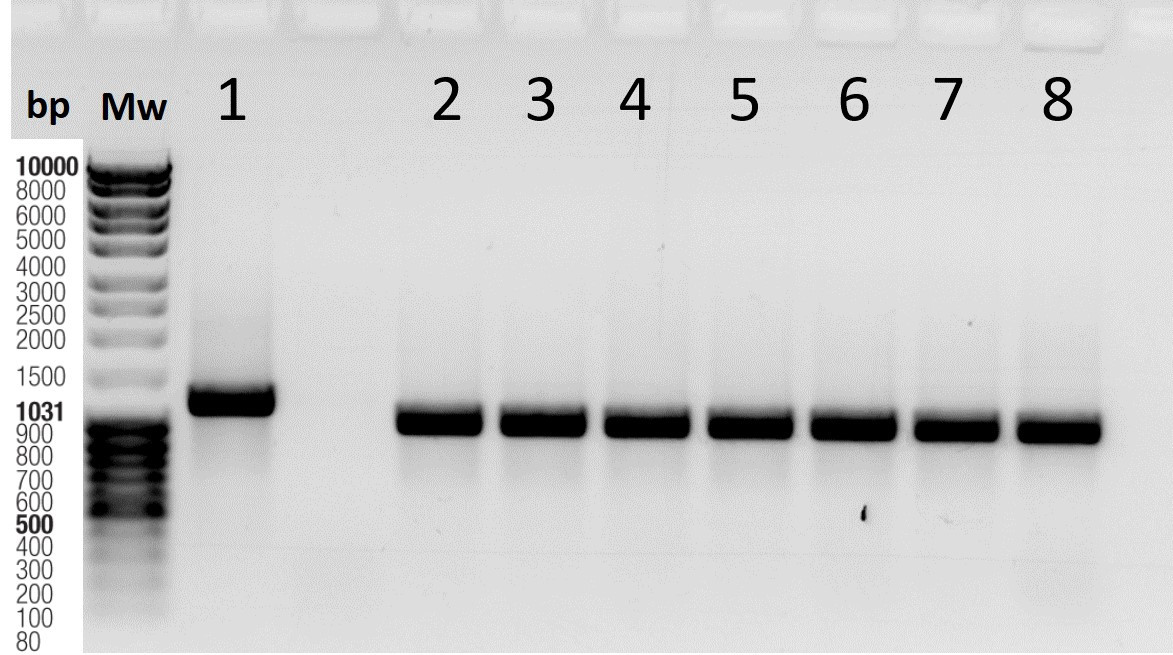
Figure 2. Agarose gel showing colonies with deletions. 1: non-mutated plasmid; 2-8: colonies with deletions; Mw: molecular weight; bp: base pairs. - Incubate for 16-18 h at 37 °C with shaking at 200–250 rpm.
- Isolate plasmids using the GenEluteTM Plasmid Miniprep Kit (Sigma) following manufacturer’s instructions with a few modifications:
- Centrifuge 5 ml of overnight culture in a 15 ml Falcon tube for 5 min at 4,500 rpm at room temperature.
- Elution step:
Add 30 µl of nuclease-free water to the spin column. Incubate for 10 min at room temperature. For plasmids ≥ 10 kb or ≥ 20 kb, use water preheated to 70 °C or 80 °C, respectively. Centrifuge at top speed for 2 min at room temperature.
- Measure plasmid concentration with the NanoDropTM 1000 Spectrophotometer. The expected yield range is 1.5-4.5 µg, and the expected purity ranges for the indicator ratios A260/A230 and A260/A280 are 1.8-2.4 and 1.8-2.1, respectively.
- Send samples for Sanger sequencing to confirm the right sequence (company: Eurofins Scientific). Use a 5' PCR primer upstream of the mutation site and a 3' PCR primer downstream of the mutation site. The obtained sequence should be 100% identical at the amino acid level to the expected sequence including the planned modification. Silent mutations are acceptable.
- After confirming the sequence, prepare 50 µg of each plasmid by using the same protocol and combining 5-10 mini-, midi-, or maxi-preps.
Note: For plasmid isolation, the Monarch Plasmid Miniprep Kit (NEB, see Quick Protocol Card) can also be used.
