- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for Peptide Synthesis on Spectrally Encoded Beads for MRBLE-pep Assays
Published: Vol 10, Iss 13, Jul 5, 2020 DOI: 10.21769/BioProtoc.3669 Views: 6483
Reviewed by: Samantha E. R. DundonLouise Jane WalportArunaksharan Narayanankutty
Original research article
The authors used this protocol in:

Jul 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Every living cell relies on signal transduction pathways comprised of protein-protein interactions (PPIs). In many cases, these PPIs are between a folded protein domain and a short linear motif (SLiM) within an unstructured region of a protein. As a result of this small interaction interface (3-10 amino acids), the affinities of SLiM-mediated interactions are typically weak (Kds of ~1-10 µM), allowing physiologically relevant changes in cellular concentrations of either protein partner to dictate changes in occupancy and thereby transmit cellular signals. However, these weak affinities also render detection and quantitative measurement of these interactions challenging and labor intensive. To address this, we recently developed MRBLE-pep, a technology that employs peptide libraries synthesized on spectrally encoded hydrogel beads to allow multiplexed affinity measurements between a protein and many different peptides in parallel. This approach dramatically reduces both the amount of protein and peptide as well as the time required to measure protein-peptide affinities compared to traditional methods. Here, we provide a detailed protocol describing how to: (1) functionalize polyethylene glycol diacrylate (PEG-DA) MRBLE beads with free amine groups, (2) synthesize peptide libraries on functionalized MRBLEs, (3) validate synthesized peptide sequences via MALDI mass spectrometry and quantify evenness of peptide coverage on MRBLEs, (4) use MRBLE-bound peptide libraries in multiplexed protein binding assays, and (5) analyze binding data to determine binding affinities. We anticipate that this protocol should prove useful for other researchers seeking to use MRBLE-pep in their own laboratories as well as for researchers broadly interested in solid-phase peptide synthesis and protein-protein binding assay development.
Background
Cellular signal transduction pathways rely on protein-protein interactions (PPIs). The majority of these interactions are mediated by short linear motifs (SLiM) in disordered regions of proteins and a globular domain on another protein (Neduva and Russell, 2006; Dinkel et al., 2016). Low affinities and prevalent regulation by post-translation modifications have left this type of interaction relatively unexplored. Screening methods like yeast-2-Hybrid (Y2H), phage display (PD) and protein microarrays (PMA) have proven successful for identifying SLiM-based interactions (Mu et al., 2014; Meyer et al., 2018; Ueki et al., 2019); however, they do not return quantitative information about binding affinities. In addition, these methods cannot report negative information (e.g., mutations that ablate PPIs) and in Y2H and PD do not easily allow incorporation of modified or unnatural amino acids. Conversely, current methods designed specifically to measure binding affinities, such as ITC, SPR and fluorescence polarization, are low throughput and require large amounts of reagents (Fodor et al., 1991; Gfeller et al., 2011; Zhou, 2012; Tinti et al., 2013; Ivarsson and Jemth, 2019). An ideal workflow for identification and characterization of SLiM-based interactions would allow both high-throughput identification of interesting PPIs and medium-throughput characterization of their binding affinities.
The recently published MRBLE-pep technology allows the measurement of binding affinities for many peptides in parallel, thereby facilitating protein-SLiM interaction characterization (Nguyen et al., 2019). In line with one-bead-one-compound libraries, the method synthesizes peptides directly on beads via solid-phase peptide synthesis to generate one-code-one-peptide libraries (Lam et al., 1991 and 1997; Nguyen et al., 2016, 2017 and 2019). However, unlike standard one-bead-one-compound libraries which require downstream isolation of bound beads and either mass spectrometry, Edman degradation or sequencing to identify bound analytes of interest, MRBLE-pep relies on libraries of peptides synthesized directly on spectrally encoded beads with a 1:1 relationship between the sequence of the displayed peptide and the embedded spectral codes (Brown et al., 2010; Liu et al., 2011; MacConnell et al., 2017; Nguyen et al., 2019). These spectrally encoded beads (MRBLEs, for Microspheres with Ratiometric Barcode Lanthanide Encoding) are generated by embedding lanthanide nanoparticles (LNP) ratiometrically into polyethylene glycol diacrylate (PEG-DA), as described previously. Each LNP species emits light in a series of narrow and well-separated spectral bands upon excitation with deep UV light, allowing codes to be read out by imaging beads at ~9 different wavelengths and using linear unmixing to determine the ratiometric combination of LNP concentrations most likely to have produced the observed spectra and thus the embedded spectral code (Gerver et al., 2012; Nguyen et al., 2016 and 2017; Harink et al., 2019).
Bead-based assays are extremely useful for multiplexed measurements of protein-peptide affinities and have the potential to directly quantify affinities using small amounts of material. However, successful binding assays depend critically on considerations of reagent concentration, binding equilibria and detection of the binding partners. Moreover, it is essential that binding assays take place at appropriate concentrations. Therefore, high-quality measurements depend critically on a variety of additional technical details, including robust and even functionalization of beads, gentle yet thorough mixing of beads during functionalization and binding assays, careful consideration of peptide density at the bead surface, and the number of beads added to each reaction. Here, we provide a detailed protocol describing best practices for: (1) functionalization of MRBLEs prior to peptide synthesis, (2) solid-phase peptide synthesis directly on functionalized MRBLEs, (3) peptide sequence validation and peptide density measurements, (4) MRBLE-pep protein binding assays, and (5) data analysis. While these results are presented here in the context of the MRBLE-pep assay, we anticipate that this detailed protocol may provide useful information for researchers optimizing any bead-bound binding assay.
Materials and Reagents
- Quartz Coverslip, Square 1" x 1" (Electron Microscopy Sciences, catalog number: 72256-02 )
- Quartz Microscope Slide 75 x 25 mm ~1 mm (Electron Microscopy Sciences, catalog number: 72250-03 )
- Deep Well Plates (Life Science, Axygen, catalog number: P-DW-20-C-S )
- Eppendorf 200 μl tubes (Eppendorf, catalog number: 00 30124707 )
- Eppendorf 5 ml tubes (Eppendorf, catalog number: 00 30119401 )
- Pipette Tips for Multichannel, ClipTip 1250, filter reload, sterile (Thermo Fisher Scientific, catalog number: 94420818 )
- Pipette Tips for Repeater (Eppendorf, Combi Tips advanced, 5 ml, catalog number: 00 30089456 ; 25 ml, catalog number: 00 30089472 ; 50 ml, catalog number: 00 30089480 )
- PP-Reactor 2 ml with PTFE frit (Biotage, catalog number: V020TF051 )
- Inert Gas Cover Plate, 48 x 2 ml reactor vials (MultiSynTech, catalog number: Z002IC048 )
- N,N Dimethylformamide (DMF) (Certified ACS) (Fisher, catalog number: D119-4 )
