- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Superresolution Microscopy of Drosophila Indirect Flight Muscle Sarcomeres




Published: Vol 10, Iss 12, Jun 20, 2020 DOI: 10.21769/BioProtoc.3654 Views: 5582
Reviewed by: Imre GáspárGülçin ÇAKAN AKDOĞANNicanor González-Morales
Original research article
The authors used this protocol in:

Jan 2020
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Sarcomeres are extremely highly ordered macromolecular assemblies where proper structural organization is an absolute prerequisite to the functionality of these contractile units. Despite the wealth of information collected, the exact spatial arrangement of many of the H-zone and Z-disk proteins remained unknown. Recently, we developed a powerful nanoscopic approach to localize the sarcomeric protein components with a resolution well below the diffraction limit. The ease of sample preparation and the near crystalline structure of the Drosophila flight muscle sarcomeres make them ideally suitable for single molecule localization microscopy and structure averaging. Our approach allowed us to determine the position of dozens of H-zone and Z-disk proteins with a quasi-molecular, ~5-10 nm localization precision. The protocol described below provides an easy and reproducible method to prepare individual myofibrils for dSTORM imaging. In addition, it includes an in-depth description of a custom made and freely available software toolbox to process and quantitatively analyze the raw localization data.
Keywords: Drosophila flight musclesBackground
Structure of the sarcomeres has been studied in details by X-ray crystallography as well as with various EM methods leading to quasi-atomic models of the thin and thick filaments from numerous species. However, although these examinations resulted in a remarkably good understanding of the actin-myosin overlap region, spatial arrangement of the I-band and H-zone complexes remained largely unknown. Recent advances in fluorescent super-resolution microscopy (also called nanoscopy) provide spatial resolutions that are well below the diffraction limit. Most notably, single-molecule localization microscopy (SMLM) can deliver localization maps of multiprotein complexes with very high precision, virtually attaining a single protein size resolution (Sigal et al. , 2018).
We recently developed a standardized, powerful nanoscopic approach, combined with a structure averaging algorithm, that allowed us to determine the position of 27 muscle proteins in 1 day old adult Drosophila indirect flight muscles (IFMs) with a quasi-molecular, ~5-10 nm localization precision. By using this protein localization atlas, and by means of template based protein structure modeling, we assembled a refined I-band and H-zone model with an unparalleled scope and resolution (Szikora et al. , 2020). For our approach individual myofibrils were isolated from the IFM of Drosophila as described previously (Burkart et al. , 2007; Weitkunat and Schnorrer, 2014), with minor modifications. Individual muscle fibers of the flight muscle are ideal candidates for SMLM because they exhibit an extremely regular structural organization; their intrinsic auto-fluorescence is negligible, and they can be isolated very easily in large numbers. In addition, thickness of the fibers is below ~1.5 µm, therefore the fluorophores can be easily pumped into their dark, non-fluorescent state. As the focal plane is parallel to the myofibrils we can acquire two-dimensional projections of the sarcomeres which reveal the lateral distribution of proteins at the H-zones and I-bands. Since highly specific antibodies are available against different components of the sarcomere, and the organic fluorophores exhibit certain advantageous characteristics, i.e. , high brightness and photostability, we recommend the use of conventional immunofluorescent labeling. Typically, an SMLM measurement generates tens of thousands of microscopic images, to record hundreds of thousands of ‘single molecule events’. To evaluate these large datasets we developed a standardized data-flow protocol. For processing and quantification of the raw localization datasets, we developed a user friendly and freely available software tool (IFM Analyzer) with a detailed user guide including a sample dataset. We trust that with some minor modifications, our approach and the software tools developed can also be used for structural studies of similarly regular or quasi-regular molecular assemblies, such as synaptonemal complexes or chemical synapses.