- Introduce mutation into the plasmid (PVY cDNA clone PVY-N605(123), Bukovinszki et al., 2007) by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent technologies).
- Biolistic bombardment using the Helios Gene Gun system (Bio-Rad) to infect plants with the PVY cDNA clones (WT and mutated)
Here, the gold nanoparticles that help transfer the plasmids into the plants are prepared. First, the plasmids are bound to the gold nanoparticles, and then the Gene Gun cartridges are loaded with the plasmid-bound nanogold particles. The cartridges are inserted into the Gene Gun, and the plasmid-bound gold nanoparticles are shot into the plant leaf tissue.- Precipitation of plasmid DNA onto microcarriers.
- Prepare a stock solution of 20 mg/ml PVP in 100% ethanol. Dilute this solution with 100% ethanol to prepare 3 ml of PVP solution with a concentration of 0.05 mg/ml. Keep these solutions tightly capped when not in use. Only use freshly prepared dilutions.
- Weigh gold microcarriers in a 1.5 ml microfuge tube (size of gold particles: 0.6 µm, use 6.25 mg/plasmid).
- Add 0.05 M spermidine to the 6.25 mg of gold in the tube. The volume of 0.05 M spermidine should be equal to the volume of 50 µg of plasmid.
- Vortex the gold and spermidine mixture for a few seconds, and then sonicate with an ultrasonic cleaning bath for 3-5 s to break up gold clumps.
- Add 50 µg of plasmid to the gold and spermidine mixture.
- Mix DNA, spermidine, and gold by vortexing for ~5 s.
- While vortexing the mixture at a moderate rate, add 1 M CaCl2 dropwise to the mixture. The volume added should be equal to the volume of plasmid. The maximal volume of the entire mixture (gold, spermidine, plasmid, and CaCl2) should not exceed 1,200 µl.
- Allow the mixture to precipitate at room temperature for 10 min.
- Most of the gold will now be in the pellet, but some may remain on the sides of the tube. Spin (10,000 rpm) the microcarrier solution in a microfuge for ~30 s to pellet the gold. Discard the supernatant.
- Resuspend the pellet in the remaining supernatant by vortexing briefly. Wash the pellet three times with 1 ml of fresh 100% ethanol each time, sonicate if you see aggregates of gold particles (only for a short time to avoid DNA damage); spin for ~30 s in a microfuge between each wash. Discard the supernatant.
- After the final ethanol wash, resuspend the pellet in 200 µl of ethanol solution containing 0.05 mg/ml of PVP (prepared in Step B1a). Transfer the suspension to a 15 ml disposable polypropylene centrifuge tube with a screw cap. Rinse the microfuge tube with 200 µl of the same ethanol/PVP solution and add to the 15 ml centrifuge tube. Rinse again until all gold is washed from the tube. Add up to 3 ml of ethanol/PVP solution to the 15 ml centrifuge tube.
- Preparation of cartridges
- Load the DNA/microcarrier suspension into the Gold-Coat tubing using the Tubing Prep Station.
- Prior to cartridge preparation, ensure that the Gold-Coat tubing is completely dry by purging with nitrogen. Insert an uncut piece of tubing into the Tubing Prep Station. Connect the Tubing Prep Station to a nitrogen gas tank. Using the knob on the flowmeter of the Tubing Prep Station, turn on the nitrogen and adjust the flow to 0.3-0.4 LPM (liters per minute). Allow nitrogen to flow through the Gold-Coat tubing for at least 15 min immediately prior to using it in the following steps.
- Remove the Gold-Coat tubing from the Tubing Prep Station.
- Turn off the nitrogen flow to the Tubing Prep Station using the knob on the flowmeter.
- Cut a piece of dried Gold-Coat tubing with a length of the Tubing Station plus 10 cm. Insert one end of the Gold-Coat tubing into the end of the adaptor tubing fitted to the 10 ml syringe.