- Dichloromethane (DCM) (GR ACS) (Millipore, catalog number: DX0835-3 )
- Methanol (Certified ACS) (Fisher, catalog number: A412-4 )
- Cysteamine hydrochloride (Sigma, BioXtra, catalog number: 30078-25G ), backflush with nitrogen
- Ethyl Ether Anhydrous (Certified ACS) (Fischer, catalog number: E138-1 ), backflush with nitrogen (explosive Hazard)
- Fmoc-N-hydroxysuccinimide ester (Fmoc-OSU), ≥ 98% (Sigma, catalog number: 46920-5G-F ), at 4 °C in desiccator
- Pyridine anhydrous, anhydrous 99.8% (Sigma, catalog number: 270970-1L )
- N,N Diisopropylethylamine (DIPEA), purum 99.5% biotech grade (Sigma, catalog number: 496219-100ML ), backflush with nitrogen
- N,N'-Diisoipropylcabodiimide (DIC), purum ≥ 98% (Sigma, catalog number: 38370-25ML ), backflush with nitrogen
- Acetic Anhydride, Reagent Plus ≥ 99% (Sigma, catalog number: 320102-100ML )
- 4-Methylpiperidine (Sigma, catalog number: M73206-500ML )
- Acetyl Glycine, Reagent Plus 99% (Sigma, catalog number: A16300-100G ), desiccator
- Glycine (Fmoc-Gly-OH) (Novabiochem, catalog number: 8.52001.0100 ), desiccator
- HCTU (Anaspec, catalog number: 62626-250 ), 4 °C
- 4-Methylmorpholine (NMM) (Sigma, catalog number: M56557-100ML )
- Trifluoroacetic acid (TFA) (Fischer, catalog number: O4902-100 100ML ), certified for peptide and protein analysis
- Phenol (Sigma, catalog number: 185450-100G )
- Triisopropylsilane (TIPS) (Sigma, catalog number: 233781-50G )
- Alanine (Fmoc-Ala-OH) (Novabiochem, catalog number: 8.52003.0100 ), desiccator
- Cysteine (Fmoc-Cys(Trt)-OH) (Novabiochem, catalog number: 8.52008.0025 ), -20 °C
- Aspartic Acid (Fmoc-Asp(OtBu)-OH) (Novabiochem, catalog number: 8.52005.0025 ), -20 °C
- Glutamic Acid (Fmoc-Glu(OtBu)-OH) (Novabiochem, catalog number: 8.52009.0025 ), -20 °C
- Phenylalanine (Fmoc-Phe-OH) (Novabiochem, catalog number: 8.52016.0100 ), -20 °C
- Histidine (Fmoc His(Trt)-OH) (Novabiochem, catalog number: 8.52032.0025 ), -20 °C
- Isoleucine (Fmoc-Ile-OH) (Novabiochem, catalog number: 8.52010.0100 ), desiccator
- Lysine (Fmoc-Lys(Boc)-OH) (Novabiochem, catalog number: 8.52012.0025 ), -20 °C
- Leucine (Fmoc-Leu-OH) (Novabiochem, catalog number: 8.52011.0100 ), desiccator
- Asparagine (Fmoc-Asn(Trt)-OH) (Novabiochem, catalog number: 8.52044.0025 ), -20 °C
- Glutamine (Fmoc-Gln(Trt)-OH) (Novabiochem, catalog number: 8.52045.0025 ), -20 °C
- Methionine (Fmoc-Met-OH) (Novabiochem, catalog number: 8.52002.0100 ), desiccator
- Proline (Fmoc-Pro) (Novabiochem, catalog number: 8.52017.0025 ), -20 °C
- Arginine (Fmoc-Arg(Pbf)-OH) (Novabiochem, catalog number: 8.52067.0025 ), -20 °C
- Serine (Fmoc-Ser(tBu)-OH) (Novabiochem, catalog number: 8.52019.0025 ), -20 °C
- Threonine (Fmoc-Thr(tBu)-OH) (Novabiochem, catalog number: 8.52000.0025 ), -20 °C
- Valine (Fmoc-Val-OH) (Novabiochem, catalog number: 8.52021.0100 ), desiccator
- Tyrosine (Fmoc-Tyr(tBu)-OH) (Novabiochem, catalog number: 8.52020.0025 ), -20 °C
- Biotin (Sigma, catalog number: B4501-1G ), 4 °C
- p-Threonine (Fmoc-Thr(PO-(OBzl)-OH)-OH) (Novabiochem, catalog number: 8.52070.0001 ), -20 °C
- p-Serine (Fmoc-Ser(PO-(OBzl)-OH)-OH) (Novabiochem, catalog number: 8.52069.0001 ), -20 °C
- Natural Streptavidin protein (DyLight 650) (Abcam, catalog number: Ab134341 ), 4 °C
- Rink amide (4-[(2,4-Dimethoxyphenyl)(Fmoc-amino)methyl]phenoxyacetic acid) (Sigma, catalog number: 38784-5G ), 4 °C
- Ammonium citrate dibasic (DHAC) (Sigma, catalog number: C1883-100G ), desiccator
- Trihydroxyacetophenone monohydrate (THAP) (Sigma, catalog number: 91928-1G )
- Acetonitrile (Sigma, catalog number: 271004-1L )
- Bovine Serum Albumine (Sigma, catalog number: A3294-50G )
- Tris (Sigma, catalog number: T3253-500G )
- NaCl (Fisher, catalog number: BP358-10 )
- TWEEN 20 (Sigma, catalog number: P7949-500ML )
- Phosphate Buffered Saline (Gibco, catalog number: 10010-023 )
- CNA binding buffer (see Recipes)
- PBS-T (see Recipes)
- Reagent B (see Recipes)
Equipment
- Syro II Peptide Parallel Peptide Synthesis System (MultiSynTech, model: S2PS )
- U-Block Reactor, 48 position, for 2 ml reactor vials (MultiSynTech, catalog number: Z048UB020 )
- Vortex Mixer, Stand-Alone, Holds 2-Reactor Blocks (MultiSynTech, catalog number: S006VO002 )
- Empty Head, 48 position (MultiSynTech, catalog number: Z048AK000 )
- Mounting Tool–Peptide synthesis (MultiSynTech, catalog number: Z000VW000 )
- Transfer head 48 pos (MultiSynTech, catalog number: Z048TK048 )
- Transfer rack 48/15 (MultiSynTech, catalog number: Z048TR015 )
- 96 Well Nitrogen flow evaporator (Glas-Col, model: 099A=Ev96 )
- Thermo multichannel pipette adjustable spacing (Thermo Fischer Scientific, catalog number: 4672090BT )
- Eppendorf repeater (Eppendorf, catalog number: 4982000322 )
- Water bath sonicator (Shenzhen Jie Tai Ultrasonic Cleaning Equipment Co., LTD, model: 10A )
- MSP 96 target polished steel BC (Bruker, catalog number: 8280800 )
- Nikon Ti Eclipse automated inverted microscope (Nikon, model: TE2000 )
- Motorized XY translation stage (Applied Scientific Instrumentation, Eugene, OR)
- SOLA light engine (Lumencor, Beaverton, OR)
- Xenon arc lamp and ozone scrubber (Lambda LS, Sutter Instrument, Novato, CA)
- UV-liquid light guide (Sutter Instrument, Novato, CA)
- Programmable filter wheel, Lambda 10-2 (Sutter Instruments, Novato, CA)
- EMCCD camera, Andor Zyla 4.2 (Oxford Instruments, UK)
Note: For more information about the microscope used for imaging, please see Harink et al., 2019.
Software
- Micromanager (to control microscope used for imaging MRBLEs, can be substituted as necessary): https://micro-manager.org/ (Edelstein et al., 2010)
- MRBLE Software package (an open-source software analysis package for analyzing images of MRBLEs), https://github.com/FordyceLab/MRBLEs (Harink et al., 2019)
- GraphPad Prism (for analyzing binding data, can be substituted as necessary) (https://www.graphpad.com/)
- ProteoWizard See MS (for analyzing MALDI mass spectrometry data): http://proteowizard.sourceforge.net/download.html (Chambers et al., 2012)
- Microsoft Excel
- Syro II Peptide Synthesizer Software (MultiSynTech, comes with peptide synthesizer)
Procedure
This complete protocol describes (Figure 1): (1) Functionalization of PEG-DA hydrogel beads in preparation for peptide synthesis, (2) solid-phase Fmoc peptide synthesis on PEG-DA beads, (3) peptide validation and peptide density measurements, (4) multiplexed protein-peptide binding assays using bead-bound peptide libraries, and (5) final data analysis to extract quantitative binding affinity information (Kd). In this protocol, all amounts are calculated for 48 tubes containing one code each (a 48-code peptide library) with an estimated 1 mg of MRBLEs for each code (about 10,000 beads) and a loading capacity of 0.32 mmol/g. Therefore, 1 equivalent ~50 mg ~0.016 mmol.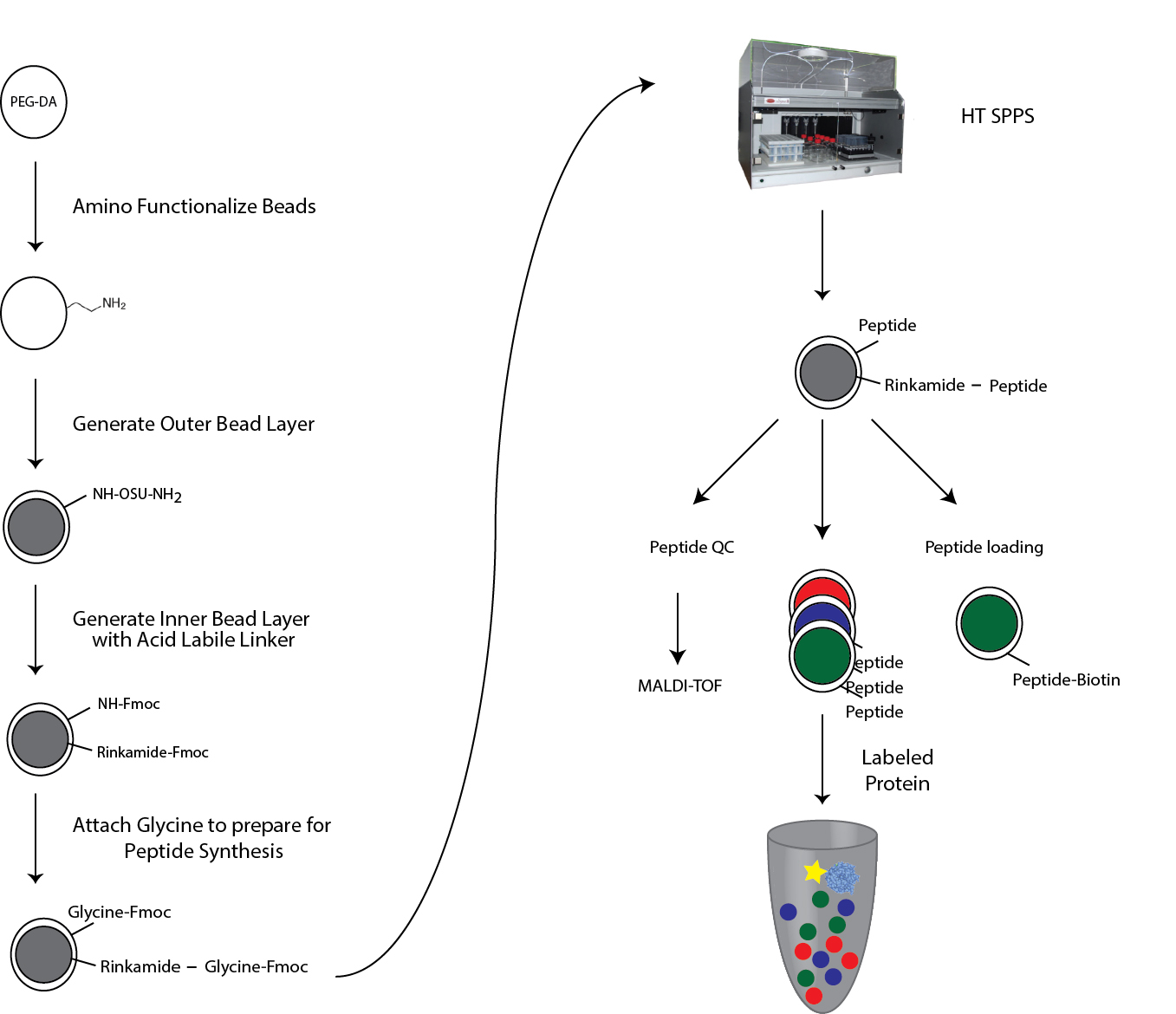
Figure 1. Overview of MRBLE functionalization. PEG-DA beads are functionalized with free amine groups for subsequent solid-phase peptide synthesis via Michael addition. The outer shell and inner core are differentially functionalized with acid-labile and acid-sensitive linkers by sequential immersion in water and solvents. The acid labile Rink amide linker at the inner core is thus cleaved during final amino acid side chain deprotection, allowing collection of eluted peptides for quality control and validation via mass spectrometry. The linker in the outer shell stays attached to the bead and is used for downstream measurement of protein-peptide interactions in MRBLE-pep assays.
