- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Assessment of Pathogen Effector Binding to Host Proteins by Surface Plasmon Resonance
Published: Vol 10, Iss 13, Jul 5, 2020 DOI: 10.21769/BioProtoc.3676 Views: 5290
Reviewed by: Chiara AmbrogioAbhinit NagarSneha Ray
Original research article
The authors used this protocol in:

Sep 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
The mechanisms of virulence and immunity are often governed by molecular interactions between pathogens and host proteins. The study of these interactions has major implications on understanding virulence activities, and how the host immune system recognizes the presence of pathogens to initiate an immune response. Frequently, the association between pathogen molecules and host proteins are assessed using qualitative techniques. As small differences in binding affinity can have a major biological effect, in vitro techniques that can quantitatively compare the binding between different proteins are required. However, these techniques can be manually intensive and often require large amounts of purified proteins. Here we present a simplified Surface Plasmon Resonance (SPR) protocol that allows a reproducible side-by-side quantitative comparison of the binding between different proteins, even in cases where the binding affinity cannot be confidently calculated. We used this method to assess the binding of virulence proteins (termed effectors) from the blast fungus Magnaporthe oryzae, to a domain of a host immune receptor. This approach represents a rapid and quantitative way to study how pathogen molecules bind to host proteins, requires only limited quantities of proteins, and is highly reproducible. Although this method requires the use of an SPR instrument, these can often be accessed through shared scientific services at many institutions. Thus, this technique can be implemented in any study that aims to understand host-pathogen interactions, irrespective of the expertise of the investigator.
Keywords: Surface Plasmon ResonanceBackground
As part of their colonization process, pathogens can deploy an array of molecules, termed effectors, inside the host cell (Win et al., 2012). Frequently, these effectors bind to host targets to modify them, and/or re-direct their activities, which ultimately allows pathogens to overcome host immune defenses and subvert host cell pathways in their favor (Dodds and Rathjen, 2010). On the other hand, plants and animals have evolved a set of diverse intracellular immune receptors (NLRs) that detect the presence of pathogen effectors (Jones et al., 2016). This can be by direct binding (Kourelis and van der Hoorn, 2018). This recognition event triggers host immune signaling and restricts pathogen growth. Therefore, understanding the association between effectors and their host targets, as well as between effectors and immune receptors, have major implications in the study of host-pathogen interactions.
The biology of pathogen effectors and their mechanisms of action can be investigated by a combination of methods including cell biology, molecular biology and biophysics (Varden et al., 2017). The biochemical study of the interactions between effectors and their targets often employs qualitative techniques such as Yeast-Two-Hybrid (Y2H) (Mukhtar et al., 2011; Weßling et al., 2014) and co-immunoprecipitation (co-IP) (Fujisaki et al., 2015; Dagdas et al., 2016). However, small differences in binding between effectors and host proteins can have major impacts in function (De la Concepcion et al., 2018). Therefore, techniques that can quantitatively determine the binding between an effector and a given host protein are increasingly being required.
Isothermal Titration Calorimetry (ITC) has been commonly used as the gold standard to measure interactions between pathogen effectors and host virulence targets (Dagdas et al., 2016; Maqbool et al., 2016). This technique has also been used to investigate the binding between effectors and immune receptors, and how this is translated into immune recognition (Zhang et al., 2017). However, in many cases, multiple allelic variants of both effectors and host proteins are involved in the virulence/immunity process (Zess et al., 2019), increasing the number of combinations to test and the labor-intensity of this approach. This, together with the requirement of relatively large amounts of purified proteins, can make the study of interactions by ITC impractical in some cases.
Surface Plasmon Resonance (SPR) has several advantages over ITC. First, the microfluidic nature of the technique allows the use of very small volumes of proteins at often nanomolar concentration, reducing the amount of purified protein required for the experiments compared with ITC. Also, as SPR is a high-throughput and automatable technique, multiple interactions and their respective controls can be tested at the same time under the same conditions, increasing the robustness and reproducibility of the data.
We have successfully used SPR to understand how direct binding of a domain from the rice NLR immune receptor Pik to an effector from the rice blast fungus (Magnaporthe oryzae), leads to immune recognition (Maqbool et al., 2015; De la Concepcion et al., 2018 and 2019). The rice blast effector AVR-PikD is recognized by the rice NLR Pikp via direct binding to the integrated Heavy Metal Associated (HMA) domain of the plant receptor (Maqbool et al., 2015; De la Concepcion et al., 2018). However, polymorphic variants of AVR-Pik that escape immune recognition are present in nature (Kanzaki et al., 2012; Bialas et al., 2018). Although these natural variants can also bind to the integrated HMA domain, they do so with lower affinity, resulting in lack of immune response (De la Concepcion et al., 2018 and 2019).
Although SPR can be used to calculate binding affinities and kinetics, this was not possible for some Pik-HMA/AVR-Pik combinations due to weak binding (De la Concepcion et al., 2018 and 2019). However, the SPR protocol presented here ranks the binding of different AVR-Pik variants to different alleles and mutants of the integrated HMA domain of the Pik receptor in the absence of precise quantification of binding affinities (expressed as equilibrium dissociation or KD values), allowing us to overcome these issues with respect to biological function (De la Concepcion et al., 2018 and 2019). Therefore, this method presents a quick way to screen and quantitatively rank interactions, which will be informative to understanding the biological implications of the interactions.
