- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunofluorescent Staining of Claudin-2 in Cultured Kidney Tubular Cells
Published: Vol 10, Iss 14, Jul 20, 2020 DOI: 10.21769/BioProtoc.3678 Views: 4697
Reviewed by: Christina Yan Ru TanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Members of the claudin family of tight junction proteins regulate paracellular permeability and modulate cell signaling. During junction remodeling, these proteins are selectively inserted into or retrieved from the tight junctions, but the control and coordination of these processes remain incompletely understood. Visualization of claudins allows the assessment of changes in their localization and abundance. We use the described protocol to stain claudin-2, but it can also be adapted to stain any tight junction protein. We found that using methanol for fixing allows the best preservation of claudin-2 both at the membrane and in cytoplasmic vesicles. Staining is done using a claudin-2 specific primary and a fluorescently labelled secondary antibody, along with DAPI to label nuclei. The samples are then imaged using confocal microscopy, and a z-stack is obtained allowing visualization of both junctional and intracellular claudin-2. Total claudin-2 signal can be quantified after 3D reconstruction of the images using the Imaris software.
Keywords: ClaudinBackground
Tight junctions (TJ) are multiprotein complexes localized at the apical-most portion of the intercellular junctional complexes that connect epithelial cells (Van Itallie and Anderson, 2014). These structures generate a permeability barrier and ion-specific paracellular pathways, maintain apico-basal polarity, provide input for various vital functions and modulate signaling pathways. The proteins located at the TJs can be divided into transmembrane and associated cytoplasmic proteins (reviewed in (González-Mariscal et al., 2003). The transmembrane proteins can be further subdivided into three major categories: claudins, tight junction-associated Marvel proteins (e.g., occludin) and single span proteins (e.g., Junctional Adhesion Molecules). TJ-associated cytosolic proteins include a large array of adaptors connected to signaling and cytoskeletal proteins. Collectively, these proteins are referred to as the cytoplasmic plaque. Claudins are small molecular weight (20-34 kDa) tetraspan membrane proteins that are essential components of the TJs (Tsukita et al., 2019). They incorporate into the TJ strands that typically contain a mosaic of various claudin isoforms that determine permselectivity (Van Itallie and Anderson, 2004). Interestingly, claudins have also recently emerged as key modulators of signaling, an effect that is likely due to the interactions with cytosolic adapters.
In this protocol we focus on claudin-2 (Cldn-2), a 230 amino acid member of the family, with a calculated molecular mass of 24.5 kDa. Cldn-2 was first described by Dr Shoichiro Tsukita and coworkers (Furuse et al., 1998a and 1998b) (for review see Venugopal et al., 2019). It is highly enriched in the kidney proximal tubules (Enck et al., 2001) and in intestinal and liver cells (e.g., Sakaguchi et al., 2002; Escaffit et al., 2005). It generates a cation selective paracellular channel, and therefore, its presence corresponds to elevated paracellular permeability (e.g., Amasheh et al., 2002). Multiple studies have revealed that in various cells Cldn-2 can be found not only at the membrane, i.e., in the TJs, but also in intracellular vesicles (Dukes et al., 2012; Lu et al., 2014; Amoozadeh et al., 2015), and in the nucleus (Ikari et al., 2014; Amoozadeh et al., 2018).
Many studies have used immunofluorescent staining in fixed cells followed by confocal microscopy to visualize TJ proteins and analyze their localization and abundance. These approaches allow investigation of the mechanisms that regulate TJ protein trafficking. The protocols differ in the fixation and blocking method and use primary antibodies targeted against specific proteins. Here we describe the method used in our lab to stain Cldn-2 in cultured kidney tubular cells based on (Amoozadeh et al., 2015 and 2018; Dan et al., 2019). We also detail our protocol for 3D reconstitution and analysis of changes in staining intensity following treatment, e.g. with cytokines. While we focus specifically on claudin-2, the same approach can be used for other TJ proteins in any epithelial or endothelial cells.