Part I: Production of MRBLEs
Prior to the start of the protocol, produce PEG-DA hydrogel beads using either a multilayer PDMS ‘bead synthesis’ microfluidic device (as described previously [Gerver et al., 2012]) or using syringe pumps and standard droplet generators.
Preparation after bead synthesis:
- Transfer MRBLEs containing each spectral code (here, a 48-code library) into 48 x 2 ml reactor vessels placed in a U-Block Reactor mounted in a vacuum manifold for easy bead washing without bead loss (Figure 2 and see Notes).
- Wash MRBLEs by placing U-Block Reactor assemblies onto the manifold and sequentially washing 3 x with Dichlomethane (DCM), 3 x with Methanol (MeOH), and 3 x with Dimethylformamide (DMF). For each wash step, use a repeater pipette to fill each vessel with 1 ml of solvent and remove solvent by pushing it through the reactor vessel using the gas cover plate with nitrogen flow (Figure 3).
- After washing, add 400 µl DMF to each vessel, move the entire U-Block Reactor assembly to a water bath sonicator, and sonicate for 30 min. To ensure thorough sonication without contamination, water levels in the sonicator should match the level of the fluid in the tubes. Depending on the size of the water bath sonicator, the reactor vessels may need to be transferred to a different rack.
In prior work, we have found that pushing solvents through vessels using this gas cover plate leads to more even washing than simply drawing solvents through using a vacuum manifold; however, this may depend on the precise setup used. To ensure even functionalization, during this and all subsequent steps, it is important to verify that the beads are uniformly distributed and don’t clump. Check this frequently during sonication by observing the floating beads; if necessary, perform additional sonication steps to break up remaining clumps.
Figure 2. U-Block Reactor for Peptide Synthesis used for washing beads prior to chemical functionalization. Each U-Block Reactor holds 48 x 2 ml reaction vessels.
Figure 3. Nitrogen cover plate to push liquid through reaction vessel into waste bottle. The U-Block reactor is attached to empty head to collect the liquid from all 48 tubes into one tube going to the waste (glass bottle in the picture). The head can be exchanged in case it is necessary to keep the content of each tube separate (see Figure 6). Wash solutions are added using a repeater. Liquid from all 48 tubes is pushed into waste bottle. This set up can be replaced by an alternate configuration; however, it makes the multiple wash steps both more convenient and more reproducible between runs.
Part II: Functionalization of MRBLEs
Prior to peptide synthesis, the PEG-DA polymers that make up MRBLEs hydrogel beads must be chemically functionalized with free amine groups to allow direct coupling of Fmoc-protected amino acid building blocks. In this protocol, the outer shell of the MRBLEs is selectively functionalized with an acid-resistant linker (which remains intact upon exposure to the TFA required for final side chain deprotection during Fmoc peptide synthesis) while the inner core is selectively functionalized with an acid-labile linker that is cleaved upon exposure to TFA (Kunys et al., 2012). In this manner, peptides from the inner core can be eluted during the final steps of peptide synthesis for direct analysis via mass spectrometry while peptides coupled to the outer shell remain attached for downstream binding assays. This procedure takes place in 5 main steps: (A) Michael addition, (B) selective functionalization of the outer shell, (C) selective functionalization of the inner core, (D) capping of any remaining free amino groups, and (E) addition of a glycine residue to all uncapped functional groups to allow downstream couplings in peptide synthesis.
- Michael Addition to add amine functional groups to acrylate groups on PEG-DA beads using cysteamine and pyridine (Reaction 1)
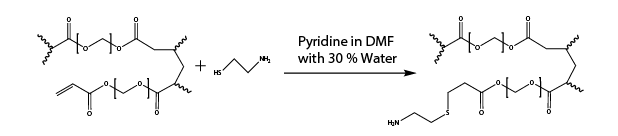
Reaction 1. Michael addition to add amine handles to acrylate groups on PEG-DA based MRBLEs using cysteamine and pyridine
- Add 400 µl of 20 ml DMF/30% Water containing 20 eq Cysteamine (36 mg) and 20 eq Pyridine (24 µl) to each reaction vessel.
- Place the U-Block assembly on the Vortex Mixer (Figure 4) and incubate reactions for > 20 h at room temperature (1,600 rpm with cycles of 15 s shaking and 30 s paused).
- Repeat reaction by removing liquid and add fresh reagents and incubating beads as before.
- Wash beads 3 x with 1 ml each of DMF, MeOH, and DCM using the inert gas cover plate as described previously in Part I: Production of MRBLEs.

Figure 4. Vortex mixer with 2 U-Block Reactors mounted
- Selective functionalization of outer shell with acid-stable Fmoc-protected hydroxysuccinimide esters (Reaction 2)
Note: To ensure that only the outer shell will be functionalized in this step, the inner core is first swelled with water and then the reaction is performed in water-insoluble solvent with Fmoc-OSU at 0.1 equivalence.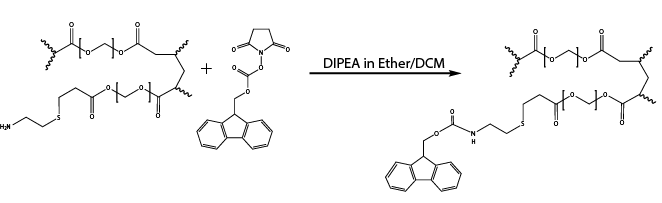
Reaction 2. Selective functionalization of outer surface with Fmoc-N-hydroxysuccinimide (Fmoc-OSU) in ether/DCM after MRBLEs were swelled in water
- Wash all reaction vessels with 1 ml DMF and 1 ml ddH2O.
- Add 400 µl ddH2O to each reaction vessel and incubate for at least 20 h on vortex mixer (15 s shaking, and 30 s pause at 1,600 rpm).
Note: This step ensures that beads are swelled with water prior to performing the reaction in non-aqueous solvent. - Drain water carefully and make sure all the water around the beads is gone. There should still be water inside the beads for the next reaction and it is therefore important not to drain the beads for too long to prevent the inside of the beads from drying out.
- Add 400 µl of a 40 ml solution of 55% DCM and 45% Ether to each reaction vessel containing:
0.1 eq Fmoc OSU (2 mg)
1 eq DIPEA (2.68 µl) - Incubate all reaction vessels for 20 min on vortex mixer (15 s shaking 30 s pause at 1,600 rpm).
- Wash beads 3 x with 1 ml each of DMF, MeOH, and DCM using the inert gas cover plate as described previously in Part I: Production of MRBLEs.
Note: This washing step is essential to remove all water from bead cores prior to the next functionalization step.
- Functionalization of inner core with acid labile Fmoc protected Rink amide linker (Reaction 3)
Note: The extensive washing at the end of the previous step should have removed all water from bead cores, now allowing functionalization of the inner core with acid-labile Rink amide using DMF as solvent.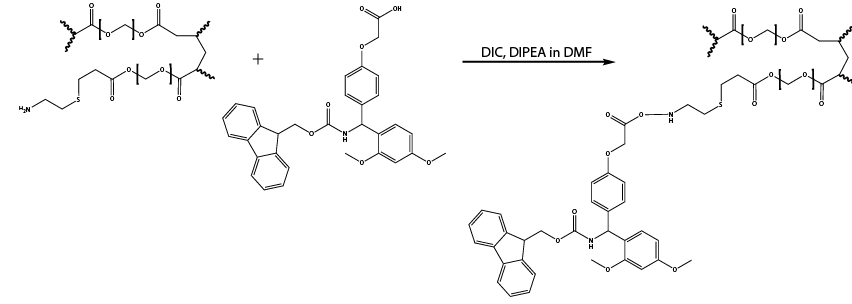
Reaction 3. Functionalization of inner core with Rink amide using DIPEA and DIC in DMF
- To each reaction vessel, add 400 µl of a 20 ml solution of DMF containing:
5 eq Rink amide (41 mg)
5 eq DIC (12 µl)
10 eq DIPEA (28 µl) - Shake all reaction vessels for at least 16 h (15 s shaking 30 s pause at 1,600 rpm).