Materials and Reagents
- Pipette tips
- 6-well plate
- Parafilm
- 1.5 ml microcentrifuge tube
- Precision cover glasses (#1.5, 24 x 24 mm; e.g., Marienfeld-superior; catalog number: 0 117640 )
- Standard fly food media in glass vials (e.g., BDSC Cornmeal Food)
- Cell Culture Multiwell Plate (e.g., Greiner Bio-One, catalog number: 657160 )
- Drosophila (isogenized w1118 strain)
- Glycerol for molecular biology, ≥ 99.0% (Sigma-Aldrich, catalog number: G5516-100ML )
- Adenosine triphosphate (ATP) (Alfa Aesar, catalog number: J10585 )
- Triton X-100 (Sigma-Aldrich, catalog number: X100-500ML )
- Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), molecular biology grade (Merck, catalog number: 324626-25GM )
- Magnesium chloride (MgCl2) (Merck, catalog number: M8266-100G )
- Potassium chloride (KCl) (Merck, catalog number: P9541-500G )
- 1,4-dithiothreitol (DTT) (Merck, catalog number: 10197777001 )
- Phenylmethylsulfonyl fluoride (PMSF) (Merck, catalog number: 10837091001 )
- Paraformaldehyde (Fisher scientific, 16% w/v aq. soln., methanol free, Alfa Aesar, catalog number: 433689M )
- Goat serum (Sigma-Aldrich, catalog number: G9023-10ML )
- Primary antibodies (see Table 1 for examples)
- Secondary antibodies (goat anti-rabbit, anti-mouse or anti-rat IgG highly cross-absorbed antibodies coupled to Alexa647; Thermo Fisher, catalog numbers: A-21245 , A-21235 , A-21247 )
- GFP antibodies (e.g., Thermo Fisher, catalog number: A6455 )
- Nanobodies (e.g., Chromotek, catalog number: gb2AF647 )
- FLAG antibodies (e.g., Sigma-Aldrich, catalog number: F7425 )
- Alexa FluorTM 488 Phalloidin (Thermo Fisher, catalog number: A12379 )
- Glucose (Sigma-Aldrich, catalog number: 49139 )
- Glucose-oxidase (Sigma-Aldrich, catalog number: G2133-50KU )
- Catalase (Sigma-Aldrich, catalog number: C100 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 204439 )
- Tris(hydroxymethyl)aminomethane (Sigma-Aldrich, catalog number: T5941 )
- Tris(2-carboxyethyl)phosphine (TCEP) (Sigma-Aldrich, catalog number: C4706 )
- β-mercaptoethylamine (MEA) (Sigma-Aldrich, catalog number: M6500 )
- Twinsil speed (Picodent, catalog number: 1300 1002 )
- Immersion oil (Olympus Type-F)
- 2x Relaxing solution (see Recipes)