Although the protocol presented here has been optimized for the interaction between two proteins where one partner is immobilized on a Nitrilotriacetic Acid (NTA) chip through a hexa-histidine tag, it is equally applicable if any other form of capture is used. For example, effector-DNA interactions can be easily tested by immobilizing biotinylated DNA to a streptavidin (SA) chip (Stevenson et al., 2013).
Materials and Reagents
- MF-MilliporeTM Membrane Filter, 0.22 µm pore size (Merck, catalog number: GSWP04700 )
- Proteins of interest
The proteins to be tested must be purified and concentrated prior dilution into the running buffer. One of the proteins whose interaction wants to be tested must contain a histidine tag while the other requires no tag.
Notes:- Protein concentrations must be adjusted in every case: In this protocol we provide a particular example to which working protein concentrations have been optimized. As starting point, we recommend using two different protein concentrations (e.g., 50 nM and 500 nM). A range of dilutions could be tested in subsequent experiments.
- Consider which protein is to be tagged: Consider which protein will be immobilized on to the chip surface (ligand), and which one will be flowed over (analyte). Many factors can affect the choice for immobilization like purity, amount available, stoichiometry and theoretical response. Ideally both proteins could be produced tagged and untagged and both orientations tested. Whether the hexa-histidine tag is placed in the N-terminus or C-terminus of the protein should be also considered (e.g., if the proteins are predicted to bind near to the C-terminus, proteins should be attached to the chip in the N-terminus). It is also crucial that the untagged protein does not bind directly to the chip.
- HEPES (Melford, catalog number: H75030-1000.0 )
- NaCl (Merck, catalog number: 1064041000 )
- Tween® 20 (Merck, catalog number: P9416-100ML )
- NiCl2·6H2O (Merck, catalog number: 203866-5G )
- EDTA (VWR Chemicals, catalog number: 20302.26 0)
- NaOH (Merck, catalog number: 221465 )
- Running buffer (see Recipes)
- 0.5 mM NiCl2 (see Recipes)
- Regeneration solution (see Recipes)
Equipment
- BiacoreTM T200 SPR instrument (GE Healthcare, catalog number: 28975001 )
Note: This method can be used with different instruments: Although the method presented here has been implemented using a BiacoreTM T200 SPR instrument, it could be adapted to run with any SPR instrument. - BiacoreTM Sensor Chip Series S NTA (GE Healthcare, catalog number: 28994951 )
Note: Different brands of NTA chips can be used: We use GE Healthcare NTA chips. However, this method can use any compatible NTA chip. - Direct Detect® Infrared Spectrometer (Merck, catalog number: DDHW00010-WW )
Note: Different methods to measure the concentration of the proteins can be used: As some of the proteins we used in the example do not contain aromatic residues, we used a Direct Detect® Infrared Spectrometer to determine concentrations. However, other standard methods such as Bradford or absorbance at 280 nm can be used. Also, the concentration of the His-tagged proteins does not necessarily have to be known as their concentrations can be assessed by the response on binding to the chip.
Software
- BiacoreTM T200 evaluation software (GE Healthcare)
- R studio (R Core Development Team, 2018) with ggplot2 package (Wickham, 2016)
Procedure
For all experiments, dock an NTA chip into the SPR instrument and prime it with running buffer. Prior to docking, the NTA chip needs to equilibrate at room temperature for 10-30 min to prevent condensation. Once docked there are four flow cells available with a Biacore T200. In this experiment, use two of these flow cells, with flow cell 1 as the reference (FCref) without immobilize-d ligand and flow cell 2 as the test cell (FCtest). For all experiments, load the tubes with the appropriate amount of solution and placed in the rack as detailed in the BiacoreTM T200 Control Software.
Note: Leave the reference cell blank: In this protocol, the reference flow cell is blank and the analyte injected over both cells, this will reveal any non-specific binding of the analyte to the chip and will be subsequently subtracted from the final result. Ideally very little or no binding of the analyte to the chip surface should occur.