- Repeat by draining reaction mix and adding fresh reagents. Shake for at least 2 more hours.
- Wash beads 3 x with 1 ml each of DMF, MeOH, and DCM using the inert gas cover plate as described previously in Part I: Production of MRBLEs.
- To each reaction vessel, add 400 µl of a 20 ml solution of DMF containing:
- Capping of remaining free amino groups
Note: To prevent coupling of amino acids to any remaining free amino groups in later peptide synthesis steps, which would lead to truncated peptides, amine groups that have not reacted with Fmoc-OSU (during Procedure B) or Rink amide (during Procedure C) must be capped.- Cap remaining NH2 groups by adding 400 µl of a mixture of Acetic-Anhydride with 25% Pyridine (prepare 25 ml) to each reaction vessel.
- Incubate all reaction vessels for 15 min (and not longer to avoid possible side reactions) on vortex mixer (15 s shaking 30 s pause at 1,600 rpm).
- Wash beads in each reaction vessel 3 x with 1 ml each of DMF, MeOH, and DCM using the inert gas cover plate as described previously in Part I: Production of MRBLEs.
- Glycine coupling: Deprotect all inner and outer Fmoc-protected amine groups and attach Fmoc-protected glycine (Reaction 4 and Reaction 5) in preparation for downstream peptide synthesis.
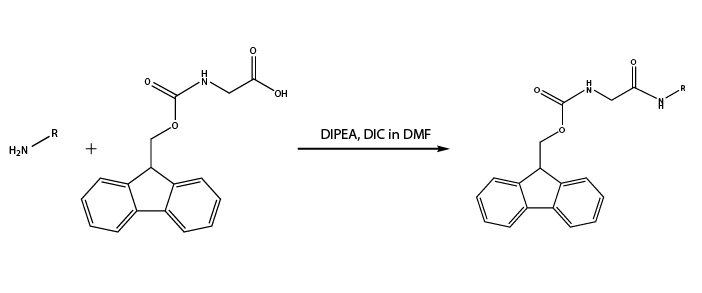
Reaction 4. Reaction of outer shell amine groups with Fmoc-Glycine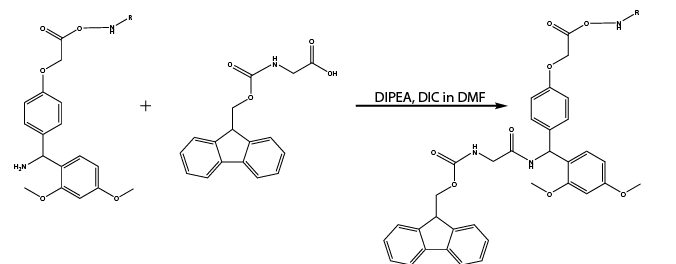
Reaction 5. Reaction of inner core Rink amide with Fmoc-Glycine
- For all reaction vessels, deprotect Fmoc-protected amine groups by adding 400 µl of a solution of 20% 4-Methylpiperidine in DMF and shaking for 10 min (to prevent unwanted side reactions) on the vortex mixer (15 s shaking 30 s pause at 1,600 rpm).
- Stop deprotection reaction by washing beads with 3x DMF, 3x MeOH and 3x DCM.
- To each reaction vessel, add 400 µl of a 20 ml solution of DMF containing:
0.5 eq Fmoc Glycine (2.38 mg)
4.5 eq Acetyl-Glycine (8.43 mg)
5 eq DIC (12.4 µl)
10 eq DIPEA (28 µl) - Incubate all reaction vessels for at least 16 h on vortex mixer (15 s shaking 30 s pause at 1,600 rpm).
- Remove solution and repeat Steps E3 and E4.
- Wash beads 3 x with 1 ml each of DMF, MeOH, and DCM using the inert gas cover plate as described previously in Part I: Production of MRBLEs.
Part III: Solid-phase peptide synthesis on MRBLEs
Following functionalization of the outer shell of MRBLEs with free amino groups, peptides can be synthesized directly on MRBLEs using standard solid-phase peptide synthesis techniques (e.g., Fmoc peptide synthesis). Here, we provide a brief protocol for peptide synthesis using the Syro II high-throughput peptide synthesizer, which includes the associated U-Block reactor and vortex mixer used here. For additional detailed information about instrument setup and use, please consult the manufacturer’s instructions. In addition, we have found these on-line resources describing peptide synthesis to be particularly helpful: source 1 and source 2.
- Peptide Synthesis
Note: Automated solid phase peptide synthesis (SPPS) on functionalized MRBLEs using the Syro II high-throughput peptide synthesis robot (or similar).- Deprotect all Fmoc groups on MRBLEs in each reaction vessel using 400 µl 20% 4-Methylpiperidine in DMF for 10 min.
- Wash each reaction vessel with DMF 3 times with 400 µl DMF.
- Prepare peptide synthesizer for synthesis run.
- For typical MRBLE-pep runs, we use the following synthesis protocol:
- Amino acid coupling: 2 x 20 min coupling for each amino acid (10 eq) in the presence of HCTU (9.8 eq) and NMM (20 eq).
- Wash each reaction vessel 3 times with 400 µl DMF.
- Add 200 µl of 40% 4-Methylpiperidine in DMF and incubate for 2 min and drain.
- Add 200 µl of 20% 4-Methylpiperine in DMF and incubate for 2 min.
- Wash each reaction vessel 3 times with 400 µl DMF.
- After synthesis program is complete, add 400 µl of DMF to each reaction vessel. This step is to ensure that none of the reaction vessels dry out completely at the end of the run.
- Prior to beginning peptide synthesis, ensure that there are no air bubbles in the fluid lines that draw the solutions. Air bubbles might result in tubes getting uneven amounts of liquid. If any air is found, wash lines to eliminate air bubbles.
- Select 48 x 2 ml reaction vessels in peptide synthesis software (or other as appropriate).
- Load a text document specifying the sequence of each peptide to synthesize, making sure there are no spaces. Set reactions for each step as necessary (e.g., you may consider different coupling conditions for specific steps dependent on your amino acid). On the Syro II program, we make sure that ‘amino acid priority’ and ‘volume check’ are marked and that ‘wash all’ is not.
- Calculate amounts at a concentration of 0.16 mmol using 0.54 mmol/g resin load, 2 mg resin amount and 15 equivalents. Prior to preparing solutions, save the table the software generates to guide the amount of each reagent required.
- Save the molecular mass for each peptide calculated by the software for future reference during MALDI mass spectrometry.
- Prepare amino acids, HCTU and NMM solutions in DMF and sonicate if necessary, according to the table the Syro II software generated. The table shows the amount of amino acids, HCTU and NMM needed and in how much DMF they need to be dissolved. It will also show how much DMF is necessary for all the washing steps as well as the amount of 40% 4-Methylpiperidine. We generally make all the solutions in 10% excess to make sure there is enough (e.g., to account for evaporation during prolonged peptide synthesis). Prepare amino acids in 50 ml tubes. Fill HCTU and NMM in special glass peptide synthesizer bottles according to manufacturer’s instructions. Fill DMF and 40% 4-Methylpiperidine glass bottles connected to peptide synthesizer.
- Start synthesizer. Synthesizing 48 peptides with 15 amino acids each takes approximately 24 to 48 h on the Syro II synthesizer, depending on coupling times and complexity of your library (e.g., whether a library contains systematic single amino acid changes of a particular peptide sequence vs. a collection of peptides with completely different sequences).
- For typical MRBLE-pep runs, we use the following synthesis protocol:
Part IV: Peptide loading density testing and characterization
Accurate multiplexed measurement of protein-peptide binding affinities requires that all reagents be present at approximately the same concentrations. Given that peptides are displayed on beads in the MRBLE-pep assay, this means that beads from each code are all approximately the same size, that equal numbers of beads are added to each assay, and that the surface density of displayed peptides is approximately constant. While microfluidically-produced MRBLE beads are significantly more monodisperse than most commercially available bead-based resins, care must be taken to ensure that each peptide sequence is displayed at approximately the same density. To directly visualize synthesized peptide density (and estimate peptide loading per bead), we biotinylate peptides to allow visualization using fluorescently labeled streptavidin via the following steps: (1) biotinylate the peptides after synthesis, (2) detect the biotinylated peptides using fluorescently labeled streptavidin and (3) decode the beads and measure fluorescence intensities (Figure 5). Importantly, this step can be performed at any point in the synthesis protocol to allow troubleshooting of individual steps (e.g., after initial MRBLE functionalization or after particularly challenging coupling steps). In all cases, comparing fluorescence signals after a given protocol with appropriate negative controls can test that streptavidin binding is specific.
