- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A CRISPR Competition Assay to Identify Cancer Genetic Dependencies
Published: Vol 10, Iss 14, Jul 20, 2020 DOI: 10.21769/BioProtoc.3682 Views: 10055
Reviewed by: Pilar VillacampaJaira Ferreira de VasconcellosChhuttan L MeenaAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Mar 2017
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The CRISPR/Cas9 system is a powerful tool for genome editing, wherein the RNA-guided nuclease Cas9 can be directed to introduce double-stranded breaks (DSBs) at a targeted locus. In mammalian cells, these DSBs are typically repaired through error-prone processes, resulting in insertions or deletions (indels) at the targeted locus. Researchers can use these Cas9-mediated lesions to probe the consequences of loss-of-function perturbations in genes of interest. Here, we describe an optimized protocol to identify specific genes required for cancer cell fitness through a CRISPR-mediated cellular competition assay. Identifying these genetic dependencies is of utmost importance, as they provide potential targets for anti-cancer drug development. This protocol provides researchers with a robust and scalable approach to investigate gene dependencies in a variety of cell lines and cancer types and to validate the results of high-throughput or whole-genome screens.
Keywords: CancerBackground
The CRISPR/Cas9 system is believed to have evolved as an adaptive prokaryotic viral defense (Mojica et al., 2005; Makarova et al., 2006). Soon after its discovery, it was co-opted by researchers and modified for laboratory use in genome editing (Doudna and Charpentier, 2014; Hsu et al., 2014). By transgenically expressing the Cas9 nuclease along with a short guide RNA (sgRNA) complementary to the target sequence, double-stranded breaks (DSBs) can be introduced at a locus of interest in a variety of cells and organisms (Cong et al., 2013). Mammalian cells typically repair these DSBs through the error-prone nonhomologous end-joining (NHEJ) pathway, resulting in insertions or deletions (indels) at the targeted locus (Bothmer et al., 2017). These indels can disrupt coding sequences and result in the generation of null alleles.
A key goal of preclinical cancer research is to identify and characterize cancer “genetic dependencies”, or genes that are required for cancer cell proliferation or fitness. These genes, sometimes called “cancer addictions”, are attractive targets for anti-cancer drug development. Historically, the first set of cancer dependencies to be identified were recurrently-mutated oncogenes, and inhibitors of these driver genes like BRAF and EGFR have proven to be highly successful clinical agents (Paez et al., 2004; Luo et al., 2009; Chapman et al., 2011). The advent of systemic screening approaches with RNA interference (RNAi) provided researchers with a powerful new methodology to identify other potential genetic dependencies in cancer cells. Through the use of short hairpin and small interfering RNA constructs complementary to target transcripts, researchers could knock-down the expression of a gene of interest to test whether its loss blocked cancer growth (Hannon and Rossi, 2004). While numerous potential drug targets have been identified with these methodologies, RNAi-based perturbations have been found to cause significant off-target effects, and often silence the expression of many other genes that were not intentionally targeted (Jackson et al., 2003; Birmingham et al., 2006). In particular, experiments conducted by our lab and several others have demonstrated that cancer cells can tolerate the loss of many putative dependencies previously discovered using RNAi and have implicated off-target gene knockdown as one potential cause of these discrepant results (Lin et al., 2017 and 2019; Huang et al., 2017; Giuliano et al., 2018; Thomenius et al., 2018). In contrast, head-to-head comparisons have demonstrated that CRISPR/Cas9 constructs exhibit significantly fewer off-target effects compared to RNAi-based approaches, underscoring the significant potential that this technology harbors to identify true cancer dependencies (Morgens et al., 2016; Smith et al., 2017).