- Vortex the microcarrier suspension and, if necessary, briefly sonicate to achieve an even suspension of gold. Invert the tube several times to resuspend the gold. Immediately remove the cap and quickly draw the gold suspension into the Gold-Coat tubing to cover the tube. Avoid drawing bubbles into the Gold-Coat tubing. Remove the tubing from the suspension and continue drawing the suspension into the tubing for another ~3 cm to leave some space at each end. Cut the part of the tube that was soaked in the liquid to avoid contamination of Tubing Prep Station.
- Immediately bring the Gold-Coat tubing to a horizontal position and slide the loaded tube, with syringe still attached, into the tubing support cylinder in the Tubing Prep Station until the tubing passes through the O-ring.
- Allow the microcarriers to settle for 5 min.
- Remove the ethanol by slowly drawing the syringe plunger outwards at a rate of 1-2 cm/s.
- Detach the syringe from the Gold-Coat tubing and attach a disposable filter (0.2 µm) at the end of the tube. Immediately turn the Gold-Coat tubing by 180° while in the groove and allow the gold to begin coating the inside surface of the tubing for 3-4 s.
- Start rotating the Tubing Prep Station. Allow the gold to spread within the tube for 20-30 s and then open the valve on the flowmeter to allow 0.35-0.4 LPM of nitrogen to dry the Gold-Coat tubing, while it continues to rotate.
- Continue drying the Gold-Coat tubing while rotating for 5 min.
- Turn off the motor of the Tubing Prep Station. Turn off the nitrogen by closing the valve on the flowmeter.
- Remove the tubing from the tubing support cylinder.
- Prepare cartridges using the tubing cutter. Store the cartridges in a cartridge storage vial containing a desiccant pellet.
- Load the DNA/microcarrier suspension into the Gold-Coat tubing using the Tubing Prep Station.
- Bombardment with the Gene Gun.
- Experimental set-up:
- Use N. clevelandii plants at the 6 to 10 leaf stage.
- Bombard seven plants (five leaves per plant) for one construct.
- Use a yellow fluorescent protein-expressing plasmid or some other plasmid that lacks any PVY-related sequence as a negative bombardment control (to check for any background signal due to PVY carry over during the bombardment procedure).
- The WT virus should always be used last to bombard the leaves (to avoid contamination).
- Bombardment:
Notes:
1)Use a disposable coat and glasses and work in a chamber with UV light.
2)After bombardment and before proceeding with the next plasmids, carefully clean with DNA remover and turn on the UV light in the chamber.- Prepare the Gene Gun: insert the battery, tighten the Barrel Liner on the gun, and insert the Barrel O-ring in the Barrel Liner.
- Connect the gun to the Helium tank (open the first valve fully and set the second valve to a pressure of 200 psi).
Note: Check after the first bombardment that the pressure is still 200 psi. - Fill cartridge holder with cartridges (use tweezers).
- Insert the cartridge holder into the gun (open all locks), set number 12 on the top, and press the Cylinder Advance lever (see Figure 3 for clarification).
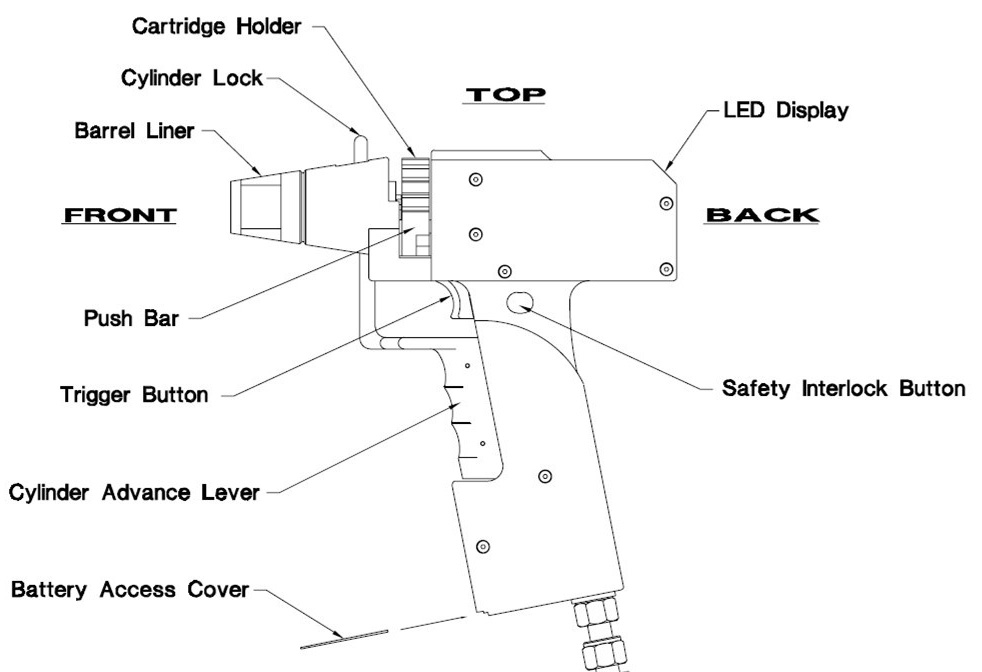
Figure 3. Components and controls on the Helios Gene Gun (manual for Helios Gene Gun System) - Touch the leaf with the gun (with the Barrel Liner), hold the Safety Interlock Button, and press the Trigger button to bombard the leaf (see Figure 4). Press the Cylinder Advance lever to switch to a new cartridge.