- Biotinylation of MRBLE-bound peptides.
- Take out about 10% of beads from each code and pool them in a new reaction vessel (e.g., draw 40 µl out of 400 µl after vigorous pipetting).
- Deprotect MRBLE-bound peptides with 400 µl 20% 4-Methylpiperidine in DMF for 10 min rotating at 20 rpm.
- Prepare deprotection solution by adding the following to 800 µl of DMF and add all to the beads:
10 eq Biotin (39 mg)
10 eq DIC (24 µl)
20 eq DIPEA (56 µl) - Rotate at room temperature for at least 16 hours at 20 rpm.
- Drain liquid and repeat Steps G3-G4 with fresh solution.
- Deprotect as described (see Part V: Peptide side chain deprotection).
- Binding Assay with Streptavidin Dylight 650
- Take out ~5,000 beads in PBS-T (wash thoroughly when stored in DMF) and block with 100 µl 5% BSA in PBS-T for 1 h at room temperature on rotator at 20 rpm. To estimate the number of beads, consider dilution steps and initial target numbers. For example, if you started with ~10,000 beads per code and used 10% of each code for biotinylation, you should have about 48 beads/µl and would need 100 µl to yield 4,800 beads. If you find large variations in bead number after analyzing the data, you can adjust the number of beads per code you need for the experiments.
- Exchange the buffer by centrifuging beads at 200 x g for 10 s, adding 2% BSA in PBS-T with 1:100 Streptavidin and incubating for 30 min at 4 °C while rotating at 20 rpm.
- Wash beads 3 x with 100 µl PBS-T by centrifuging beads (200 x g, 10 s), discarding the supernatant and adding fresh PBS-T.
- Image beads on a cover slip in 20 µl PBS-T. See Image acquisition section for more details.
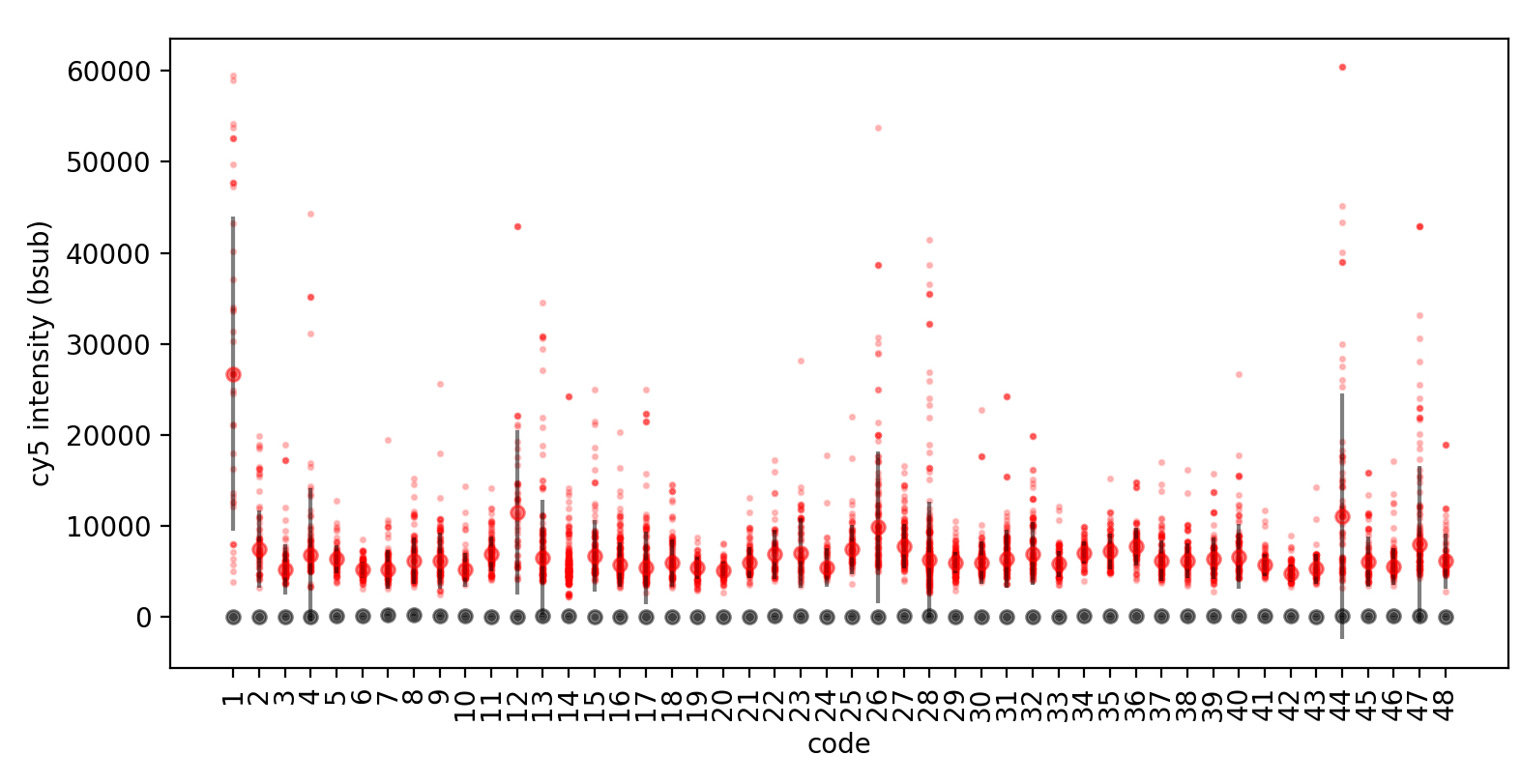
Figure 5. Streptavidin-Dylight 650 detection of biotinylated (red) and non-biotinylated (grey) peptides. For this acquisition, MRBLEs were imaged in the Cy-5 fluorescence channel using a SOLA LED light engine at 5% intensity with a 100 ms exposure time. Images were processed and beads decoded using the MRBLE software package as described previously (Harink et al., 2019). For each code, all Cy5 intensities for each bead were plotted (small markers) along with the median intensity and standard deviation (large markers, error bars) using custom software written in a Python Jupyter notebook.
Part V: Peptide side chain deprotection and inner core peptide elution for sequence validation
A crucial advantage of bead-bound peptide libraries is that peptides can be eluted from beads and analyzed via mass spectrometry to ensure that any peptides showing decreased protein binding reflect a truly disrupted interaction rather than failure to synthesize the correct peptide. In this step, we describe elution of peptides from the inner core (by exposing this acid-labile linker to strong acid while removing side chain protecting groups during the final peptide synthesis step) followed by sequence analysis via MALDI mass spectrometry.
- Deprotection and simultaneous cleavage of peptides attached to the MRBLE core via an acid-labile linker (Figure 6)
- Wash each reaction vessel with 3x DMF, 3x MeOH and 3x DCM.
- Add 400 µl freshly prepared Reagent B (TFA is volatile) to each reaction vessel on the U-Block.
- Incubate all reaction vessels for 1-3 h on the Vortex Mixer (15 s shaking 30 s pause at 1,600 rpm).
Note: Time depends on amino acids used and the side protecting groups. - Drain eluted peptides into 5 ml eppendorf tubes using nitrogen (cut off lids to make sure all tubes fit or use 15 ml tubes) (see Figure 6). At this step, label all tubes with the bead code number or expected peptide sequence.
- Add an additional 500 µl of TFA to each vessel.
- Drain again into the same tubes.
- Transfer all solutions into a 96 well deep well plate using a multichannel pipette with adjustable spacing and allow peptide solutions to evaporate completely under Nitrogen flow (tubes should be completely dry).
- Wash the peptide coated beads that remain in the reaction vessels 3 x with 1 ml DCM.
- Wash beads 2 x with 1 ml 10% DIPEA in DMF.
- Wash beads 4 x with 1 ml DMF.
- If bead-bound peptides will ultimately be stored in aqueous buffer, continue washing 3 times with 1 ml water and 1 ml PBS-T. Otherwise, bead-bound peptides can remain in DMF. Peptide-conjugated beads stored in PBS-T or DMF have been successfully used up to one year after synthesis. After long term storage we recommend imaging a small aliquot of beads to check if beads can still be decoded.

Figure 6. Global deprotection of side protecting groups of peptides and collection of peptides from inner core in individual tubes. Beads are incubated with deprotection reagent mix for 1 to 3 h (depending on amino acid composition). The deprotected and cleaved amino acids from the inner core are then pushed into individual tubes using the U-Block reactor attached to the transfer head for downstream mass spectrometry (The transfer head allows the transfer from each individual reaction vessel to individual tube).
- Transfer beads to 96 deep well plates or combine for downstream analysis. In most cases, we suggest leaving beads in separate vessels when possible to allow synthesis and substitution of individual additional peptides as required.
Part VI: Mass spectrometry to check quality of synthesized peptides
Peptide synthesis can be challenging and factors such as peptide length, amino acid repeats and choice of coupling reagents influence the success. In addition, quality control of synthesized peptides is especially important when peptides are synthesized directly on beads subsequently used for binding experiments since chromatographic purification steps are not possible. Here, we describe analysis of eluted peptides via mass spectrometry. We use MALDI-TOF for peptide analysis, as it has a high sensitivity and generates mostly single charged ionized peptides species that make downstream identification simple (Nadler et al., 2017). However, other mass spectrometry-based methods can be used as well.