As with RNAi, the highly programable nature of CRISPR facilitates targeted screening approaches to uncover genetic addictions in cancer cell lines (Shalem et al., 2014; Wang et al., 2014). Through rational guide design, researchers can disrupt protein function and interrogate potential drug targets. In this protocol, we describe an optimized screening methodology first described by Shi et al. (2015) in which cells that constitutively express the Cas9 nuclease are transduced with a vector that co-expressed a sgRNA and GFP. The resulting population is comprised of both transduced (GFP+) and untransduced cells (GFP-), and the relative abundance of each population is tracked via flow cytometry over the course of several passages (Figure 1). Guides that target the functional protein domains of genes essential to cellular fitness are consistently outcompeted by untransduced cancer cells, which is easily detectable as a decreasing abundance of GFP+ cells over time.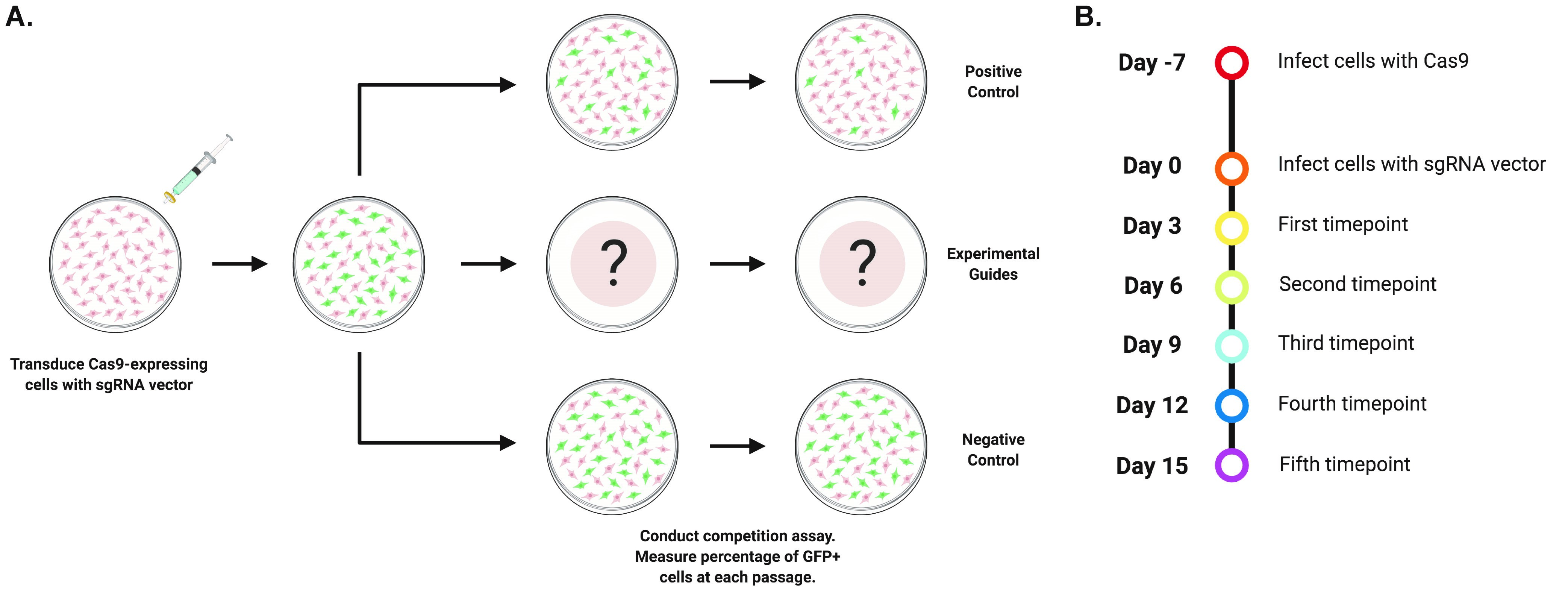
Figure 1. Overview of the competition assay. A. Cas9-expressing cells are transduced with a sgRNA vector at a low multiplicity-of-infection and the relative abundance of the transduced population is measured over the course of five passages via flow cytometry. B. Timeline of the competition assay.
Here we describe a straightforward protocol to determine whether a gene of interest is required for the viability or fitness of a particular cancer cell line. This protocol can be easily scaled-up and offers robust results in as little as three weeks. We have extensively validated this approach through the use of control gRNAs targeting known essential genes and non-essential loci. Researchers can investigate potential dependencies through this GFP competition assay by utilizing guides targeting their gene of interest in parallel with appropriate positive and negative controls. We have found that the results of these GFP competition assays are consistent with other in vitro assays measuring cellular fitness, including 2-D proliferation assays and soft-agar assays to measure anchorage-independent growth (Lin et al., 2017 and 2019). This protocol also represents a straightforward approach to validate individual hits recovered in high-throughput or whole-genome CRISPR screens.
Materials and Reagents
- 5 ml Polystyrene Round-Bottom Tubes (Falcon, catalog number: 352054 )
- 5 ml Polystyrene Round-Bottom Tubes, with Cell Strainer Snap Cap (Falcon, catalog number: 352235 )
- BD 10 ml Syringe Luer-Lok Tip (BD, catalog number: 309604 )
- Millex-HV Syringe Filter Unit, 0.45-µm (Millipore, catalog number: SLHVM33RS )
- 100 x 15 mm LB + 100 µg/ml ampicillin culture plates (VWR, catalog number 25384-342)
- Standard tissue culture plates (6 and 12-well plates recommended)
- One Shot Stbl3 Chemically Competent E. coli (Invitrogen, catalog number: C737303 )
- HEK293T cells (ATCC, catalog number: CRL-3216 )
- Recipient cancer cell line (from ATCC or other sources)
- LentiV-Cas9-Puro plasmid (Addgene, catalog number: 108100 )
- psPAX2.0 plasmid (Addgene, catalog number: 12260 )
- VSVG plasmid (Addgene, catalog number: 12259)
- Lenti-XTM qRT-PCR Titration Kit (Takara, catalog number: 631235 )
- gRNA plasmid backbone (see Table 1 for suggested plasmids and their respective Addgene numbers)
Table 1. Suggested CRISPR Plasmids
- LB + 100 µg/ml ampicillin medium (Sigma-Aldrich, catalog number: A9518 )
- DMEM medium supplemented with 10% FBS, 1% Pen/Strep, and glutamine (Life Technologies, catalog number: 11995-073 )
- Appropriate cell culture medium for the cancer cell line of interest
- 50% (v/v) glycerol
- Anti-FLAG antibody (Sigma-Aldrich, catalog number: F1804 )
- 2 M CaCl2
- 2x HEPES-buffered saline
- 100 mM chloroquine (Cayman Chemical, catalog number: 14194 )
- Polybrene (Santa Cruz Biotechnology, catalog number: sc-134220 )
- Puromycin (Gibco, catalog number: A11138-03 )
- BsmBI restriction endonuclease with NEB buffer 3.1 (NEB, catalog number: R0580 )
- Alkaline phosphatase, calf intestinal (CIP) stock (10,000 U/ml; NEB, catalog number: M0290 )
- 100 µM gRNA oligos (IDT or preferred oligonucleotide synthesis provider)
- T4 Polynucleotide Kinase (NEB, catalog number: M0201 )
- T4 DNA Ligase with T4 DNA Ligase Buffer (NEB, catalog number: M0202 )
Equipment
- Pipettes
- Mammalian cell culture equipment
- CO2 incubator
- Laminar flow cabinet
- CO2 incubator
- Fluorescent Microscope
- Thermocycler
- -20 °C freezer
- -80 °C freezer
- Miltenyi Biotec MACSquant VYB Flow Cytometer (Miltenyi Biotech, catalog number: 130-096-116 )
Alternative: Luminex Guava easyCyte or comparable flow cytometer capable of measuring GFP fluorescence
Software
- Benchling (https://www.benchling.com/)
- Microsoft Excel
- Prism 8
Procedure
Note: In this section, we will introduce the Cas9 transgene into the cell line of interest. If researchers already have stable Cas9-expressing cell lines, they can proceed directly to Procedure B.