- Repeat the procedure on five leaves per plant.

Figure 4. Bombardment of the plant leaf
- Collect the material in Fast-Prep tubes.
- At 7 days post-bombardment, separately collect three bombarded leaves per each plant and immediately freeze them in liquid N2.
- Using the same approach, sample systemic infected leaves at 14 days post-bombardment.
- Experimental set-up:
- Precipitation of plasmid DNA onto microcarriers.
- PCR analysis (following amplification of the virus in plants)
Here, the total RNA is purified from plant leaves bombarded with mutant PVY clones, and the presence of introduced mutations in the viral progeny is confirmed by classic RT-PCR and agarose gel electrophoresis. Then, the viral load in plants bombarded with WT and mutated virus clones is relatively quantified using a PVY universal RT-qPCR assay.- RNA isolation
- Use the MagMAX-96 Total RNA Isolation kit (Thermo Fisher Scientific). The RNA can be isolated manually using a magnetic stand and orbital shaker for 96-well plates or automatically using the KingFisher instrument (Thermo Fisher Scientific).
- Follow manufacturer’s instructions with modifications regarding homogenate preparation: Add 450 µl of Lysis Binding solution concentrate and 50 µl of Plant RNA Isolation Aid to each frozen leaf and immediately start homogenisation with the FastPrep instrument (6 m/s, 60 s). After complete homogenisation, centrifuge samples at 14,000 x g for 10 min. Discard the pellet. Transfer 50 μl of homogenised sample to the wells of the processing plate and follow the manufacturer’s protocol.
- Include a negative control for the extraction procedure.
- The optimal RNA yield range is 1-9 µg, and the optimal purity ranges for the indicator ratios A260/280 and A260/230 are 1.7-2.2 and 1.0-2.1, respectively. In our experience, values that are not within these ranges also yield optimal qPCR results.
- Set up classic one-step RT-PCR to confirm that the introduced mutation is maintained in the viral progeny in planta using the Qiagen OneStep RT-PCR kit. Use reagents specified in Table 3 and the cycling program shown below. Load the PCR product on 1% agarose gel made in 1x TAE buffer with ethidium bromide (final concentration 0.5 µg/ml). Inspect the shift on the agarose gel to confirm the mutation.
Table 3. Reaction components for One-Step RT-PCR
Thermal cycler conditions50 °C 30 min 95 °C 15 min 94 °C 30 s Tm °C 30 s 35 cycles 72 °C 1 min/Kb 72 °C 10 min - Set up the qPCR reaction (single-step quantitative RT-PCR) using the AgPath-IDTM One-Step RT-PCR kit (Thermo Fisher Scientific) for virus detection.
- Quantify the PVY-N605 virus.
- Use the amplicon targeting the CP to detect the PVY clone (PVY) and cytochrome oxidase transcript (COX) as the endogenous control (see primers and probes in Table 4).
Note: Ensure that the amplicon for the PVY clone is not at the position of the introduced mutation (e.g., deletion). This amplicon quantifies WT virus and mutants and enables their comparison.