- Preparation of the ligand protein
The method uses a hexa-histidine tagged protein (Magnaporthe oryzae AVR-Pik, C-terminally tagged in our example), which is captured to the surface on a standard NTA chip (Figure 1). We purified the proteins as described in Maqbool et al. (2015) and De la Concepcion et al. (2018 and 2019).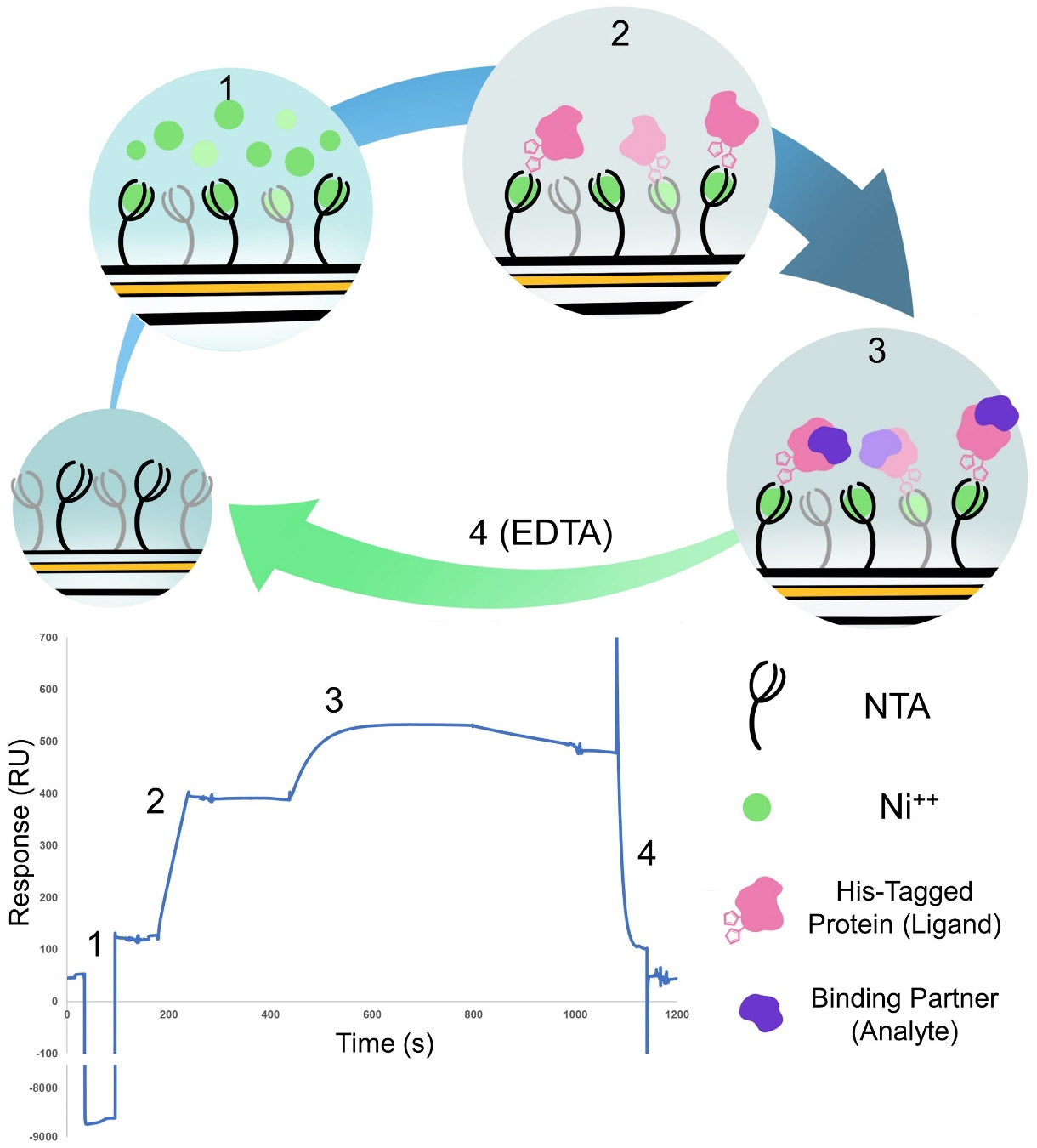
Figure 1. Cartoon representation and example result of the four steps of the binding experiment. (1) Activation of the NTA chip with nickel. (2) Binding of the his-tagged protein ligand to the chip. (3) Binding of the protein analyte. (4) Regeneration of the chip with EDTA to remove everything bound to start the cycle again. Steps 1 and 2 are only carried out on Flow cell 2 whereas steps 3 and 4 are carried out over both flow cells. The sensorgram shown represent a typical trace of the subtracted FC2-1 sensorgram.- Preparation of stock dilution of His-tagged (ligand) protein
The concentration of the protein that will be immobilized on the chip is measured and diluted in buffer (20 mM HEPES, 150 mM NaCl, pH 7.5) to obtain 2 ml of stock solution at 2 μM.
Note: The stock solution can vary depending on the experiment: The amount of stock solution we prepare in our example is larger than needed. This is because the proteins used in this example can be produce in large amounts, are stable, and can be kept on ice for a few days. If the protein to be tested is not produced in enough quantities and/or is not stable, we recommend preparing a fresh stock solution with a lower volume each time before running. - Preparation of working solution of ligand protein
From the stock solution, we take 25 μl and dilute it with 975 μl of running buffer to obtain a 50 nM dilution. The volume of the ligand solution will vary depending on the number of cycles that are set up, and will be indicated by the BiacoreTM T200 SPR control software.
- Preparation of stock dilution of His-tagged (ligand) protein
- Manual run to estimate the amount of protein immobilized onto the chip
Once the proteins that will serve as ligand are prepared, test the binding to the chip by performing a manual run. In our experiments, we aimed to use a final capture level of ligand of 250 ± 50 Response Units (RU) for each ligand to be tested.