Materials and Reagents
- Micro cover glasses, round, No. 1, 18 mm (VWR® International, catalog number: CA-48380-046 ), sterilize coverglasses using autoclave
- Microscope slides (Fisherbrand, Fisher Scientific, catalog number: 12-550-15 )
- Clear tissue culture-treated 12-well microplates, (Corning Costar®, Millipore-Sigma, catalog number: CLS3513 )
- Parafilm
- Paper towel
- Cell line: LLC-PK1 kidney tubule epithelial cell line (European Collection of Authenticated Cell Cultures (Salisbury, UK), catalog number: 86121112 )
- For cell culture:
- Dulbecco's Modified Eagle Medium (D-MEM), low glucose, with L-glutamine
- 110 mg/L sodium pyruvate (Thermo Fisher, Gibco, catalog number: 11885084 )
- 10% Fetal Bovine Serum (FBS) (Thermo Fisher, Gibco, catalog number: 12483-020 )
- 1% Penicillin-Streptomycin (Penicillin-Streptomycin, 100x, sterile-filtered, cell culture tested) (Millipore-Sigma Aldrich, catalog number: P-4333 )
- Trypsin-EDTA (0.05% Trypsin with EDTA 4Na) (Thermo Fisher, Gibco, catalog number: 25300062 )
- Phosphate buffered saline (PBS) (Thermo Fisher, Gibco, catalog number: 10010023 )
- Dulbecco's Modified Eagle Medium (D-MEM), low glucose, with L-glutamine
- Methanol (BioShop Canada, catalog number: Met302 ), cool to -20 °C before using
- 10x PBS Buffer (pH 7.4 Sterile, w/o Ca, Mg) (BioShop Canada, catalog number: PBS415 ), open in hood to maintain sterility for storage
- Bovine serum albumin (BSA) (fraction V, heat shock isolation, > 98%) (BioShop Canada, catalog number: ALB001 )
- Polyclonal rabbit anti-Claudin-2 antibody (Thermo Fisher Scientific, Invitrogen, catalog number: 28530 ), aliquot to avoid repeated thawing, store at -20 °C
- Fluorophore-labeled secondary antibody:
Donkey anti-Rabbit IgG labelled with Alexa Fluor 488 (green) (Thermo Fischer Scientific, catalog number: A-21206 )
or
Donkey anti-Rabbit IgG labelled with Alexa Fluor 555 (red) (Thermo Fischer Scientific, catalog number: A- A31572 ) - 2,4-diamidino-2-phenylindole (DAPI) (Thermo Fischer Scientific, catalog number: D1306 )
Make a 5 mg/ml DAPI stock solution by dissolving the contents of one vial (10 mg) in 2 ml of deionized water. Aliquot the stock, protect from light. DAPI stock solution can be stored at 4 °C for up to 6 months, or at -20 °C for longer periods. - Fluorescent mounting medium (Dako, Agilent catalog number: S3023 ), store at 4 °C
- Cell culture medium (see Recipes)
- 1x PBS (see Recipes)
- Blocking buffer (see Recipes)
- Antibody buffer (see Recipes)
Equipment
- Tweezers
- Nikon Eclipse TS100 microscope
- WaveFX spinning-disk confocal microscope (Quorum Technologies, Guelph, Canada) with an ORCA-flash4.0 digital camera with Gen II sCMOS image sensor
Software
- Metamorph Image Analysis Software, Molecular Devices
- Imaris software 8.0.2 (Bitplane)
Procedure
- Seeding cells on cover glasses
- Grow LLC-PK1 cells in a T75 flask with culture medium (low glucose DMEM supplemented with 10 % FBS and 1% Pen-Strep, see Recipes) in an incubator with 5% CO2. Use flask of LLC-PK1 cells grown to about 90% confluence. Rinse with 5-8 ml PBS and add 1-2 ml trypsin according to standard cell culture procedures. Place back into incubator for about 10 min, until cells are floating. Note that details of culturing have to be adjusted for specific cell lines.
- While cells are in trypsin, place autoclaved coverglass in multi-well plates using sterile tweezers.
- When cells are floating, resuspend them in 8 ml fresh medium, mix well and seed an equal number of cells onto coverglasses. Use 2 x 105-5 x 105/ml cells in 1 ml medium/well (12-well plate).
- Gently shake the culture dish to evenly distribute the cells and place into culture incubator.
- Grow cells to 100% confluency (about 48 h). Of note, we found that expression of Cldn-2 is low in subconfluent cell cultures and increases as the cells become confluent (Amoozadeh et al., 2018). Therefore, it is important to allow the cells to become fully confluent and generate mature TJs, before performing the immunostaining (see Figure 1).