- Generation of Cas9-expressing cell line
- Select a Cas9 expression vector
We suggest using LentiV-Cas9-Puro (Addgene) to introduce Cas9 into the cell line of interest, although any constitutive SpCas9 expression vector with a mammalian selection marker should suffice. - Produce Cas9 virus
- We suggest using the lentiviral packaging plasmids psPAX2 and VSVG (Addgene).
- We provide a brief overview of lentivirus production below. Our lab has made available a more detailed protocol for this process, which can be found in Giuliano et al. (2019).
- Calcium phosphate transfect HEK293T packaging cells with LentiV-Cas9-Puro, psPAX2, and VSVG.
- Replace media on the packaging cells approximately 8-14 h post transfection with fresh media.
- Harvest virus 24 h post-transfection. We recommend filtering the supernatant with a 0.45-µM filter to separate cell debris. Virus can be collected up to three times in the 72-h period post-transfection.
- Virus can be used immediately or stored at -80 °C.
- Calcium phosphate transfect HEK293T packaging cells with LentiV-Cas9-Puro, psPAX2, and VSVG.
- We suggest using the lentiviral packaging plasmids psPAX2 and VSVG (Addgene).
- Transduce cell line of interest
- Plate cells at approximately 50% confluence.
- Mix viral supernatant with fresh media in a 1:1 ratio.
- Add polybrene to the diluted viral supernatant to a final concentration of 8 µg/ml. We find this concentration improves transduction efficiency for nearly all cell lines commonly used in our lab. However, this concentration can be adjusted if needed.
- Replace media on cells to be transduced with the polybrene-containing viral supernatant.
- Change media on the transduced cells with fresh media 24 h post-transduction.
- Plate cells at approximately 50% confluence.
- Select for Cas9-expressing cells
- The Cas9 expression vector used in our lab (LentiV-Cas9-Puro) contains a puromycin-resistance marker. The ideal concentration of puromycin for selection is cell-line dependent and should be optimized prior to the start of the experiment.
- To optimize the concentration of puromycin for selection, we suggest plating wild-type cells at approximately 50% confluence on 6 or 12-well plates.
- Replace media on the cells 24 h later with media containing puromycin at a range of 1-4 µg/ml. We generally test in increments of 0.5 μg/ml, with each well containing a different concentration of puromycin.
- Assess cell survival 3-5 days post drug selection. The optimal concentration of puromycin is the lowest concentration at which there are no surviving cells. For adherent cell lines, dead/dying cells generally detach from the plate, which facilitates identifying surviving cells. Suspension cell lines may require a viability dye for this purpose.
- While this range of puromycin concentrations is sufficient for most cell lines, some may require higher working concentrations. In that case, test additional concentrations up to 10 µg/ml.
- To optimize the concentration of puromycin for selection, we suggest plating wild-type cells at approximately 50% confluence on 6 or 12-well plates.
- Replace media on the transduced cells 48 h post-transduction with media containing puromycin at a concentration of 1-4 µg/ml or as determined to be optimal for the cell line of interest.
- Maintain selection until Cas9 is stably expressed. We find that for most cell lines, only one round of transduction, followed by 3 to 5 days of puromycin selection, is sufficient for generating stable Cas9 expression. However, some cell lines may require more than one round of transduction and/or a longer selection period.
- The Cas9 expression vector used in our lab (LentiV-Cas9-Puro) contains a puromycin-resistance marker. The ideal concentration of puromycin for selection is cell-line dependent and should be optimized prior to the start of the experiment.
- Verify Cas9 expression
- Researchers can utilize a variety of approaches to verify expression of Cas9, including:
- Quantitive PCR (qPCR). Through the use of PCR primers targeting the Cas9 mRNA transcript, expression of the Cas9 nuclease can be confirmed. The qPCR primers our lab utilizes for detecting Cas9 expression are forward: 5′ GGCCTACCACGAGAAGTACC 3′ and reverse: 5′ CTGGCGTTGATGGGGTTTTC 3′.
- Western Blot. The Cas9 expression vector used in our lab (LentiV-Cas9-Puro) contains a FLAG-epitope tag fused to the N-terminus of Cas9. As such, researchers can make use of an anti-FLAG antibody in place of a Cas9 specific antibody if needed. The anti-FLAG antibody our lab utilizes can be found in the Materials and Reagents section (Sigma-Aldrich).
- Transduction with guides targeting essential genes. While this may not be suitable for the first GFP competition assay you conduct, our lab routinely performs control competition assays with several positive and negative control guides (as described later in this protocol) to verify Cas9 functionality in a cell line of interest.
- Quantitive PCR (qPCR). Through the use of PCR primers targeting the Cas9 mRNA transcript, expression of the Cas9 nuclease can be confirmed. The qPCR primers our lab utilizes for detecting Cas9 expression are forward: 5′ GGCCTACCACGAGAAGTACC 3′ and reverse: 5′ CTGGCGTTGATGGGGTTTTC 3′.
- It is of note to mention that qPCR and western blotting for Cas9 can confirm Cas9 expression but not functionality. Transduction with guides with known biological activity can serve to confirm Cas9 functionality. For these reasons, we recommend using a combination of these approaches to verify Cas9 expression and functionality in any Cas9-expressing cell lines you generate.