Table 4. Primers and probes for the COX and PVY amplicons
- Set up a One-Step quantitative RT-PCR with the reagents shown in Table 5 and cycling conditions below.
Table 5. Reaction components for One-Step quantitative RT-PCR
Thermal cycler conditions48 °C 10 min 95 °C 10 min 95 °C 15 s 40 cycles 60 °C 1 min
- Use the amplicon targeting the CP to detect the PVY clone (PVY) and cytochrome oxidase transcript (COX) as the endogenous control (see primers and probes in Table 4).
- Prepare a standard curve: combine 2 µl of each sample. Dilute 5x, 25x, 125x, and 625x.
- Include a non-template control.
- If an automated liquid handling station will be used to pipet onto 384-well plates, two technical replicates are sufficient, otherwise, three are recommended.
- RT-qPCR can detect both viral RNA and residual plasmid DNA. To quantify only viral RNA, the contribution of the plasmid-derived signal to the concentration of PVY RNA must be eliminated. To detect any plasmid-derived signals, a qPCR reaction without RT is run for each sample. For this, denature the RT enzyme from the AgPath-ID One-Step RT-PCR kit by heating the 25x RT-PCR Enzyme Mix at 95 °C for 10 min before the qPCR reaction. To calculate the quantity of viral RNA, the plasmid-derived signal is subtracted from the overall signal (viral RNA + plasmid DNA) (see Data analysis, step 2).
- Calculate relative copy numbers of PVY RNA using the quantGenius software (http://quantgenius.nib.si/user/login, Baebler et al., 2017), see Data analysis.
- Quantify the PVY-N605 virus.
- RNA isolation
- Sample observation with transmission electron microscopy and image acquisition
Here, extracts from plant leaves bombarded with WT and mutated virus clones are observed by electron microscopy to visualise virion formation.- Cut 3-4 pieces (of 1-2 mm2) of the lower leaves of N. clevelandii 7 days post-bombardment, place them into Eppendorf tubes, and macerate them with 300 µl of 0.1 M phosphate buffer.
- Place a formvar- and carbon-coated grid on a drop of sample for 5 min.
- Soak away the sample by carefully touching the edge of the grid with filter paper and wash the grid with 5-10 drops of deionised water.
- Stain the sample with one drop of 1% (w/v) uranyl acetate.
- Immediately remove excess uranyl acetate by touching the edge of the grid with filter paper.
- Dry the sample at room temperature.
- Observe the grids with a transmission electron microscope and acquire the images with a couple-charged device camera (Figure 5).
- Use Digital Micrograph software to process the images.

Figure 5. Negative-stain transmission electron microscopy of clone PVY-N605(123) in bombarded leaves. PVY-N605(123) flexuous virions are indicated by arrows.
Data analysis
- Check for any background signal due to PVY carry over during the bombardment procedure (viral RNA from plants with plasmid expressing yellow fluorescent protein). All samples with Cq (cycle quantification) values above the background (e.g., Cq = 32) should be considered negative.
- Calculate relative copy numbers of PVY RNA using quantGenius software (http://quantgenius.nib.si/user/login, Baebler et al., 2017). With this software, relative gene quantification is calculated using the standard curve. Enter Cq-s for the reference gene (COX) and set extraction quality control to 28. A Cq value of < 28 indicates efficient RNA extraction. Then enter Cq-s for the target gene (PVY-N605 CP). The final outputs from the software are quality-controlled relative copy numbers for each sample. Subtract the copy numbers calculated from qPCR without RT (presence of plasmid DNA in the sample) from the copy numbers calculated from RT-qPCR. The final results are relative copy numbers of viral RNA.
- Perform one-way analysis of variance with Bonferroni post-hoc analysis to statistically evaluate the differences in virus accumulation between the PVY clones. P-values of less than the selected value indicate significant differences among the groups. Post-hoc analysis reveals which PVY mutants accumulate differently than the WT PVY. For each group, include at least 20 samples (i.e., seven plants and three leaves per plant) and perform the complete experiment at least twice.