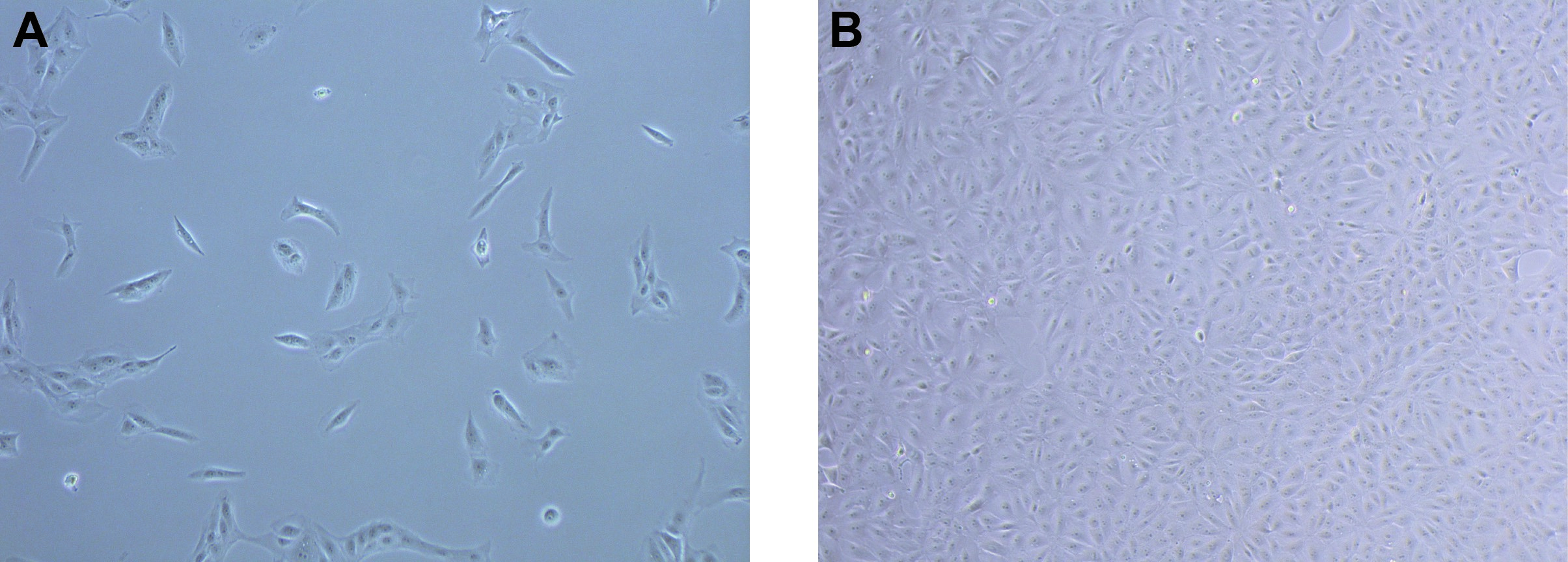
Figure 1. Subconfluent (A) and confluent (B) LLC-PK1 cultures. Phase contrast images of live cells grown on cover glasses were obtained using a Nikon eclipse TS100 microscope (10x objective).
- Fixation
- Treat cells according to experimental needs, e.g., with cytokines. At the end of the treatment, aspirate the medium and quickly rinse with 1x PBS. The steps after rinsing do not need to be performed under sterile conditions. The multi-well dish used for culturing cells can be used for the staining protocol under non-sterile conditions.
- Aspirate PBS and immediately fix with 0.5-1 ml methanol (stored at -20 °C) for 5 min. Make sure the methanol fully covers the cover glasses. These 2 steps should be done as fast as possible.
- Wash with 1x PBS 5 times. After this step the samples can be stored overnight at 4 °C if needed. However, we prefer to proceed to the next steps immediately. Longer storage is not recommended.
- Blocking, staining and mounting
- Block with 0.5-1 ml blocking buffer in PBS for 1 h at room temperature. Make sure the buffer fully covers the cover glasses.
- Primary antibody incubation:
- Dilute antibody 1:100 in antibody buffer.
- To reduce antibody use, place 50 μl of diluted antibody on parafilm and place the cover glass with cells facing the drop. Avoid bubbles and don’t let cover glass dry out. Incubate for 1h at room temperature.
- Non-specific secondary antibody binding control: use one of the cover glasses to demonstrate specificity of the secondary antibody. Omit primary antibody but do all other steps similarly.
- Control sample for primary antibody specificity: use samples where Cldn-2 was silenced or knocked down (Dan et al., 2019).
- Dilute antibody 1:100 in antibody buffer.
- At the end of the incubation use tweezers to place cover glasses back into the same culture dish turning the cells upside. Wash with 1x PBS 5 times.