Note: Ligand RUs value: In this experiment, we aim for a ligand immobilization level of ~250 RU as the value that gives a response sufficiently large enough to be measured even for weak binders. This value was selected by using the Rmax calculation in the data analysis section and this theoretical response obtained is related to the molecular weight of the two proteins and the amount of the His-tagged protein immobilized on the chip. Therefore, we first recommend determining the optimal level of immobilization for each ligand. If the level of binding of the ligand differs too much from the desired RU, it can be corrected by reducing or increasing the concentration of ligand in the working solution. If multiple His-tagged protein are to be tested, the concentration to use to capture on the chip will need to be optimized for each protein.- Select flow cells
Carry out the experiments using flow cells 1 and 2. The ligand is captured in flow cell 2 and the analyte is injected over both flow cells. The final result (sensorgram) is a subtraction of the response from flow cell 2 min cell 1 (FC2-1). - Activation of the chip
Inject 30 µl of 0.5 mM NiCl2 with a flow rate of 30 μl/min to activate the chip. This step only applies to flow cell 2. - Immobilization of the ligand
Inject 30 µl of C-terminally His-tagged AVR-Pik in flow cell 2 with a flow rate of 30 μl/min. Binding to the Ni2+ on the surface of the chip is recorded as (RU). An additional check should be carried out without activation with NiCl2 to ensure that the protein does not non-specific binding to the chip surface. - Regeneration
Regenerate the sensor chip by removing anything bound to the chip, with an injection of 30 μl of 0.35 M EDTA with a flow rate of 30 μl/min over both the flow cells.
- Select flow cells
- Preparation of the protein analyte
Once the manual run is completed and the binding of each ligand to the chip is around 250 ± 50 RUs, prepare the working dilutions of the protein analyte. In our case, we produced purified Pik-HMA domain as described in De la Concepcion et al. (2018 and 2019).- Preparation of stock dilution of analyte protein
After measuring the concentration of the protein, dilute it in buffer (20 mM HEPES, 150 mM NaCl, pH 7.5) to obtain 2 ml of stock solution at 2 μM as described above for the protein ligand. - Preparation of working solution of analyte protein
Make serial dilutions of the protein stock to obtain final concentrations of 4 nM, 40 nM and 100 nM. The total volume depends on the number of cycles and will be indicated by the BiacoreTM T200 SPR control software.
Note: Adjust working concentration of analyte: The working concentration of the analyte will vary in each experimental case depending the strength of interaction between proteins. We recommend starting with concentrations of 50 nM and 500 nM to find the appropriate concentrations to use. Ideally a top concentration should be used where the binding is saturated for the strongest interaction to be tested.
- Preparation of stock dilution of analyte protein
- Set up cycle parameters and Rmax run
For the experiment, dock the chip in the instrument and prime it with running buffer. We carried out the experiment at 25 °C but the samples are stored at 4 °C. We used a flow rate of 30 μl/min.
Note: Adjusting experimental temperature and flow rate: The optimal temperature and flow rate can vary depending on the proteins to be tested. The parameters used by this protocol are standard, but some proteins might require a different temperature and/or a higher contact time (achieved by a lower flow rate) for a successful interaction. Likewise, the time that the protein stock can be stored at 4 °C is protein-dependent. As a standard, we do not recommend freeze-thaw the protein aliquots (working with small, single-use protein aliquots is preferred).
Each experimental cycle consists of 4 steps described below and represented in Figure 1. This cycle can be repeated multiple times in an automated fashion using different concentrations of analyte tested over different ligands.
- Chip activation
As a first step, inject a solution of 0.5 mM NiCl2 with a flow rate of 30 μl/min over Flow cell 2 to activate the chip. - Ligand immobilization
After chip activation, inject the His tagged protein (ligand) to be tested (in our case C-terminally tagged AVR-Pik effector) over the flow cell 2 (FCtest). Sixty seconds are used as the injection time to achieve a desired response at this concentration. After the ligand has been immobilized buffer is flowed over FC1 and 2 to ensure any non-specific his tagged protein is removed and a stable baseline should be achieved prior to analyte injection. - Analyte injection
Once the ligand is bound to the chip surface, inject the test analyte at a given concentration (or buffer-only control) over both flow cells (FCref and FCtest). A contact time of 120 s is used as a standard to make sure the maximum concentration is (ideally) reaching the steady state. After injection of the protein, the system switches back to buffer only flow and the bound protein will start to dissociate. Generally, 120 s of buffer only is used to see this dissociation. This region of the sensorgram can be used to evaluate differences between analyte proteins as generally tighter interactions take longer to dissociate. - Chip regeneration
After the analyte has been injected in the flow cell, pass a regeneration solution of 0.35 M EDTA over both flow cells (FCref and FCtest) at a flow rate of 30 µl/min. This strips the Ni2+ from the chip, and the proteins bound to it. After this step, the response should return to similar levels to those prior to Step D1.
For each analyte, we set 3 replicates of each cycle at working concentration of 4 nM, 40 nM and 100 nM. In addition, two start up cycles were carried out using buffer only as the analyte. The total running time of 11 cycles is around 4 h for each ligand to be tested and once initiated it does not require further user intervention. Multiple runs involving different ligands and analytes can be stack together in a single run.
When the experiment is completed, the NTA chip can be removed from the instrument and stored in buffer at 4 °C until next use. The chip can be re-used multiple times for different experiments. Each time a chip is re-used the capture of nickel and his-tagged protein is checked. As long as this is what is expected the chip can be used again. On the rare occasions a chip fails it is obvious as the nickel and his tagged protein is no longer captured.