- MALDI-MS quality control
- If peptides have not already been dried during the Part V: Peptide side chain deprotection validation step, fully dry eluted peptides in a 96 deep well plate under nitrogen flow.
- Wash dried peptides with ice cold ether to remove any residual sidechain protecting groups.
- Add 1 ml ether to each well.
- Incubate plate for 1 h at -20 °C.
- Spin for 30 min at 3,000 x g and 4 °C in a plate-compatible centrifuge.
- Remove ether without disturbing the pellet (which might not be visible).
- Dry again under nitrogen flow.
- Dissolve peptides in a suitable solution in which peptides are soluble. A good starting point is to use 70% Acetonitrile in ddH2O, although this may have to be adjusted depending on the peptide solubility.
- Add 0.5 µl of a 250 mmol DHAC water solution to matrix plate and let dry to a point where liquid water is no longer visible (see Figure 7).
Note: The choice of matrix influences the detection of the peptide and needs to be determined experimentally.
Figure 7. Preparation of samples for MALDI-TOF. Mix peptides with matrix on parafilm and transfer to MALDI plate. - Prepare 200 µl 250 mmol THAP solution in Acetonitrile.
- Create a 1:1 solution of DHAC:THAP.
- Mix 1 µl of the DHAC:THAP solution with 1 µl of dissolved peptide on parafilm (Figure 7) for each well.
- Add 1 µl of this mixture to each well of the matrix plate and leave to dry.
- Analyze with MALDI-TOF. Here, we use the Bruker microflex (Billerica, MA, USA) spectrometer, but this is easily adapted to other instruments. Some tips for ensuring high-quality spectra include:
- Always use a peptide standard for calibration.
- Run the instrument in positive-ion reflector mode with a laser setting of 1,810 V.
- For each run, record the average over 100 scans.
- Try different laser powers and spots on the plate as necessary to obtain a good spectrum.
- Analyze data using the SeeMS software or other commercially available software.
- Ideally, the resultant spectra should appear as a single peak at the expected molecular weight (Figure 8). MALDI ionization mostly generates single charged ions, simplifying interpretation of measured spectra. The glycine added before solid phase peptide synthesis is not necessarily visible and you might see variations of your molecular weight from salt or other impurities. To interpret more complex spectra, consider possible truncations resulting from elimination of the first or last amino acid (where synthesis efficiencies might be lower). We recommend a useful MS mass adduct calculator published by the Fiehn lab at UC Davis (https://fiehnlab.ucdavis.edu/staff/kind/Metabolomics/MS-Adduct-Calculator) (Huang et al., 1999).
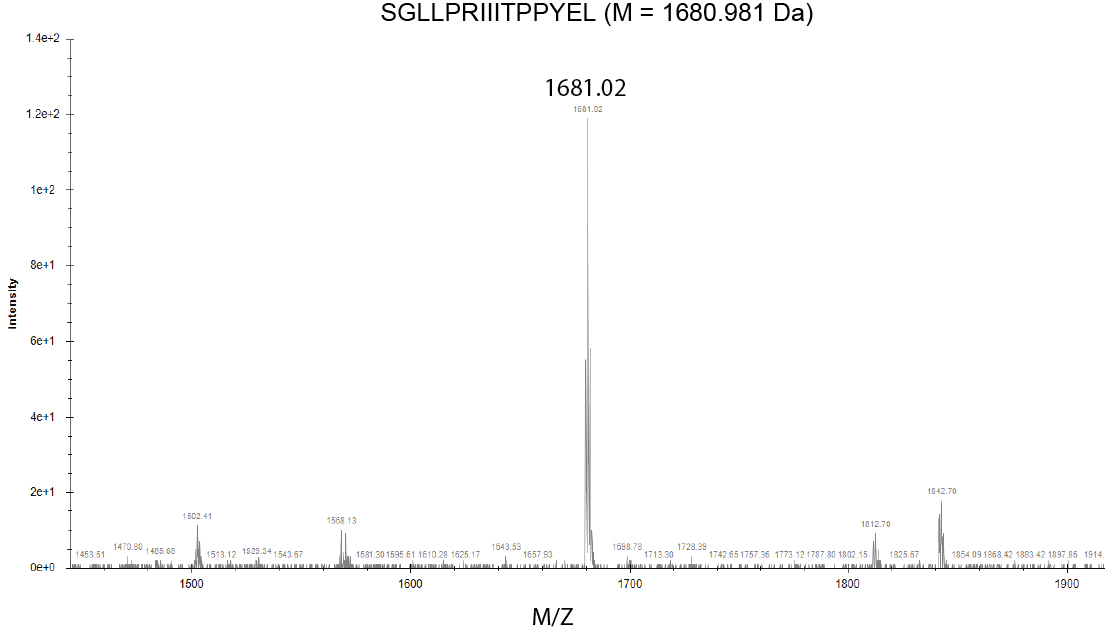
Figure 8. MALDI-MS spectra using SeeMS program
Part VII: MRBLE-pep binding assay
Binding assays designed to quantify affinities between bead-bound peptides and proteins in solution take place in 3 main steps: (1) blocking (to reduce non-specific binding), (2) binding (to test for specific binding between a protein of interest and bead-bound peptides), and (3) imaging to identify the peptide sequence displayed by each bead and quantify the amount of bound protein. MRBLE-pep allows multiplexed measurement of binding affinities between 48 peptides and a protein of interest using only 4 µM of protein in a 100 µl volume, thus requiring a total of 40 µg of a 100 kDa protein per assay. However, several parameters must be considered carefully to ensure a successful binding experiment.
- The number of beads added per code must be held constant within and between experiments. Empirically, we find that ~100 beads per code ensures that the concentration of available peptide is sufficiently low relative to the concentration of available protein while simultaneously reducing variability due to stochastic Poisson error in the number of beads present in each volume. In addition, having 100 replicates of each measurement allows robust detection of small but statistically significant differences in binding between peptides via signal averaging.
- Accurate quantification of binding requires that bound protein be detected via imaging. In past MRBLE-pep experiments, we detected binding of epitope-tagged proteins using an antibody (Nguyen et al., 2019). Here, we describe quantitation of binding for proteins that are directly tagged with a fluorescent indicator, e.g., YFP. In general, any tagging strategy can be used as long as appropriate negative controls are included to eliminate spurious fluorescence that derives from non-specific interactions. For binding assays using fluorescent protein tags, an appropriate negative control should include the fluorescent protein alone. When using fluorescently labeled antibodies, binding assays should: (1) incubate the protein of interest 1:1 with antibody prior to incubation with beads to reduce direct binding of free labeled antibody to beads, and (2) directly measure binding of the labeled antibody alone to peptide-bead libraries.
- The amount of protein added should include a concentration range that is 10-fold higher and lower than the expected interaction Kd whenever possible. We typically use a range of protein concentrations spanning from ~30 nM to 2 µM. However, this may need to be adjusted depending on expected affinities or the presence of protein aggregation at high concentrations.
- Recovering quantitative absolute and relative binding affinities requires that interactions be measured at thermodynamic equilibrium. The use of hydrogel beads results in slow association and dissociation kinetics, facilitating measurement of even weak and transient interactions difficult to detect via other methods. However, accurate affinity measurements require initial experiments to ensure that the reaction has come to equilibrium. This can be accomplished by initially monitoring protein association and dissociation rates as a function of time and then using these incubation times in all downstream experiments.
- Binding assay for multiplexed measurement of protein-peptide affinities, using the calcineurin phosphatase tagged with YFP as an example
Note: Calcineurin-YFP was expressed and purified from E. coli as described in Nguyen et al. (2019).- For each experiment, use ~100 beads per code. In the protocol described here, this corresponds to ~1% of the beads from each code (e.g., if beads of 1 code are resuspended in 1 ml of buffer, draw up 10 µl of beads after extensive mixing). In typical assays, this means measurements take place on ~4,800 beads for 48 codes in a total volume of 20 µl.
- Spin down beads, remove PBS-T and block to prevent excess nonspecific binding by incubating with 100 µl 5% BSA in PBS-T for 1 h rotating at room temperature at 20 rpm.
- Wash beads once with 100 µl PBS-T by centrifuging beads (200 x g, 10 s), discarding the supernatant and adding fresh PBS-T.
- Use 100 µl binding buffer with protein and add to beads after removing PBS-T.
- When measuring a dilution series, ensure that the final concentration of all components in the final binding assay remain constant for each protein. Differences in the concentration of crowding agents (e.g., glycerol) can change diffusion kinetics; similarly, differences in salt concentrations can change electrostatic interactions between proteins and peptides. Take care to consider all components of the buffer in which your purified protein and/or antibodies are dissolved.
- Incubate beads for 6 h (time it takes to reach equilibrium should be determined experimentally, since it varies for each protein) with protein at indicated concentrations in binding buffer at 4 °C while rotating at 20 rpm.
- If protein detections are performed using indirect labeling, i.e., fluorescently labeled antibodies, antibody and protein must be preincubated and then added to the beads. For example, 1 µmol protein is incubated with 1 µmol antibody at 4 °C for 1 h. Note that this step was not performed here as a YFP-tagged protein was used.