- Researchers can utilize a variety of approaches to verify expression of Cas9, including:
- Select a Cas9 expression vector
Note: In this section, we will design guides targeting the gene of interest, and clone these guides, alongside appropriate positive and negative control guides, into guide plasmids for sgRNA and fluorescent protein expression. We discuss considerations for ideal guide design and provide control guides extensively validated by our lab and our methods for plasmid cloning.
- Design of sgRNAs and guide plasmids
- Identify target sites within the gene of interest
- Use an online tool such as Benchling (https://www.benchling.com/) to design suitable guides targeting the gene-of-interest.
- For this protocol, we use single guides with 20 base pairs of homology to the target sequence, as this is best-suited for CRISPR-knockout.
- The PAM sequence should be NGG on the 3′ side of the guide sequence, as the Cas9 strain we use is SpCas9. The PAM sequence is necessary for Cas9-mediated DNA cleavage and is found 3-4 base pairs downstream of the cut site.
- For this protocol, we use single guides with 20 base pairs of homology to the target sequence, as this is best-suited for CRISPR-knockout.
- Guides targeting functional protein domains produce the most-efficient knockout of the target gene. For this reason, we highly recommend cross-referencing exons with protein domains to identify which guides are best-suited to disrupt protein function, as guides targeting the first exon alone can lead to false negatives (Figure 2A) (Shi et al., 2015).
- Additionally, we recommend designing 3 or more guides per gene, ideally targeting different exons.
- Order oligonucleotides through IDT or your preferred commercial source for DNA synthesis.
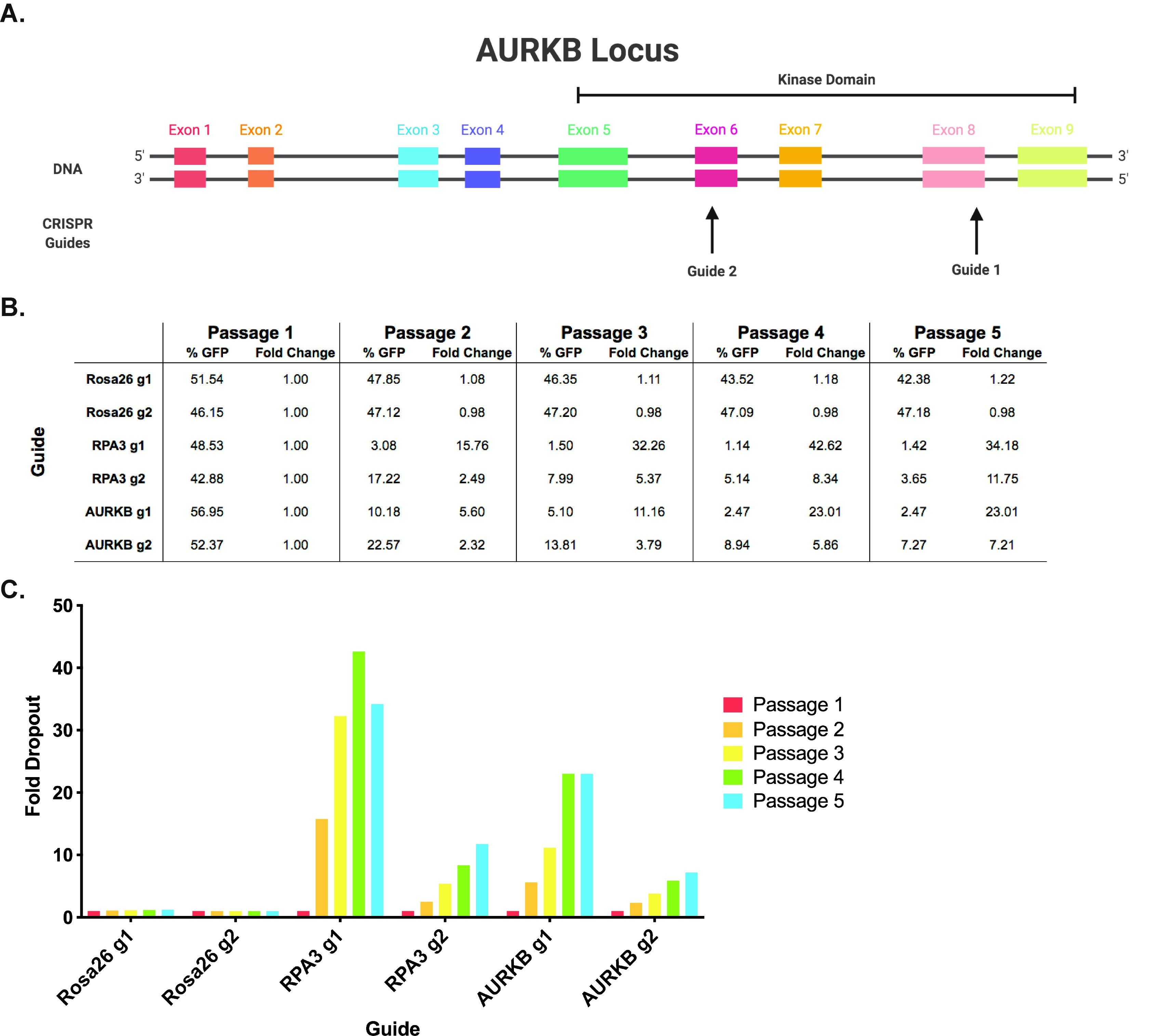
Figure 2. Example experimental design and data analysis for the competition assay. A. Example guide RNA design. AURKB (Aurora B) encodes a kinase with key roles in mitosis. Guides are shown targeting multiple exons within Aurora B’s kinase domain. B. Example dropout data. The relative abundance of the transduced population is indicated by % GFP and the calculated fold dropout is shown over the course of five passages. C. Example dropout figure. The fold dropout over the course of five passages is shown for each guide. It can be seen that the negative control guides targeting Rosa26 exhibit minimal depletion and the positive control guides targeting RPA3 display over ten-fold dropout at the final timepoint. Consistent with prior knowledge, these results verify that AURKB is a cancer cell dependency.