- 1x Relaxing solution (see Recipes)
- Dissection solution (see Recipes)
- Fixative solution (see Recipes)
- Blocking solution (see Recipes)
- dSTORM imaging buffer (see Recipes)
Table 1. Examples of primary antibodies, labeling Drosophila IFM components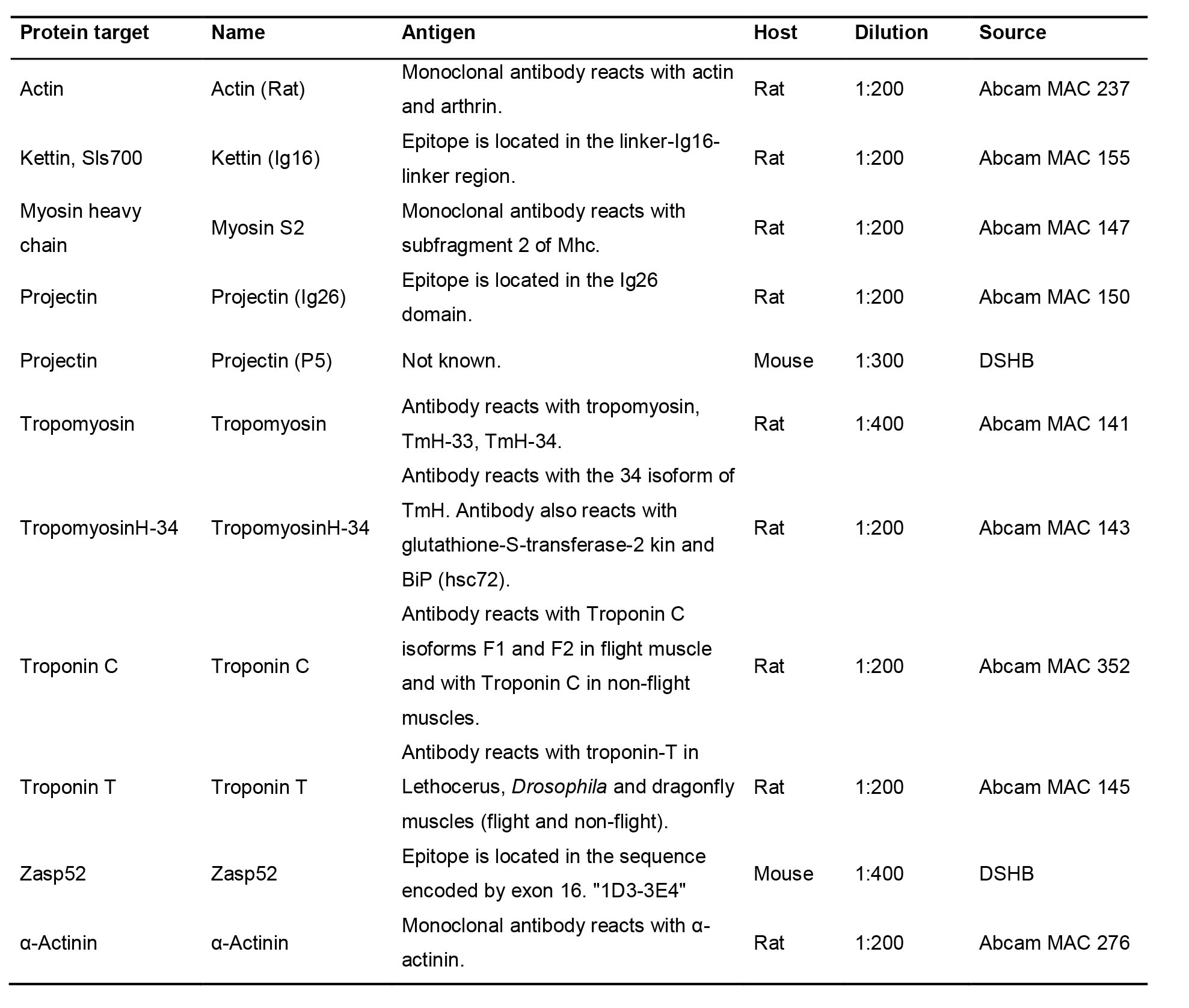
Equipment
- Standard fly husbandry equipment
- CO2 pad (e.g., FlyStuff flypad, Genesee Scientific, catalog number: 59-114 )
- Incubator, set to 25 °C
- Stereo microscope (e.g., Leica microsystems, model: M80 )
- Home-made dissection pad or a commercially available equivalent (see Procedures for details)
- Precision tweezers, straight with fine tips, 0.06 mm (Carl Roth DUMONT, catalog number: K342.1 )
- P2, P20, P100 and P1000 Micro Pipettes (e.g., Gilson, catalog numbers: F144801 , F123600 , F123615 and F123602 )
- Refridgerated centrifuge (e.g., Eppendorf, model: 5424 R , catalog number: 5404000010 )
- Humidity chamber (see Procedures for details)
- 647 nm laser source for fluorescence excitation (647 nm, Pmax = 300 mW; MPB Communications)
- 405 nm laser source for reactivation (405 nm, Pmax = 60 mW; Nichia)
- Nikon LU-N4 laser unit
- Nikon Ti-E inverted microscope frame with oil-immersion objective (CFI Apo 100x, 1.49 NA Nikon)
- Nikon Perfect Focus System
- Quad channel fluorescence filter set (Semrock LF405/488/561/635-A-000, Semrock FF01-446/523/600/677-25)
- Single channel fluorescence filter set (Semrock, catalog numbers: Di03-R635-t1 , BLP01-647R-25 )
- Filter Wheel for emission filters (Thorlabs, catalog number: FW102C )
- Bandpass emission filter 690/70 H (AHF, catalog number: F49-691 )
- 4f optical telescope in detector path, 2* (Thorlabs, catalog number: TTL200 )
- EM-CCD camera (Andor iXon3 897 BV EMCCD)
Software
- NIS Elements AR 5.02 (Nikon)
- Matlab R2018b (Mathworks) or newer with Image Processing Toolbox, Optimization Toolbox, Parallel Computing Toolbox and Curve Fitting Toolbox
- rainSTORM v3.1.7 (available here)
- Minimum System Requirements: 4-core CPU, 32GB RAM
Note: The User Guide is available on the download page. - IFM Analyzer v2.0 (available here)
Note: The User Guide is available on the download page. Example exported section files are available on the download page.