- Wash beads 1 time with 100 µl binding buffer by centrifuging beads (200 x g, 10 s), discarding supernatant and adding fresh PBS-T.
- Resuspend beads in 20 µl PBS-T.
- Image acquisition and data analysis
- Transfer sample (20 µl) onto a microscope quartz slide and cover with a quartz coverslip without trapping air. To do this, carefully seal one side by applying oil using a syringe and needle and, in the event that any air is trapped, slowly add a few microliters PBS-T from the other side. When air bubbles are gone, seal with oil.
- Image Beads using a custom built inverted Microscope Nikon TE2000 to allow UV excitation using a Xenon arc lamp, a 292/27 nm bandpass filter and a liquid light guide to illuminate beads from the top, as described previously in more detail (Gerver et al., 2012; Nguyen et al., 2016; Harink et al., 2019). However, any microscope with deep UV excitation and the ability to collect light for each lanthanide channel should work. For lanthanide emission, we currently use the following filters: 435 nm (40 nm bandpass), 473 nm (10 nm bandpass), 536 nm (40 nm bandpass), 546 nm (40 nm bandpass), 572 nm (15 nm bandpass), 620 nm (14 nm bandpass), 630 nm (92 nm bandpass), 650 nm (13 nm bandpass), and 780 nm (25 nm bandpass).
- Use microManager and multidimensional acquisition to take multiple images to cover entire area with beads at all Lanthanide channels and exposure times, brightfield and assay channel. Here, we used an YFP filter cube (Semrock, Rochester) to detect YFP-tagged Calcineurin and a Cy5 filter cube (Semrock, Rochester) to detect Dylight 650 tagged Streptavidin.
- Images were analyzed using the Python code available for download on Github (Harink et al., 2019). The images were loaded into the software analysis package and then processed to decode beads and obtain fluorescence intensities for each bead and each code. The Excel table was saved and used to further process and plot the data with Python (Figure 9).
- We used Python to plot the mean background subtracted fluorescence intensities for indicated peptides as well as fit the Langmuir isotherm (Figure 10).
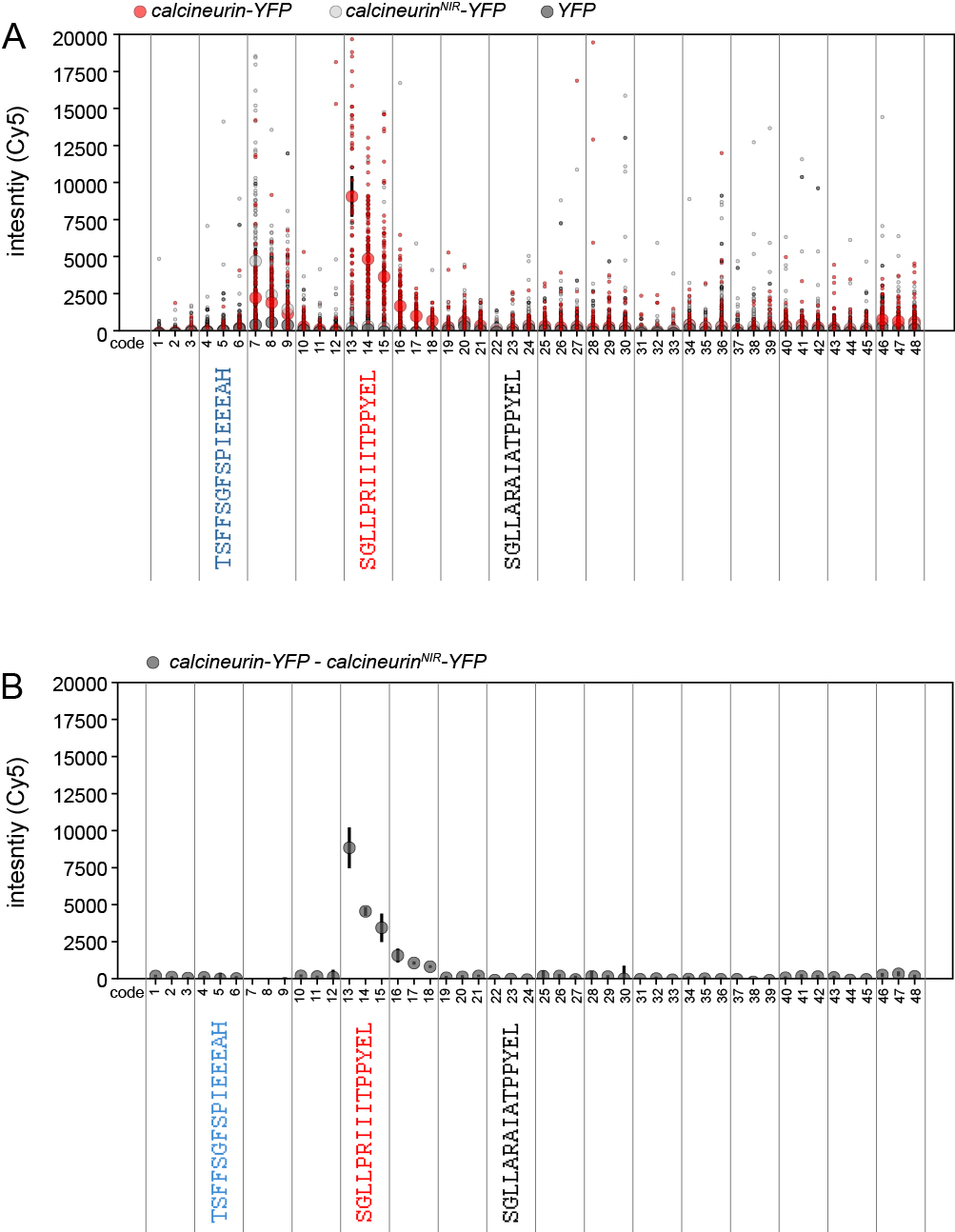
Figure 9. Measured bead-associated YFP fluorescence intensities for a MRBLE-pep library comprised of 16 different peptides each synthesized in triplicate on 3 different codes. A. Small markers indicate measured per-bead YFP signals for WT calcineurin-YFP (red), a calcineurin mutant deficient in peptide binding (NIR, negative control #1, light grey), and YFP only (negative control #2, black). Large markers and error bars indicate median intensity ± standard error of the mean for each protein. B. Median measured background-subtracted calcineurin-YFP intensity (calculated by subtracting the calcineurinNIR-YFP binding-deficient signal from the WT signal).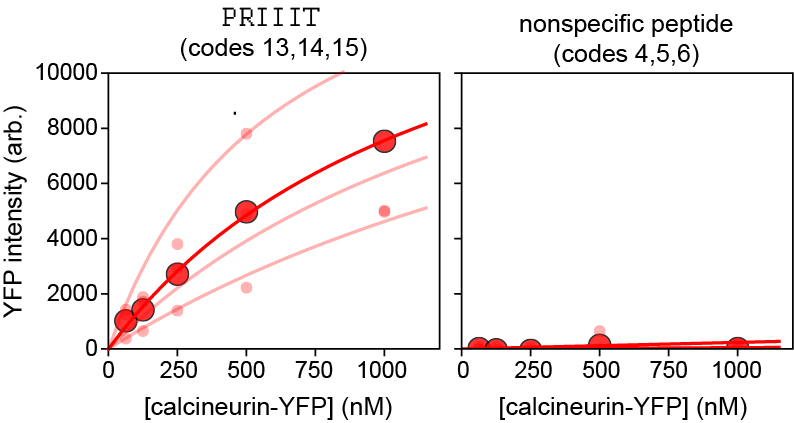
Figure 10. Concentration-dependent binding measurements and Langmuir isotherm fits for Calcineurin-YFP binding to 2 peptides from the 16-sequence MRBLE-pep library shown in Figure 9. Median background-subtracted fluorescence intensities for all beads with each code displaying a given peptide sequence are shown as light red dots; associated lines show Langmuir isotherm fits used to extract binding affinities. Dark red dots and lines indicate median bead intensities measured across all 3 codes and associated Langmuir isotherm fit to these median values. The left plot shows curves for a known calcineurin substrate (PRIIIT, Kd = 1.67 ± 0.86 µM calculated as the mean ± standard deviation of the returned values from each code using a global fit); the right plot shows curves for a negative control peptide.
Notes
We use 48 x 2 ml reaction vessels and a U-Block reactor equipment, which are directly compatible with the Syro II peptide synthesizer used for high-throughput peptide synthesis. However, this protocol can be adapted to any reaction vessels and mounting plates. The reaction vessels are simple columns with a filter. The U-Block holds the reaction vessel and the bottom of each vessel holder is connected to tubing which allows washing of the vessels. The vessels can be washed by either applying a vacuum or pushing the liquid through the vessel by using the inert gas plate.