- Use an online tool such as Benchling (https://www.benchling.com/) to design suitable guides targeting the gene-of-interest.
- Identify suitable positive and negative control guides
- Negative control guides should target noncoding loci or genes dispensable for cellular fitness such as Rosa26 or AAVS1. These loci are considered canonical safe-harbor sites, and thus introducing indels at these loci will have minimal effects on cancer cell fitness.
- Positive control guides should target essential genes, such as PCNA or RPA3. These genes are critical for DNA replication, and thus introducing indels at these loci will be detrimental to cancer cell fitness.
- We have provided our control guides in Table 2.
Note: The sequence provided is the 20 bp spacer sequence and does not include the 4-5 bp adapter sequence for cloning into the guide vector. The adapter for our guide vector is CACC, and a G must be added if the first nucleotide of the spacer sequence is not a guanine. For example, the complete guide sequence for Rosa26 g1 listed below with the adapter added is: 5’- CACCGACAGCAAGTTGTCTAACCCG (adapter in bold).
Table 2. Suggested Control Guides
- Negative control guides should target noncoding loci or genes dispensable for cellular fitness such as Rosa26 or AAVS1. These loci are considered canonical safe-harbor sites, and thus introducing indels at these loci will have minimal effects on cancer cell fitness.
- Clone sgRNAs into guide vector
- We recommend the use of a lentiviral GFP sgRNA expression vector (Addgene), although any comparable sgRNA expression vector with a fluorescent marker is suitable for this assay. Additionally, our lab has developed and validated several derivatives of this sgRNA expression vector with different fluorescent protein markers, which can be used if needed.
- We provide a brief overview of the guide cloning process below. Our lab has made available a more detailed protocol for this process, which can be found in Giuliano et al. (2019).
- Linearize the guide vector backbone with a BsmBI digest.
- Anneal and phosphorylate guide oligos with T4 PNK.
- Ligate annealed and phosphorylated guides into guide vector backbone.
- Transform ligation product into competent cells.
- Sequence-verify the guide plasmid using the U6 primer (5′- GAGGGCCTATTTCCCATGATTCC).
- Linearize the guide vector backbone with a BsmBI digest.
- We recommend the use of a lentiviral GFP sgRNA expression vector (Addgene), although any comparable sgRNA expression vector with a fluorescent marker is suitable for this assay. Additionally, our lab has developed and validated several derivatives of this sgRNA expression vector with different fluorescent protein markers, which can be used if needed.
- Identify target sites within the gene of interest
Note: In this section, we will introduce the guide plasmids encoding the sgRNA and fluorescent protein into the Cas9-expressing cell line. We discuss methods of lentiviral production, viral titration, and some considerations for the competition assay set-up.
- Transduction of Cas9-expressing cell line with guide plasmids
- Produce lentivirus with guide plasmids
See Step A2 for a brief overview of the virus production process, or our more comprehensive virus production protocol in Giuliano et al. (2019). - Determine viral titer
- As the guide plasmids used in this assay encode fluorescent proteins, viral titer can be approximated by transducing cells with differing amounts of virus and measuring the percentage of cells exhibiting fluorescence three days post-transduction.
- Researchers can also utilize a qPCR-based approach to determine viral titer if they prefer (e.g., Lenti-XTM qRT-PCR Titration Kit [Takara], see manufacturer’s protocol for more information). While such an approach may provide a more definitive answer as to the number of virus particles per microliter of supernatant, we find that the approximation provided by measuring the percentage of cells exhibiting fluorescence is sufficient for this assay.
- As the guide plasmids used in this assay encode fluorescent proteins, viral titer can be approximated by transducing cells with differing amounts of virus and measuring the percentage of cells exhibiting fluorescence three days post-transduction.
- Transduce Cas9-expressing cell line with titrated viral supernatant.
- The ideal transduction efficiency is 50%. However, this assay is flexible in its requirements such that transduction efficiencies ranging from 10-80% are suitable for this assay.
- See Step A3 for a brief overview of the viral transduction process.
- We recommend conducting this assay on 12 or 24-well plates. This assay can be scaled up or down if needed. Generally, conducting this assay on larger plates reduces background variation, but if there are constraining factors at play (i.e., not enough virus), this assay can be conducted on a 48 or 96-well plate as well.
- The ideal transduction efficiency is 50%. However, this assay is flexible in its requirements such that transduction efficiencies ranging from 10-80% are suitable for this assay.
- Produce lentivirus with guide plasmids
Note: In this section, we will take our first time-point of the competition assay. We discuss methods for measuring the percentage of cells expressing the GFP marker through flow cytometry, and considerations for conducting the competition assay.
- Start of competition assay
- Harvest cells
Generally, we set our first time-point for three days post-transduction to provide ample time for transgene expression. - Seed cells at appropriate confluency for the next passage
- Perform an appropriate cell split (i.e., 1:2 to 1:10) depending on the cell line’s proliferative capacity.
- Depending on the choice of cell line and its doubling time, researchers may choose to passage the cells every three or four days.
- For example, we generally split the breast cancer cell line MDA-MB-231 1:4 every three days.