Recipes
- S.O.C. Medium
Set pH to 7.0Component amount/L Bacto Tryptone 20 g Yeast extract 5 g NaCl 580 mg KCl 186 mg MgCl2 952 mg Glucose 3.603 g
Autoclave 15 min at 121 °C - LB medium liquid (low salt)
Set pH to 7.5Component amount/L Bacto Tryptone 10 g Yeast extract 5 g NaCl 5 g
LB agar (1 L): Add 15 g of Bacto agar to LB medium liquid
Autoclave 15 min at 121 °C - Antibiotic
Filter sterilise using a 0.22 μm disposable filterAntibiotic Final concentration (µg/ml) Ampicillin 100 - 0.1 M phosphate buffer
Set pH to 6.8Component amount/100 ml NaH2PO4·2H2O 6.24 g Na2HPO4 7.50 g - 50x TAE buffer
Add 100 ml of 50x TAE buffer to 4900 ml of dH2O to obtain 1x TAE bufferComponent amount/L Trizma base 242 g Acetic acid 57.1 ml 0.5 M EDTA 100.0 ml - 0.5 M EDTA
Set pH to 8.0Component amount/L EDTA disodium salt, dihydrate 186.1 g
Acknowledgments
This work was supported by the Slovenian Research Agency (P1-0391, J7-7248, P4-0165, P2-0145, and P1-0010, PhD grants for A.K.).
We thank all the authors of the research paper Kežar et al. (2019) and J. Lenarčič, M. Srnko, L. Mourrain, M. Kisovec, A. Blejec, B. Dušak, and R. Vollmeier for support, and F. Cillo for the PVY clone. The protocol for biolistic bombardment was adapted and optimised from Dobnik et al. (2013).
Competing interests
The authors declare no conflicts of interest within the work.
References
- Baebler, Š., Svalina, M., Petek, M., Stare, K., Rotter, A., Pompe-Novak, M. and Gruden, K. (2017). quantGenius: implementation of a decision support system for qPCR-based gene quantification. BMC Bioinformatics 18(1): 276.
- Bukovinszki, Á., Götz, R., Johansen, E., Maiss, E. and Balázs, E. (2007). The role of the coat protein region in symptom formation on Physalis floridana varies between PVY strains. Virus Res 127(1): 122-125.
- Chung, B. Y., Miller, W. A., Atkins, J. F. and Firth, A. E. (2008). An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci U S A 105(15): 5897-5902.
- Dobnik, D., Baebler, Š., Kogovšek, P., Pompe-Novak, M., Štebih, D., Panter, G., Janež, N., Morisset, D., žel, J. and Gruden, K. (2013). β-1,3-glucanase class III promotes spread of PVY(NTN) and improves in planta protein production. Plant Biotechnol Rep 7: 547-555.
- Fakhfakh, H., Vilaine, F., Makni, M. and Robaglia, C. (1996). Cell-free cloning and biolistic inoculation of an infectious cDNA of Potato virus Y. J Gen Virol 77 (Pt 3): 519-523.
- Grech-Baran, M., Witek, K., Szajko, K., Witek, A. I., Morgiewicz, K., Wasilewicz-Flis, I., Jakuczun, H., Marczewski, W., Jones, J. D. G. and Hennig, J. (2020). Extreme resistance to Potato virus Y in potato carrying the Rysto gene is mediated by a TIR-NLR immune receptor. Plant Biotechnol J.
- Jakab, G., Droz, E., Brigneti, G., Baulcombe, D. and Malnoë, P. (1997). Infectious in vivo and in vitro transcripts from a full-length cDNA clone of PVY-N605, a Swiss necrotic isolate of Potato virus Y. J Gen Virol 78 (Pt 12): 3141-3145.
- Kerlan, C. (2006). Potato virus Y. Descriptions of plant viruses. Am Appl Biol.
- Kežar, A., Kavčič, L., Polák, M., Nováček, J., Gutiérrez-Aguirre, I., Žnidarič, M. T., Coll, A., Stare, K., Gruden, K., Ravnikar, M., Pahovnik, D., Žagar, E., Merzel, F., Anderluh, G. and Podobnik, M. (2019). Structural basis for the multitasking nature of the Potato virus Y coat protein. Sci Adv 5(7): eaaw3808.