Recipes
- CNA binding buffer
50 mM Tris pH = 7.5
150 mM NaCl
0.1% TWEEN 20 - PBS-T
PBS with 0.1% TWEEN 20 - Reagent B
88% TFA
5% Phenol (if Cysteine is present)
5% ddH2O
2% TIPS
Acknowledgments
This work was funded by NIGMS grants DP2GM123641 and R01GM107132. JBH was funded by grant NNF17OC0025404 from the Novo Nordisk Foundation and the Stanford Bio-X Program. PMF gratefully acknowledges the support of a Bio-X seed grant, an Alfred P. Sloan Foundation fellowship, and is a Chan Zuckerberg Biohub Investigator. We thank Jagoree Roy, Adam White and Yinnian Feng for help with protein and bead production. We thank Yinnian Feng for critical reading of the manuscript.
This protocol was adapted from previous work from Nguyen et al. (2017 and 2019).
Competing interests
The work described here is related to U.S. Patent Applications 61/692,618, on which P.M.F. is listed as a co-inventor.
References
- Brown, J. M., Hoffmann, W. D., Alvey, C. M., Wood, A. R., Verbeck, G. F. and Petros, R. A. (2010). One-bead, one-compound peptide library sequencing via high-pressure ammonia cleavage coupled to nanomanipulation/nanoelectrospray ionization mass spectrometry. Anal Biochem 398(1): 7-14.
- Chambers, M. C., Maclean, B., Burke, R., Amodei, D., Ruderman, D. L., Neumann, S., Gatto, L., Fischer, B., Pratt, B., Egertson, J., Hoff, K., Kessner, D., Tasman, N., Shulman, N., Frewen, B., Baker, T. A., Brusniak, M. Y., Paulse, C., Creasy, D., Flashner, L., Kani, K., Moulding, C., Seymour, S. L., Nuwaysir, L. M., Lefebvre, B., Kuhlmann, F., Roark, J., Rainer, P., Detlev, S., Hemenway, T., Huhmer, A., Langridge, J., Connolly, B., Chadick, T., Holly, K., Eckels, J., Deutsch, E. W., Moritz, R. L., Katz, J. E., Agus, D. B., MacCoss, M., Tabb, D. L. and Mallick, P. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30(10): 918-920.
- Dinkel, H., Van Roey, K., Michael, S., Kumar, M., Uyar, B., Altenberg, B., Milchevskaya, V., Schneider, M., Kuhn, H., Behrendt, A., Dahl, S. L., Damerell, V., Diebel, S., Kalman, S., Klein, S., Knudsen, A. C., Mader, C., Merrill, S., Staudt, A., Thiel, V., Welti, L., Davey, N. E., Diella, F. and Gibson, T. J. (2016). ELM 2016--data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44(D1): D294-300.
- Edelstein, A., Amodaj, N., Hoover, K., Vale, R. and Stuurman, N. (2010). Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14: Unit14 20.
- Fodor, S. P., Read, J. L., Pirrung, M. C., Stryer, L., Lu, A. T. and Solas, D. (1991). Light-directed, spatially addressable parallel chemical synthesis. Science 251(4995): 767-773.
- Gerver, R. E., Gomez-Sjoberg, R., Baxter, B. C., Thorn, K. S., Fordyce, P. M., Diaz-Botia, C. A., Helms, B. A. and DeRisi, J. L. (2012). Programmable microfluidic synthesis of spectrally encoded microspheres. Lab Chip 12(22): 4716-4723.
- Gfeller, D., Butty, F., Wierzbicka, M., Verschueren, E., Vanhee, P., Huang, H., Ernst, A., Dar, N., Stagljar, I., Serrano, L., Sidhu, S. S., Bader, G. D. and Kim, P. M. (2011). The multiple-specificity landscape of modular peptide recognition domains. Mol Syst Biol 7: 484.
- Harink, B., Nguyen, H., Thorn, K. and Fordyce, P. (2019). An open-source software analysis package for Microspheres with Ratiometric Barcode Lanthanide Encoding (MRBLEs). PLoS One 14(3): e0203725.
- Huang, N., Siegel, M. M., Kruppa, G. H. and Laukien, F. H. (1999). Automation of a Fourier transform ion cyclotron resonance mass spectrometer for acquisition, analysis, and e-mailing of high-resolution exact-mass electrospray ionization mass spectral data. J Am Soc Mass Spectr 10(11): 1166-1173.
- Ivarsson, Y. and Jemth, P. (2019). Affinity and specificity of motif-based protein-protein interactions. Curr Opin Struct Biol 54: 26-33.
- Kunys, A. R., Lian, W. and Pei, D. (2012). Specificity profiling of protein-binding domains using one-bead-one-compound Peptide libraries. Curr Protoc Chem Biol 4(4): 331-355.
- Lam, K. S., Lebl, M. and Krchnak, V. (1997). The "One-Bead-One-Compound" combinatorial library method. Chem Rev 97(2): 411-448.
- Lam, K. S., Salmon, S. E., Hersh, E. M., Hruby, V. J., Kazmierski, W. M. and Knapp, R. J. (1991). A new type of synthetic peptide library for identifying ligand-binding activity. Nature 354(6348): 82-84.
- Liu, T., Qian, Z., Xiao, Q. and Pei, D. (2011). High-throughput screening of one-bead-one-compound libraries: identification of cyclic peptidyl inhibitors against calcineurin/NFAT interaction. ACS Comb Sci 13(5): 537-546.
- MacConnell, A. B., Price, A. K. and Paegel, B. M. (2017). An integrated microfluidic processor for DNA-encoded combinatorial library functional screening. ACS Comb Sci 19(3): 181-192.
- Meyer, K., Kirchner, M., Uyar, B., Cheng, J. Y., Russo, G., Hernandez-Miranda, L. R., Szymborska, A., Zauber, H., Rudolph, I. M., Willnow, T. E., Akalin, A., Haucke, V., Gerhardt, H., Birchmeier, C., Kuhn, R., Krauss, M., Diecke, S., Pascual, J. M. and Selbach, M. (2018). Mutations in disordered regions can cause disease by creating dileucine motifs. Cell 175(1): 239-253 e217.
- Mu, Y., Cai, P., Hu, S., Ma, S. and Gao, Y. (2014). Characterization of diverse internal binding specificities of PDZ domains by yeast two-hybrid screening of a special peptide library. PLoS One 9(2): e88286.
- Nadler, W. M., Waidelich, D., Kerner, A., Hanke, S., Berg, R., Trumpp, A. and Rosli, C. (2017). MALDI versus ESI: the impact of the ion source on peptide identification. J Proteome Res 16(3): 1207-1215.
- Neduva, V. and Russell, R. B. (2006). Peptides mediating interaction networks: new leads at last. Curr Opin Biotechnol 17(5): 465-471.
- Nguyen, H. Q., Baxter, B. C., Brower, K., Diaz-Botia, C. A., DeRisi, J. L., Fordyce, P. M. and Thorn, K. S. (2016). Programmable microfluidic synthesis of over one thousand uniquely identifiable spectral codes. Adv Opt Mater 5(3).
- Nguyen, H. Q., Brower, K., Harink, B., Baxter, B., Thorn, K. and Fordyce, P. (2017). Peptide library synthesis on spectrally encoded beads for multiplexed protein/peptide bioassays. SPIE 10061, Microfluidics, BioMEMS, and Medical Microsystems XV, 100610Z.
- Nguyen, H. Q., Roy, J., Harink, B., Damle, N. P., Latorraca, N. R., Baxter, B. C., Brower, K., Longwell, S. A., Kortemme, T., Thorn, K. S., Cyert, M. S. and Fordyce, P. M. (2019). Quantitative mapping of protein-peptide affinity landscapes using spectrally encoded beads. eLife 8: 40499.
- Tinti, M., Kiemer, L., Costa, S., Miller, M. L., Sacco, F., Olsen, J. V., Carducci, M., Paoluzi, S., Langone, F., Workman, C. T., Blom, N., Machida, K., Thompson, C. M., Schutkowski, M., Brunak, S., Mann, M., Mayer, B. J., Castagnoli, L. and Cesareni, G. (2013). The SH2 domain interaction landscape. Cell Rep 3(4): 1293-1305.
- Ueki, Y., Kruse, T., Weisser, M. B., Sundell, G. N., Larsen, M. S. Y., Mendez, B. L., Jenkins, N. P., Garvanska, D. H., Cressey, L., Zhang, G., Davey, N., Montoya, G., Ivarsson, Y., Kettenbach, A. N. and Nilsson, J. (2019). A consensus binding motif for the PP4 protein phosphatase. Mol Cell 76(6): 953-964 e956.
- Zhou, H. X. (2012). Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem Sci 37(2): 43-48.
Article Information
Copyright
![]() Hein et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Hein et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hein, J. B., Nguyen, H. Q., Cyert, M. and Fordyce, P. M. (2020). Protocol for Peptide Synthesis on Spectrally Encoded Beads for MRBLE-pep Assays. Bio-protocol 10(13): e3669. DOI: 10.21769/BioProtoc.3669.
- Nguyen, H. Q., Roy, J., Harink, B., Damle, N. P., Latorraca, N. R., Baxter, B. C., Brower, K., Longwell, S. A., Kortemme, T., Thorn, K. S., Cyert, M. S. and Fordyce, P. M. (2019). Quantitative mapping of protein-peptide affinity landscapes using spectrally encoded beads. eLife 8: 40499.
Category
Biochemistry > Protein > Synthesis
Biochemistry > Protein > Interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.