Data analysis
To compare the results between multiple cycles, the data must be normalized by correcting the different capture levels and according to the molecular weight of the different proteins. To do this, calculate the theoretical maximal binding at saturation of the analyte (Rmax) value for each run (Buckle, 2001; Majka and Speck, 2007). This value is measured in Response Units (RU), which is how binding events are recorded in SPR. This is calculated following the equation:
Mw is the molecular mass of the protein bound to the chip (ligand) and the protein flowed over the chip (analyte). This is corrected with the stoichiometry of the binding between the proteins and the amount of ligand immobilized on to the chip surface measured as Response Units (RU).
Note: The stoichiometry of the binding will affect to the final result: For example, if the binding stoichiometry is 2:1 analyte: ligand, the final %Rmax could be higher than 100% compared to assuming a 1:1 binding. If the stoichiometry is not known by other techniques, we recommend assuming a 1:1 binding and correct if the final result indicates otherwise.
Once we establish the Rmax for each run, we can express the level of binding as the percentage of Rmax calculated as follows: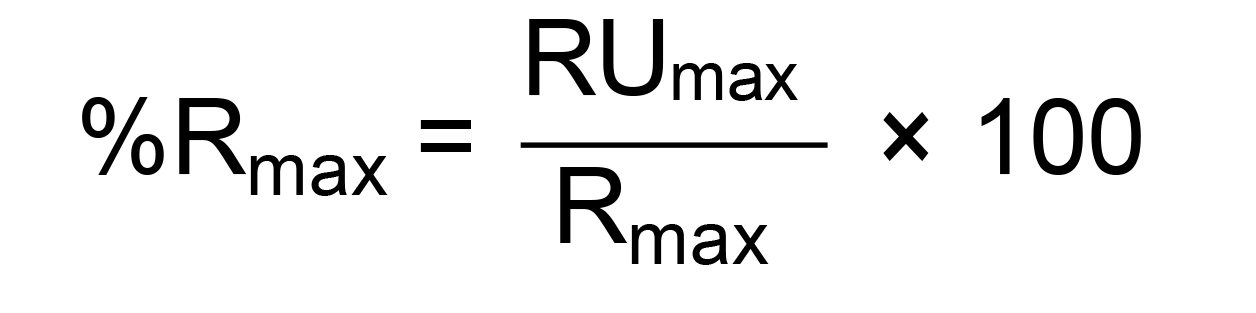
Where RUmax is the binding response measured immediately after the end of the injection of the analyte and expressed in Response Units (RU).
In the case of the results for the binding between Pikm-HMA and AVR-PikD (1:1 binding stoichiometry) presented in De la Concepcion et al. (2018 and 2019), calculations were as follows (Table 1):
Table 1. Example of binding values obtained for RMax calculation extracted from De la Concepcion et al. (2018)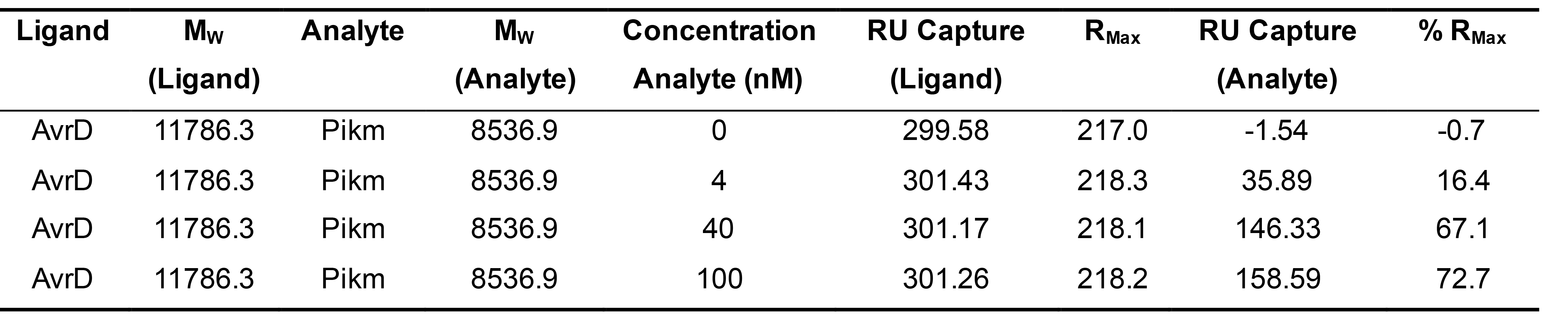
For visualization, export the SPR data and plot it using R v3.4.3 (https://www.r-project.org/) and the function ggplot2 (Wickham, 2009). SPR box plot graphs as presented in De la Concepcion et al. (2019) can be generated as follows:
- Preparation of dataset
We first prepare the dataset to generate the graph. We assigned a sample name and a position in the graph corresponding to the effector used as ligand (1, 2, 3 and 4 for AVR-PikD, AVR-PikE, AVR-PikA and AVR-PikC, respectively). We annotated the analyte concentrations (4, 40 and 100 nM) for each dataset as we generated their respective graphs separate. Rmax corresponds to the value calculated as presented above. Data belonging to each biological replicate is ranked with 1, 2 and 3, respectively. And the two analytes to be compared (Wild-type Pikp-HMA and Pikp-HMANK-KE in this case) are classified as A or B in HMA.Sample Effector Conc Rmax Replica HMA NameA 1 40 67 1A NameB 1 40 66.6 1B - Generation of the box plots