- Measure GFP expression by flow cytometry
- After plating cells for the next passage, prepare leftover samples for flow cytometry. We suggest passing cells through a single cell strainer (Falcon) prior to flow analysis. It is not necessary to utilize any flow cytometry specific resuspension buffer, as cell culture medium should be sufficient for this experiment.
- Run control GFP- cells to set appropriate voltages for forward scatter and side scatter lasers.
- Run control GFP- cells and GFP+ cells to adjust flow parameters and determine a gate for GFP-expressing cells.
- Run experimental samples. Researchers should aim to run at least 10% of the sample volume (i.e., if you harvested 1 ml of cells from a 12-well plate, run at least 100 µl). If possible, we recommend running about 25% of the sample, as a larger sample will yield a more accurate measurement of GFP expression.
- Record % GFP for all samples.
To ensure experimental consistency, we suggest saving a program with your laser voltages and analysis templates to use for successive time-points.
- After plating cells for the next passage, prepare leftover samples for flow cytometry. We suggest passing cells through a single cell strainer (Falcon) prior to flow analysis. It is not necessary to utilize any flow cytometry specific resuspension buffer, as cell culture medium should be sufficient for this experiment.
- Harvest cells
- Successive timepoints of competition assay
- Harvest cells
- Seed cells at appropriate confluency for next passage.
At the fifth timepoint, the assay is complete, and it is not necessary to re-plate the cells for the next passage. - Measure GFP expression by flow cytometry.
- Harvest cells
Data analysis
Note: In this section, we will transform our measurements of % GFP expressing cells from each of the timepoints into fold change values. These fold change values from individual guides can then be compared to one another, and to the positive and negative controls, to draw conclusions as to the essentiality of the gene of interest in the cell lines tested.
- Determine fold change
- Fold change in the context of this assay is defined as (% GFP at passage 1)/(% GFP at passage X).
- Therefore, the fold change at passage 1 should be 1. An example of these calculations can be seen in Figure 2B.
- Prepare grouped graphs
We suggest using Prism, but any software that can graph numerical data (i.e., Excel) will suffice (Figure 2C). - Set threshold for dropout
- The threshold should be greater than the fold change of the negative controls, as these guides target loci dispensable for cancer cell fitness.
- Our lab uses a threshold of 2.5-fold change for determining dependencies, as we have found that this threshold is always above the level of depletion observed with negative control guides.
- This can be adjusted based on the cell line as needed, as the dynamic range for fold change is dependent on a variety of factors including Cas9 expression, cell type differences, and GFP % at passage 1.
- If multiple guides targeting a gene exhibit minimal dropout above background levels, this gene is unlikely to be a genetic dependency in the cell line tested.
- If multiple guides targeting a gene exhibit consistent dropout above background levels, this is evidence for the gene being a dependency in that cell line.
Notes
- This protocol provides researchers with a streamlined approach to probe individual genetic dependencies in cancer cell lines. It has been variously used to identify novel dependencies in leukemia, small-cell lung cancer, pancreas cancer, and to interrogate several drug targets in clinical trials (Shi et al., 2015; Lin et al., 2017 and 2019; Huang et al., 2018; Somerville et al., 2018; Tarumoto et al., 2018). Additionally, our lab regularly deploys variations of this approach to probe for genetic dependencies in other contexts. For instance, this assay can be modified to probe for synthetically lethal relationships between two genes (i.e., gene A and gene B). If guides targeting each gene individually exhibit minimal dropout, but the researcher has evidence to suggest these genes have some degree of functional redundancy, then a dual-competition assay can be conducted instead. In this experiment, researchers can co-transduce target cells with two guides that express different fluorescent protein markers (Table 1), with guide 1 targeting gene A and guide 2 targeting gene B. By measuring the individual fluorescent protein expression, as well as the percentage of cells expressing both fluorescent proteins at each passage, researchers can gain insight into potential synthetically lethal relationships. If the double-positive population exhibits consistent depletion, but the single-positive populations do not, this is evidence for a potential synthetically lethal relationship between genes A and B.
- The development of additional CRISPR tools for transcriptional modulation provides researchers with other possible approaches for probing genetic dependencies. The CRISPR-interference (CRISPRi) system, wherein catalytically-inactive Cas9 (dCas9) is fused to a Krüppel-associated box (KRAB) domain, provides researchers the ability to suppress target gene expression without double-strand break formation (Gilbert et al., 2013). The resulting partial loss-of-function phenotype provides yet another context for researchers to investigate potential drug targets. The use of such an orthogonal approach complements the total loss-of-function phenotype generated by standard CRISPR/Cas9. Partial loss-of-function approaches, such as CRISPRi provide models of genetic perturbation more closely resembling the effects of targeted therapies, as pharmacological inhibition of any potential genetic dependency is unlikely to be absolute. This competition assay can be readily tailored to a CRISPRi approach through the use of a dCas9-KRAB expression vector in place of a Cas9 expression vector (Addgene #85969), and through the use of guides targeting promoters rather than functional exons. Our lab regularly conducts such assays, and we have found our results from this modified approach to be largely consistent with traditional Cas9 screening (Lin et al., 2019).
- While the CRISPR/Cas9 system offers numerous advantages over RNAi for investigating genetic dependencies, there are some limitations to consider. Generally, the Cas9 nuclease is utilized to produce an assortment of indels at the target site, and it is thought that the resulting premature stop codon leads to nonsense-mediated decay (NMD) of the mutant mRNA. However, the production of indels does not necessarily result in the ablation of the target gene product. CRISPR-induced genetic lesions can be bypassed by alternative splicing and downstream transcriptional initiation (Lalonde et al., 2017; Sharpe and Cooper, 2017). Additionally, an unintended consequence of CRISPR-induced knockout can be the production of aberrant protein products of unknown function (Tuladhar et al., 2019). In order to mitigate the potential confounding effect of these factors, we strongly suggest using several guides across multiple exons targeting functional protein domains. Additionally, we recommend verifying protein ablation via western blot.