- Secondary antibody incubation:
- Dilute secondary antibody (1:1,000) and DAPI (1:5,000) in antibody buffer.
- Add 500 μl antibody solution to the cover glasses.
- Incubate for 1h at room temperature in the dark.
- Dilute secondary antibody (1:1,000) and DAPI (1:5,000) in antibody buffer.
- Wash with 1x PBS 5 times over 15 min.
- Label microscope slides to identify samples. Place a drop of fluorescent mounting medium on a microscope slide. Lift up cover glasses using a tweezer and touch the edge to a paper towel to remove excess fluid. Place stained cover glass onto the drop of fluorescent mounting medium with cells facing down. Avoid air bubbles. Aspirate access fluid.
- Let the fluorescent mounting medium dry for at least 30 min before imaging or storing. Store dried slides in the dark at 4 °C.
- Visualization using confocal microscopy
- Set up the confocal microscope
- Settings (e.g., laser, objective etc) will vary depending on your experiment. We use a 63x objective, which was chosen to give the desired resolution and size of the imaged field.
- Chose the excitation/emission settings for the fluorophore of your secondary antibody (Alexa488 or 555 in our experiments) and DAPI.
- Select exposure time to avoid image saturation. Obtain z-stack sections at both wavelength through the whole sample by defining the top and bottom focal plane positions and a step size of around 2 μm (12-16 focal planes).
- Settings (e.g., laser, objective etc) will vary depending on your experiment. We use a 63x objective, which was chosen to give the desired resolution and size of the imaged field.
- A single plane at the level of the TJs of the stack can be used for display. However, LLC-PK1 cells in a monolayer often have slight height differences and can also form domes. This causes the TJs in different areas of a visual field to be at slightly different focal planes, making some areas to be out of focus. Further, TJ-localized and intracellular Cldn-2 are also in different focal planes. To visualize all Cldn-2 within the cells, use Metamorph to generate a Maximum Intensity Projection from the z-stack (see Figure 2 for an example). This process uses the brightest pixel from each layer and displays its intensity value in the final image, thus allowing visualization of a 3D image collapsed into a 2D picture. The processing must be disclosed in the manuscript.

Figure 2. Typical Claudin-2 staining in LLC-PK1 cells. LLC-PK1 cells were fixed with methanol, and Cldn-2 stained as described in the protocol. The nuclei were counterstained with DAPI. Z-stacks were obtained using confocal microscopy for each staining, maximal intensity projection pictures were generated using the Metamorph software. The images obtained for the two wavelengths were combined. The bar represents 10 μm.
- Set up the confocal microscope
Data analysis
Image processing and data analysis:
- 3D reconstruction In Imaris
To quantify intensity of total Cldn-2 staining, we use the Imaris software for a 3D reconstruction that can then be used for quantification. Of note, the above generated Maximum Intensity Projection images cannot be used for quantification. For basic familiarity with the Imaris software, users are referred to free online tutorials at http://www.bitplane.com/learning.- Upload z-stack images to the Imaris 8.0.2 software.
- Click ‘Edit/Add Channels’ and select the appropriate image.
- In the main menu, Select “Surpass/Add new surfaces” and click ‘Create Surface’.
- Select the appropriate ‘Surface Area Detail Level’. Select ‘Absolute Intensity’ for thresholding.
Note: Both settings are automatically calculated, however, users can alter these parameters. Keep these settings constant throughout image analysis. - Click ‘Finish’ to complete 3D reconstruction of the stack.
- Create a 3D spot object for quantifying the number of nuclei by adding a new spot object.
- Select ‘Different Spot Size’.
- For ‘Source Channel’, select the DAPI channel.
- For ‘Spot Detection’, enter ‘Estimated XY Diameter and Z Diameter’ of the nuclei.
- Manually adjust the ‘Spot Quality’ to include any undetected nuclei.
- Select ‘Intensity Max = masked channel number of the cell outline’ to restrict extra nuclei spots. Note that the values for all the described parameters depend on the properties of the images acquired and will differ in specific images.
- Adjust ‘Spot Region Threshold’ and ensure all nuclei spots are detected.
- Click ‘Finish’ to complete the detection of nuclei spots.
- Select the required statistic parameters (volume and total number of spots) and export the .csv files. These can be opened using a graph program.
- Data analysis
- Use a graph program (Excel or GraphPad) to calculate the sum of volumes of each surface (μm3).
- Express the data as total surface volume per cell (nuclei spots).