- We attached the dataset and defined the factors in the X and Y axis as:
> graph <- ggplot(dataset, aes(x=factor(Effector), y=Rmax)) - Then we defined the colour for each analyte and for the different biological replicas:
> colori2=c("#8faadc", "#c9a9ff")
> colori3=c("#ff85ff", "#00b4b5", "#f8f300") - The aestethic parameters for the graph are defined as follows:
Graph + geom_boxplot(aes (x=factor(Effector), y=Rmax, fill=factor(HMA)), position=position_dodge(width=0.9), lwd=0.3, fatten= 4, outlier.shape = 21, outlier.size = 0.7, outlier.stroke=0) + scale_fill_manual(values=colori2) + theme_bw() + theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(), panel.background = element_rect(fill = "transparent"), rect = element_rect(fill = "transparent")) + scale_y_continuous(breaks=seq(0,70,10), limits=c(0,70)) + geom_point(aes (x=factor(Effector), y=Rmax, fill=factor(HMA), colour=factor(Replica)), position=position_jitterdodge(jitter.width=0.7, jitter.height=0, dodge.width=0.9), size=0.7, shape=16) + scale_colour_manual(values = colori3) - As a final step, we produced the graph image with the following command:
ggsave('P-NK Rmax 35x3.png', width = 3.5, height = 3, dpi = 300, bg = "transparent") - The generated figure will then look similar to the graph presented below (Figure 2):
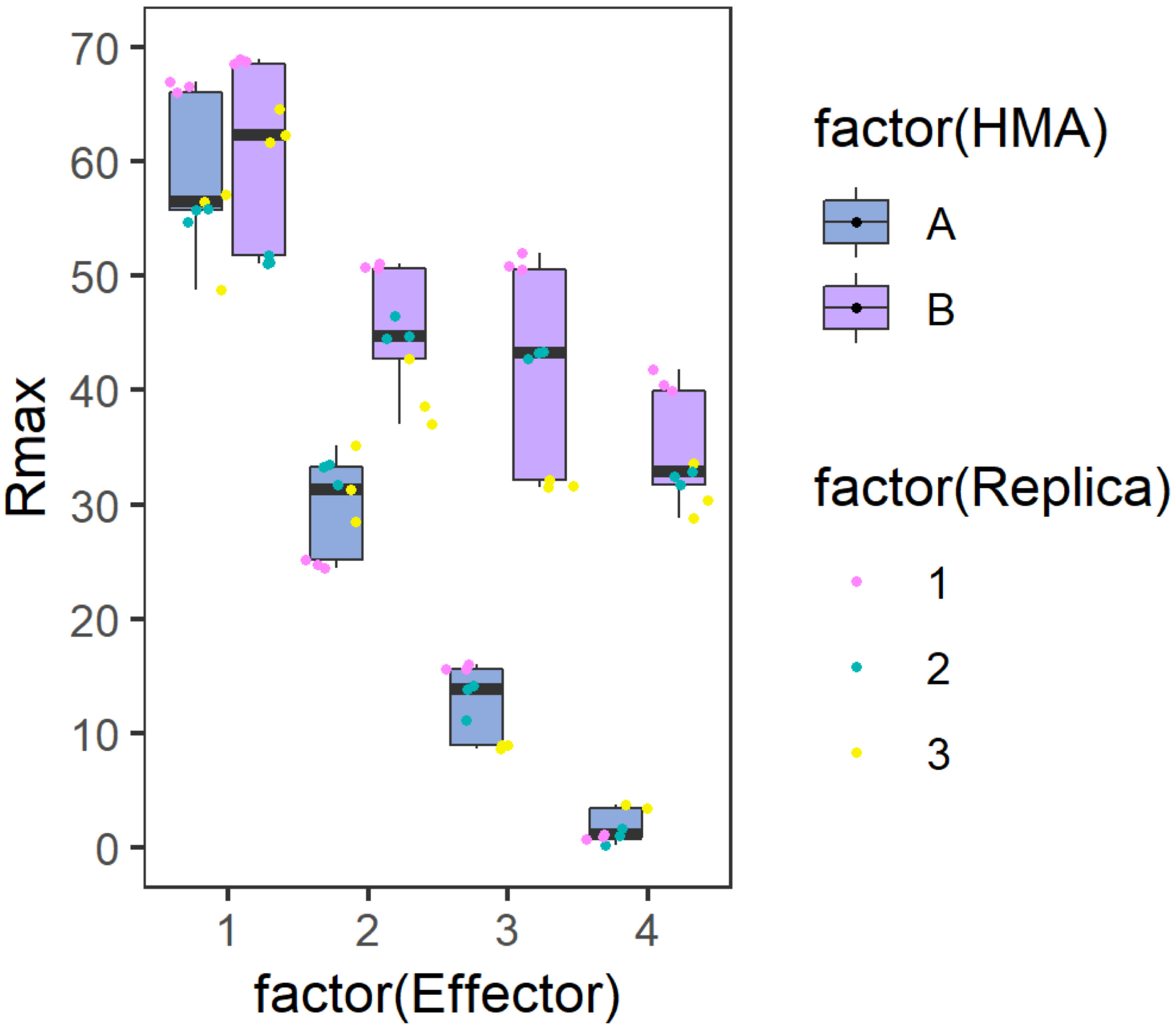
Figure 2. Example graph generated to represent Rmax results. Side-by-side comparison box plot graph for the binding of each Analyte (HMA A or B) to the different Ligands (Effector). The position of the values for each ligand are distributed on the X axis while the calculated %Rmax value in represented on the Y axis. The centre line represents the median, the box limits are the upper and lower quartiles, the whiskers extend to the largest value within Q1 - 1.5 × the interquartile range (IQR) and the smallest value within Q3 + 1.5 × IQR. All the data points are represented as dots with distinct colours for each biological replicate (with three technical replicates within each biological replicate). The graph has been generated using the results presented in De la Concepcion et al. (2019).