- Kogovšek, P., Gow, L., Pompe-Novak, M., Gruden, K., Foster, G. D., Boonham, N. and Ravnikar, M. (2008). Single-step RT real-time PCR for sensitive detection and discrimination of Potato virus Y isolates. J Virol Methods 149(1): 1-11.
- Križnik, M., Petek, M., Dobnik, D., Ramšak, Ž., Baebler, Š., Pollmann, S., Kreuze, J. F., Žel, J. and Gruden, K. (2017). Salicylic acid perturbs sRNA-gibberellin regulatory network in immune response of potato to Potato virus Y infection. Front Plant Sci 8: 2192.
- Lacomme, C. and Jacquot, E. (2017). General characteristics of Potato virus Y (PVY) and its impact on potato production: An Overview. In: Lacomme, C., Glais, L., Bellstedt, D., Dupuis, B., Karasev, A., Jacquot, E. (Eds.). Potato virus Y: biodiversity, pathogenicity, epidemiology and management. Springer, Cham, pp 1-19.
- Lukan, T., Baebler, Š., Pompe-Novak, M., Guček, K., Zagorščak, M., Coll, A. and Gruden, K. (2018). Cell death is not sufficient for the restriction of Potato Virus Y spread in hypersensitive response-conferred resistance in potato. Front Plant Sci 9: 168.
- Mäkinen, K. and Hafrén, A. (2014). Intracellular coordination of potyviral RNA functions in infection. Front Plant Sci 5: 110.
- Mäkinen, K., Lõhmus, A. and Pollari, M. (2017). Plant RNA regulatory network and RNA granules in virus infection. Front Plant Sci 8: 2093.
- Rupar, M., Florence, F., Michel, T., Gutiérrez-Aguirre, I., Agnès, D., Laurent, G., Maja, K., David, D., Kristina, G., Emmanuel, J., Ravnikar M. (2015). Fluorescently tagged Potato virus Y: a versatile tool for functional analysis of plant-virus interactions. Mol Plant Microbe Interact 28(7): 739-750.
- Stare, T., Ramšak, Ž., Križnik, M. and Gruden, K. (2019). Multiomics analysis of tolerant interaction of potato with Potato virus Y. Sci Data 6(1): 250.
- Tribodet, M., Glais, L., Kerlan, C. and Jacquot, E. (2005). Characterization of Potato virus Y (PVY) molecular determinants involved in the vein necrosis symptom induced by PVYN isolates in infected Nicotiana tabacum cv. Xanthi. J Gen Virol 86(Pt 7): 2101-2105.
- Valkonen, J. (2007). Potato viruses: economical losses and biotechnological potential. Vreugdenhil, D., Bradshaw, J., Gebhardt, C., Govers, F., MacKerron, D. K. L., Taylor, M. A. and Ross H. A. (Eds.). In: Potato Biology and Biotechnology: Advances and Perspectives (pp. 619-641). Elsevier Scientific Publ. Co.
- Weller, S. A., Elphinstone, J. G., Smith, N. C., Boonham, N. and Stead, D. E. (2000). Detection of Ralstonia solanacearum strains with a quantitative, multiplex, real-time, fluorogenic PCR (TaqMan) assay. Appl Environ Microbiol 66(7): 2853-2858.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Stare, K., Coll, A., Gutiérrez-Aguirre, I., Tušek Žnidarič, M., Ravnikar, M., Kežar, A., Kavčič, L., Podobnik, M. and Gruden, K. (2020). Generation and in Planta Functional Analysis of Potato Virus Y mutants. Bio-protocol 10(14): e3692. DOI: 10.21769/BioProtoc.3692.
- Kežar, A., Kavčič, L., Polák, M., Nováček, J., Gutiérrez-Aguirre, I., Žnidarič, M. T., Coll, A., Stare, K., Gruden, K., Ravnikar, M., Pahovnik, D., Žagar, E., Merzel, F., Anderluh, G. and Podobnik, M. (2019). Structural basis for the multitasking nature of the Potato virus Y coat protein. Sci Adv 5(7): eaaw3808.
Category
Plant Science > Plant immunity > Host-microbe interactions
Microbiology > Microbe-host interactions > Virus
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.