Notes
- Cell line choice: The LLC-PK1 cell line we use originated from the European Collection of Authenticated Cell Cultures. This LLC-PK1 clone was originally also available at ATCC (ATCC® CRL-1392TM) but has since been discontinued. ATCC distributes another LLC-PK1 cell line, however, we found that LLC-PK1 cells from various sources differ in their claudin-2 expression levels. Thus, prior to choosing a cell line for the project, good expression of the claudin of interest should be verified.
- Cell counting and confluency: TJs develop as the layer becomes confluent and therefore it is recommended that fully confluent layers are used. Claudin-2 total expression is low in subconfluent cells and increases as confluency develops. Confluency should be kept constant when comparing different conditions. We found that when the same conditions are consistently applied during culture and seeding, cell numbers can be kept constant. Thus, after establishing the protocol, routine cell counting is not necessary. However, experimental manipulations might alter cell growth and thus confluency should be routinely checked and considered as an important variable.
- Throughout the staining process, drying of the cover glasses must be avoided.
- The protocol described here is used to stain claudin-2. The same protocol can be used to visualize other junction proteins in any epithelial cell lines. We found that the mode of fixation is key for good preservation and visualization of junctional proteins. We had good results using methanol when staining claudins. For occludin we used acetone fixation. For these fixation methods additional permeabilization is not needed. For a detailed analysis of the various fixation methods used for junctional proteins, please refer to Buckley et al., 2018.
- The quantification described is not specific for TJ-localized claudin-2 but reflects total claudin-2 abundance, including cytosolic protein. In our experiments the quantification was used for a comparison between control and TNF-α-treated samples. The data were expressed as staining/cell, relative to the control (untreated) to demonstrate fold changes. In this case, the control, untreated samples serve as reference. For other type of comparisons (e.g., comparison of untreated samples), a reference protein, e.g., another TJ protein should be used (e.g., costaining for occludin or ZO-1 and expressing ratio of claudin-2 to occludin staining). The choice of the reference however can pose difficulties for cytokine-treated samples, as we found that expression of all TJ proteins we tested (e.g., other claudins, occludin), as well as the adherens junction protein E-cadherin were all altered by TNFα (Amoozadeh et al., 2015). Thus, before choosing a reference, it is important to show that it is unaltered by the treatment. Finally, quantification should be verified by other quantitative methods, such as Western blotting.
Recipes
- Cell culture media
Dulbecco's Modified Eagle Medium (D-MEM), low glucose, with L-glutamine and 110 mg/L sodium pyruvate was supplemented with 10% Fetal Bovine Serum (FBS) and sterile-filtered 1% Penicillin-Streptomycin - 1x PBS
Dilute from 10x PBS: add 50 ml 10x PBS to 450 ml distilled H2O - Blocking buffer
3% BSA in PBS- Mix 1.5 g BSA in 50 ml PBS
- Store short term (1 day) at 4 °C
- Mix 1.5 g BSA in 50 ml PBS
- Antibody buffer
0.3% BSA in PBS- Mix 0.15 g BSA in 50 ml PBS
- Store short term (1 day) at 4 °C
- Mix 0.15 g BSA in 50 ml PBS
Acknowledgments
Funding: Kidney Foundation of Canada, Canadian Institutes of Health Research (CIHR) grants PJT-149058 and MOP-142409.
This protocol was derived from: Amoozadeh et al., 2015 and 2018; Dan et al., 2019.
Competing interests
The authors declare that they have no financial or non-financial conflicts of interest.
References
- Amasheh, S., Meiri, N., Gitter, A. H., Schoneberg, T., Mankertz, J., Schulzke, J. D. and Fromm, M. (2002). Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci 115(Pt 24): 4969-4976.
- Amoozadeh, Y., Anwer, S., Dan, Q., Venugopal, S., Shi, Y., Branchard, E., Liedtke, E., Ailenberg, M., Rotstein, O. D., Kapus, A. and Szaszi, K. (2018). Cell confluence regulates claudin-2 expression: possible role for ZO-1 and Rac. Am J Physiol Cell Physiol 314(3): C366-C378.
- Amoozadeh, Y., Dan, Q., Xiao, J., Waheed, F. and Szaszi, K. (2015). Tumor necrosis factor-α induces a biphasic change in claudin-2 expression in tubular epithelial cells: role in barrier functions. Am J Physiol Cell Physiol 309(1): C38-50.