- The process of mutant mRNA NMD itself may also have unintended consequences. NMD is known to be a mechanism of mRNA regulation in many cell types, including liver and bone marrow cells (Lykke-Andersen and Jensen, 2015). A recent study suggested that CRISPR-induced NMD causes a compensatory upregulation of the homologs of a targeted gene, which could potentially obfuscate the true effects of the CRISPR knockout (El-Brolosy et al., 2019). However, experiments performed by our lab suggest that this compensatory upregulation may be rare or non-existent in cancer cells (Lin et al., 2019). If researchers are concerned about potential CRISPR induced-homolog upregulation, we suggest conducting gene expression assays in parallel with the competition assay to rule out this possible confounding factor. Notably, the limitations described thus far are consequences of the genetic lesions induced by Cas9. It follows that these limitations could potentially be minimized through use of alternative systems for perturbation that do not cleave DNA, such as CRISPR-interference.
- Beyond the limitations of the CRISPR system itself, it is important to note that the results of this GFP competition assay are by no means dispositive of a gene’s status as being necessary or dispensable for cellular fitness in other contexts. While the consistent lack of dropout for guides targeting a gene across multiple trials and cell lines indicates that a targeted gene is dispensable for cell-autonomous growth in vitro, it remains possible that the gene is a dependency in other cellular contexts, like anchorage-independent growth, low oxygen conditions, in the presence of chemotherapeutics, in vivo, etc. Thus, a gene that does not score as a genetic dependency through this assay can still provide a valuable drug target for anticancer therapies. On the other hand, if researchers do see consistent dropout for guides targeting a gene, this is strong evidence for the gene being a genetic dependency. Such results should be followed up with further investigation through orthogonal approaches.
- We believe this assay will provide researchers with a valuable approach to investigate putative cancer dependencies. Results from our lab show this assay to be robust in its findings, and its relative simplicity allows researchers to simultaneously conduct this assay in many cell types. This assay is best utilized as a first-pass approach, and complemented with thorough, in-depth investigation of any putative cancer dependencies.
Acknowledgments
Research in the Sheltzer Lab is supported by an NIH Early Independence Award (1DP5OD021385), a Damon Runyon-Rachleff Innovation Award, a Gates Foundation Innovative Technology Solutions grant, and a CSHL-Northwell Translational Cancer Research grant. Cell sorting was performed with assistance from the CSHL Flow Cytometry Shared Resource, which is supported by the CSHL Cancer Center Support grant 5P30CA045508. This protocol draws on work original published in Shi et al. (2015), Lin et al. (2017 and 2019) and Giuliano et al. (2019).
Competing interests
J.M.S. is a member of the advisory board at Tyra Biosciences and has received consulting fees from Ono Pharmaceutical Co. and Merck.
References
- Birmingham, A., Anderson, E. M., Reynolds, A., Ilsley-Tyree, D., Leake, D., Fedorov, Y., Baskerville, S., Maksimova, E., Robinson, K., Karpilow, J., Marshall, W. S. and Khvorova, A. (2006). 3' UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods 3(3): 199-204.
- Bothmer, A., Phadke, T., Barrera, L. A., Margulies, C. M., Lee, C. S., Buquicchio, F., Moss, S., Abdulkerim, H. S., Selleck, W., Jayaram, H., Myer, V. E. and Cotta-Ramusino, C. (2017). Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat Commun 8: 13905.
- Chapman, P. B., Hauschild, A., Robert, C., Haanen, J. B., Ascierto, P., Larkin, J., Dummer, R., Garbe, C., Testori, A., Maio, M., Hogg, D., Lorigan, P., Lebbe, C., Jouary, T., Schadendorf, D., Ribas, A., O'Day, S. J., Sosman, J. A., Kirkwood, J. M., Eggermont, A. M., Dreno, B., Nolop, K., Li, J., Nelson, B., Hou, J., Lee, R. J., Flaherty, K. T., McArthur, G. A. and Group, B. S. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26): 2507-2516.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Doudna, J. A. and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213): 1258096.
- El-Brolosy, M. A., Kontarakis, Z., Rossi, A., Kuenne, C., Günther, S., Fukuda, N., Kikhi, K., Boezio, G. L. M., Takacs, C. M., Lai, S. L., Fukuda, R., Gerri, C., Giraldez, A. J. and Stainier, D. Y. R. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568(7751): 193-197.
- Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., Stern-Ginossar, N., Brandman, O., Whitehead, E. H., Doudna, J. A., Lim, W. A., Weissman, J. S. and Qi, L. S. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2): 442-451.
- Giuliano, C. J., Lin, A., Girish, V. and Sheltzer, J. M. (2019). Generating single cell-derived knockout clones in Mammalian cells with CRISPR/Cas9. Curr Protoc Mol Biol 128(1): e100.
- Giuliano, C. J., Lin, A., Smith, J. C., Palladino, A. C. and Sheltzer, J. M. (2018). MELK expression correlates with tumor mitotic activity but is not required for cancer growth. Elife 7: 32838.
- Hannon, G. J. and Rossi, J. J. (2004). Unlocking the potential of the human genome with RNA interference. Nature 431(7006): 371-378.
- Hsu, P. D., Lander, E. S. and Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157(6): 1262-1278.