- We attached the dataset and defined the factors in the X and Y axis as:
Recipes
Prepare all solutions using ultrapure water and analytical grade reagents. Filter all solutions with a 0.22 µm pore size filter prior use. Solutions can be stored at room temperature.
Note: Running buffer should be modified according to the protein investigated: The buffer presented in this protocol has been optimized for a particular subset of proteins. Running buffers should be based on the buffer condition were the protein of interest is stable. It is also important to include any component necessary for the interaction, e.g., presence of metals in the solution. As a good starting point we recommend using a standard concentration of 150 mM NaCl.
- Running buffer
20 mM HEPES pH 7.5
860 mM NaCl
0.1% Tween® 20 - 0.5 mM NiCl2
- Prepare 10 ml of 100 mM NiCl2 stock solution by dissolving 0.24 g of NiCl2·6H2O in water
- 0.5 mM NiCl2 working solution is prepared before use by diluting 50 μl of stock solution into 10 ml of water
- Regeneration solution
- 0.35 M EDTA prepared by dissolving 6.5 g EDTA in 50 ml of water
- pH must be adjusted to 8.0 with NaOH for EDTA solubilization
Acknowledgments
This protocol has been developed and implemented in the John Innes Centre Biophysical Analysis Platform. We thank Julia Mundy (julia.e.mundy@gmail.com) for art work in Figure 1.
Research in the Banfield lab is supported by the BBSRC (grants BB/J004553, BB/P012574, BB/M02198X), the ERC (proposals 743165, 669926) and the John Innes Foundation. J.C. was supported by the John Innes Foundation/John Innes Centre/The Sainsbury Laboratory/Earlham Institute Rotation Program. This protocol is derived from De la Concepcion et al. (2018 and 2019).
Competing interests
The authors declare no competing interests.
References
- Bialas, A., Zess, E. K., De la Concepcion, J. C., Franceschetti, M., Pennington, H. G., Yoshida, K., Upson, J. L., Chanclud, E., Wu, C. H., Langner, T., Maqbool, A., Varden, F. A., Derevnina, L., Belhaj, K., Fujisaki, K., Saitoh, H., Terauchi, R., Banfield, M. J. and Kamoun, S. (2018). Lessons in effector and NLR biology of plant-microbe systems. Mol Plant Microbe Interact 31(1): 34-45.
- Buckle, M. (2001). Surface plasmon resonance applied to DNA-protein complexes. In: DNA-Protein Interactions: Principles and Protocols. Moss, T. (Ed.). Totowa, NJ: Humana Press, pp. 535-546.
- Dagdas, Y. F., Belhaj, K., Maqbool, A., Chaparro-Garcia, A., Pandey, P., Petre, B., Tabassum, N., Cruz-Mireles, N., Hughes, R. K., Sklenar, J., Win, J., Menke, F., Findlay, K., Banfield, M. J., Kamoun, S. and Bozkurt, T. O. (2016). An effector of the Irish potato famine pathogen antagonizes a host autophagy cargo receptor. eLife 5: e10856.
- De la Concepcion, J. C., Franceschetti, M., Maqbool, A., Saitoh, H., Terauchi, R., Kamoun, S. and Banfield, M. J. (2018). Polymorphic residues in rice NLRs expand binding and response to effectors of the blast pathogen. Nat Plants 4(8): 576-585.
- De la Concepcion, J. C., Franceschetti, M., MacLean, D., Terauchi, R., Kamoun, S. and Banfield, M. J. (2019). Protein engineering expands the effector recognition profile of a rice NLR immune receptor. eLife 8: e47713.
- Dodds, P. N. and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant–pathogen interactions. Nature Reviews Genetics 11: 539.
- Fujisaki, K., Abe, Y., Ito, A., Saitoh, H., Yoshida, K., Kanzaki, H., Kanzaki, E., Utsushi, H., Yamashita, T., Kamoun, S. and Terauchi, R. (2015). Rice Exo70 interacts with a fungal effector, AVR-Pii, and is required for AVR-Pii-triggered immunity. Plant J 83(5): 875-887.
- Jones, J. D. G., Vance, R. E. and Dangl, J. L. (2016). Intracellular innate immune surveillance devices in plants and animals. Science 354(6316): aaf6395.