- Buckley, A. G., Looi, K., Iosifidis, T., Ling, K. M., Sutanto, E. N., Martinovich, K. M., Kicic-Starcevich, E., Garratt, L. W., Shaw, N. C., Lannigan, F. J., Larcombe, A. N., Zosky, G., Knight, D. A., Rigby, P. J., Kicic, A. and Stick, S. M. (2018). Visualisation of multiple tight junctional complexes in human airway epithelial cells. Biol Proced Online 20: 3.
- Dan, Q., Shi, Y., Rabani, R., Venugopal, S., Xiao, J., Anwer, S., Ding, M., Speight, P., Pan, W., Alexander, R. T., Kapus, A. and Szászi, K. (2019). Claudin-2 suppresses GEF-H1, RHOA, and MRTF, thereby impacting proliferation and profibrotic phenotype of tubular cells. J Biol Chem 294(42): 15446-15465.
- Dukes, J. D., Whitley, P. and Chalmers, A. D. (2012). The PIKfyve inhibitor YM201636 blocks the continuous recycling of the tight junction proteins claudin-1 and claudin-2 in MDCK cells. PLoS One 7(3): e28659.
- Enck, A. H., Berger, U. V. and Yu, A. S. L. (2001). Claudin-2 is selectively expressed in proximal nephron in mouse kidney. Am J Physiol Renal Physiol 281(5): F966-F974.
- Escaffit, F., Boudreau, F. and Beaulieu, J. F. (2005). Differential expression of claudin-2 along the human intestine: Implication of GATA-4 in the maintenance of claudin-2 in differentiating cells. J Cell Physiol 203(1): 15-26.
- Furuse, M., Fujita, K., Hiiragi, T., Fujimoto, K. and Tsukita, S. (1998a). Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141(7): 1539-1550.
- Furuse, M., Sasaki, H., Fujimoto, K. and Tsukita, S. (1998b). A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143(2): 391-401.
- González-Mariscal, L., Betanzos, A., Nava, P. and Jaramillo, B. E. (2003). Tight junction proteins. Prog Biophys Mol Biol 81(1): 1-44.
- Ikari, A., Watanabe, R., Sato, T., Taga, S., Shimobaba, S., Yamaguchi, M., Yamazaki, Y., Endo, S., Matsunaga, T. and Sugatani, J. (2014). Nuclear distribution of claudin-2 increases cell proliferation in human lung adenocarcinoma cells. Biochim Biophys Acta 1843(9): 2079-2088.
- Lu, R., Johnson, D. L., Stewart, L., Waite, K., Elliott, D. and Wilson, J. M. (2014). Rab14 regulation of claudin-2 trafficking modulates epithelial permeability and lumen morphogenesis. Mol Biol Cell 25(11): 1744-1754.
- Sakaguchi, T., Gu, X., Golden, H. M., Suh, E., Rhoads, D. B. and Reinecker, H. C. (2002). Cloning of the human claudin-2 5'-flanking region revealed a TATA-less promoter with conserved binding sites in mouse and human for caudal-related homeodomain proteins and hepatocyte nuclear factor-1α. J Biol Chem 277(24): 21361-21370.
- Tsukita, S., Tanaka, H. and Tamura, A. (2019). The claudins: from tight junctions to biological systems. Trends Biochem Sci 44(2): 141-152.
- Van Itallie, C. M. and Anderson, J. M. (2004). The molecular physiology of tight junction pores. Physiology (Bethesda) 19: 331-338.
- Van Itallie, C. M. and Anderson, J. M. (2014). Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol 36: 157-165.
- Venugopal, S., Anwer, S. and Szaszi, K. (2019). Claudin-2: roles beyond permeability functions. Int J Mol Sci 20(22). Doi.org/10.3390/ijms20225655.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Anwer, S. and Szászi, K. (2020). Immunofluorescent Staining of Claudin-2 in Cultured Kidney Tubular Cells. Bio-protocol 10(14): e3678. DOI: 10.21769/BioProtoc.3678.
- Dan, Q., Shi, Y., Rabani, R., Venugopal, S., Xiao, J., Anwer, S., Ding, M., Speight, P., Pan, W., Alexander, R. T., Kapus, A. and Szászi, K. (2019). Claudin-2 suppresses GEF-H1, RHOA, and MRTF, thereby impacting proliferation and profibrotic phenotype of tubular cells. J Biol Chem 294(42): 15446-15465.
Category
Biochemistry > Protein > Fluorescence
Cancer Biology > Inflammation > Cell biology assays
Cell Biology > Cell staining > Protein
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.