- Huang, H. T., Seo, H. S., Zhang, T., Wang, Y., Jiang, B., Li, Q., Buckley, D. L., Nabet, B., Roberts, J. M., Paulk, J., Dastjerdi, S., Winter, G. E., McLauchlan, H., Moran, J., Bradner, J. E., Eck, M. J., Dhe-Paganon, S., Zhao, J. J. and Gray, N. S. (2017). MELK is not necessary for the proliferation of basal-like breast cancer cells. Elife 6: 26693.
- Huang, Y. H., Klingbeil, O., He, X. Y., Wu, X. S., Arun, G., Lu, B., Somerville, T. D. D., Milazzo, J. P., Wilkinson, J. E., Demerdash, O. E., Spector, D. L., Egeblad, M., Shi, J. and Vakoc, C. R. (2018). POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev 32(13-14): 915-928.
- Jackson, A. L., Bartz, S. R., Schelter, J., Kobayashi, S. V., Burchard, J., Mao, M., Li, B., Cavet, G. and Linsley, P. S. (2003). Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21(6): 635-637.
- Lalonde, S., Stone, O. A., Lessard, S., Lavertu, A., Desjardins, J., Beaudoin, M., Rivas, M., Stainier, D. Y. R. and Lettre, G. (2017). Frameshift indels introduced by genome editing can lead to in-frame exon skipping. PLoS One 12(6): e0178700.
- Lin, A., Giuliano, C. J., Palladino, A., John, K. M., Abramowicz, C., Yuan, M. L., Sausville, E. L., Lukow, D. A., Liu, L., Chait, A. R., Galluzzo, Z. C., Tucker, C. and Sheltzer, J. M. (2019). Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med 11(509).
- Lin, A., Giuliano, C. J., Sayles, N. M. and Sheltzer, J. M. (2017). CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife 6: 24179.
- Luo, J., Solimini, N. L. and Elledge, S. J. (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136(5): 823-837.
- Lykke-Andersen, S. and Jensen, T. H. (2015). Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16(11): 665-677.
- Makarova, K. S., Grishin, N. V., Shabalina, S. A., Wolf, Y. I. and Koonin, E. V. (2006). A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1: 7.
- Mojica, F., Díez-Villaseñor, C., García-Martínez, J. and Soria, E. (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60: 174-182.
- Morgens, D. W., Deans, R. M., Li, A. and Bassik, M. C. (2016). Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol 34(6): 634-636.
- Paez, J. G., Jänne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., Herman, P., Kaye, F. J., Lindeman, N., Boggon, T. J., Naoki, K., Sasaki, H., Fujii, Y., Eck, M. J., Sellers, W. R., Johnson, B. E. and Meyerson, M. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304(5676): 1497-1500.
- Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelson, T., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G. and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Sharpe, J. J. and Cooper, T. A. (2017). Unexpected consequences: exon skipping caused by CRISPR-generated mutations. Genome Biol 18(1): 109.
- Shi, J., Wang, E., Milazzo, J. P., Wang, Z., Kinney, J. B. and Vakoc, C. R. (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol 33(6): 661-667.
- Smith, I., Greenside, P. G., Natoli, T., Lahr, D. L., Wadden, D., Tirosh, I., Narayan, R., Root, D. E., Golub, T. R., Subramanian, A. and Doench, J. G. (2017). Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol 15(11): e2003213.
- Somerville, T. D. D., Xu, Y., Miyabayashi, K., Tiriac, H., Cleary, C. R., Maia-Silva, D., Milazzo, J. P., Tuveson, D. A. and Vakoc, C. R. (2018). TP63-mediated enhancer reprogramming drives the squamous subtype of pancreatic ductal adenocarcinoma. Cell Rep 25(7): 1741-1755 e1747.
- Tarumoto, Y., Lu, B., Somerville, T. D. D., Huang, Y. H., Milazzo, J. P., Wu, X. S., Klingbeil, O., El Demerdash, O., Shi, J. and Vakoc, C. R. (2018). lkb1, salt-inducible kinases, and mef2c are linked dependencies in acute myeloid Leukemia. Mol Cell 69(6): 1017-1027 e1016.
- Thomenius, M. J., Totman, J., Harvey, D., Mitchell, L. H., Riera, T. V., Cosmopoulos, K., Grassian, A. R., Klaus, C., Foley, M., Admirand, E. A., Jahic, H., Majer, C., Wigle, T., Jacques, S. L., Gureasko, J., Brach, D., Lingaraj, T., West, K., Smith, S., Rioux, N., Waters, N. J., Tang, C., Raimondi, A., Munchhof, M., Mills, J. E., Ribich, S., Porter Scott, M., Kuntz, K. W., Janzen, W. P., Moyer, M., Smith, J. J., Chesworth, R., Copeland, R. A. and Boriack-Sjodin, P. A. (2018). Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS One 13(6): e0197372.
- Tuladhar, R., Yeu, Y., Tyler Piazza, J., Tan, Z., Rene Clemenceau, J., Wu, X., Barrett, Q., Herbert, J., Mathews, D. H., Kim, J., Hyun Hwang, T. and Lum, L. (2019). CRISPR-Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat Commun 10(1): 4056.
- Wang, T., Wei, J. J., Sabatini, D. M. and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343(6166): 80-84.
Article Information
Copyright
![]() Girish and Sheltzer. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Girish and Sheltzer. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Girish, V. and Sheltzer, J. M. (2020). A CRISPR Competition Assay to Identify Cancer Genetic Dependencies. Bio-protocol 10(14): e3682. DOI: 10.21769/BioProtoc.3682.
- Lin, A., Giuliano, C. J., Sayles, N. M. and Sheltzer, J. M. (2017). CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife 6: 24179.
Category
Cancer Biology > Proliferative signaling > Drug discovery and analysis
Cancer Biology > General technique > Molecular biology technique
Cell Biology > Cell viability > Cell survival
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.