- Kanzaki, H., Yoshida, K., Saitoh, H., Fujisaki, K., Hirabuchi, A., Alaux, L., Fournier, E., Tharreau, D. and Terauchi, R. (2012). Arms race co-evolution of Magnaporthe oryzae AVR-Pik and rice Pik genes driven by their physical interactions. Plant J 72(6): 894-907.
- Kourelis, J. and van der Hoorn, R. A. L. (2018). Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 30(2): 285-299.
- Majka, J. and Speck, C. (2007). Analysis of protein-DNA interactions using surface plasmon resonance. Adv Biochem Eng Biotechnol 104: 13-36.
- Maqbool, A., Saitoh, H., Franceschetti, M., Stevenson, C. E., Uemura, A., Kanzaki, H., Kamoun, S., Terauchi, R. and Banfield, M. J. (2015). Structural basis of pathogen recognition by an integrated HMA domain in a plant NLR immune receptor. eLife 4: 08709.
- Maqbool, A., Hughes, R. K., Dagdas, Y. F., Tregidgo, N., Zess, E., Belhaj, K., Round, A., Bozkurt, T. O., Kamoun, S. and Banfield, M. J. (2016). Structural basis of host autophagy-related protein 8 (ATG8) binding by the Irish potato famine pathogen effector protein PexRD54. J Biol Chem 291(38): 20270-20282.
- Mukhtar, M. S., Carvunis, A.-R., Dreze, M., Epple, P., Steinbrenner, J., Moore, J., Tasan, M., Galli, M., Hao, T., Nishimura, M. T., Pevzner, S. J., Donovan, S. E., Ghamsari, L., Santhanam, B., Romero, V., Poulin, M. M., Gebreab, F., Gutierrez, B. J., Tam, S., Monachello, D., Boxem, M., Harbort, C. J., McDonald, N., Gai, L., Chen, H., He, Y., Vandenhaute, J., Roth, F. P., Hill, D. E., Ecker, J. R., Vidal, M., Beynon, J., Braun, P. and Dangl, J. L. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333(6042): 596-601.
- R Core Development Team. (2018). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
- Stevenson, C. E. M., Assaad, A., Chandra, G., Le, T. B. K., Greive, S. J., Bibb, M. J. and Lawson, D. M. (2013). Investigation of DNA sequence recognition by a streptomycete MarR family transcriptional regulator through surface plasmon resonance and X-ray crystallography. Nucleic Acids Res 41(14): 7009-7022.
- Varden, F. A., De la Concepcion, J. C., Maidment, J. H. and Banfield, M. J. (2017). Taking the stage: effectors in the spotlight. Current Opinion in Plant Biology 38: 25-33.
- Weßling, R., Epple, P., Altmann, S., He, Y., Yang, L., Henz, Stefan R., McDonald, N., Wiley, K., Bader, Kai C., Gläßer, C., Mukhtar, M. S., Haigis, S., Ghamsari, L., Stephens, Amber E., Ecker, Joseph R., Vidal, M., Jones, Jonathan D. G., Mayer, Klaus F. X., Ver Loren van Themaat, E., Weigel, D., Schulze-Lefert, P., Dangl, Jeffery L., Panstruga, R. and Braun, P. (2014). Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 16(3): 364-375.
- Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. Springer International Publishing.
- Win, J., Chaparro-Garcia, A., Belhaj, K., Saunders, D. G., Yoshida, K., Dong, S., Schornack, S., Zipfel, C., Robatzek, S., Hogenhout, S. A. and Kamoun, S. (2012). Effector biology of plant-associated organisms: concepts and perspectives. Cold Spring Harb Symp Quant Biol 77: 235-247.
- Zess, E. K., Jensen, C., Cruz-Mireles, N., De la Concepcion, J. C., Sklenar, J., Stephani, M., Imre, R., Roitinger, E., Hughes, R., Belhaj, K., Mechtler, K., Menke, F. L. H., Bozkurt, T., Banfield, M. J., Kamoun, S., Maqbool, A. and Dagdas, Y. F. (2019). N-terminal beta-strand underpins biochemical specialization of an ATG8 isoform. PLoS Biol 17(7): e3000373.
- Zhang, Z. M., Ma, K. W., Gao, L., Hu, Z., Schwizer, S., Ma, W. and Song, J. (2017). Mechanism of host substrate acetylation by a YopJ family effector. Nat Plants 3: 17115.
Article Information
Copyright
![]() Franceschetti et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Franceschetti et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Franceschetti, M., Banfield, M. J., Stevenson, C. E. M. and De la Concepcion, J. C. (2020). In vitro Assessment of Pathogen Effector Binding to Host Proteins by Surface Plasmon Resonance. Bio-protocol 10(13): e3676. DOI: 10.21769/BioProtoc.3676.
- De la Concepcion, J. C., Franceschetti, M., MacLean, D., Terauchi, R., Kamoun, S. and Banfield, M. J. (2019). Protein engineering expands the effector recognition profile of a rice NLR immune receptor. eLife 8: e47713.
Category
Biochemistry > Protein > Quantification
Molecular Biology > Protein > Protein-protein interaction
Plant Science > Plant immunity > Host-microbe interactions
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.