Procedure
A. Collecting flies for myofibril preparation
- Raise flies of the desired genotype in vials. Either make a fresh flip of the dissection stock or set a cross with at least 20 female virgin flies. Maintain bottles until the flies begin to hatch.
- Collect freshly hatched flies and keep them on fresh food until they reach the desired age.
Notes:
- Flies should be raised under standard laboratory conditions at 25 °C.
- As wild type we used an isogenized w1118 strain.
- For our analysis we dissected flies ~24 h after eclosion.
B. Dissecting flies for individual myofibril preparation
- Prepare dissection solution freshly from 2x relaxing solution and glycerol (see Recipes 1 and 3).
- Assemble the necessary equipment including two precision tweezers, a pipette, pipette tips, dissection pad, and the dissection solution on ice.
- Pipette 200 µl ice-cold dissection solution onto the dissection pad.
- Anesthetize flies and transfer them onto the dissection pad one by one. Remove the head, abdomen, wings and legs from the thorax using tweezers under a stereomicroscope while the flies are still anesthetized. Use IFMs from about 6 thoraces per tube.
- Move the thoraces into the dissection solution and tear them open.
- Incubate the hemi-thoraces for 2 h in dissection solution on ice or store them at -20 °C over night.
- Use tweezers to isolate the Dorsal Longitudinal flight Muscles (DLMs) from the thoraces and place them into an Eppendorf tube in dissection solution supplemented with 0.5% Triton X-100.
- Dissociate the DLMs by pipetting them ~10 times with medium speed using a 200 µl pipette.
- Spin at 10,000 x g for 2 min at 4 °C and wash myofibrils by re-suspending them in 200 µl 1x relaxing solution. To re-suspend the pellet pipette up and down ~10 times with medium speed.
- Repeat the centrifugation and washing step.
- Repeat the centrifugation and re-suspend the fibers in 1x relaxing solution. Use 20 µl 1x relaxing solution per DLMs isolated from 2 thoraces.
Notes:
- For a visual guide on the dissection see Xiao et al., 2017.
- Our home-made dissection pad is casted from silicon glue (black color). It offers a flat, slightly elastic, washable surface. Commercially available vinyl pads can be used instead.
- Sufficient dissociation is indicated by the disappearance of all the floating visible tissue pieces. Ideally, it is reached during the last washing/re-suspending step.
C. Fixation of the individual muscle fibers
- Prepare a humidity chamber
For the assembly of the humidity chamber use a non-transparent plastic box with a lid. It protects the samples from light, which is important in handling fluorochromes. Line the bottom of the box with a wet paper towel to increase humidity. Place the lid of a 6-well plate inside the humidity chamber and cover it with a slice of Parafilm. Use it as a clean and flat platform for the coverslips. - Prepare a 24 x 24 mm coverslip and place it on the Parafilm.
- Pipette a 20 µl drop of the myofibril suspension into the middle of the coverslip prior to fixation.
- Pipette 200 µl fixative solution (see Materials and Reagents) onto the myofibril drop and fix for 15 min at room temperature.
- Wash three times with 200 µl 1x relaxing solution.
Notes:
- Lid of the humidity chamber must seal properly to protect the samples during the overnight incubation in the refrigerator.
- Make sure that the samples never fully run dry.
- Apply normal wash intensity, the myofibrils will remain attached to the coverslip during regular washes.
D. Immunostaining for dSTORM imaging
- Block the samples in 200 µl blocking solution (see Recipe 5) for 30 min in a humidity chamber at room temperature.
- Add the primary antibodies in blocking solution and incubate in a humidity chamber overnight at 4 °C (see Table 1 for examples).
- Rinse five times with 200 µl blocking solution.
- Add the appropriate Alexa Fluor 647 conjugated secondary antibody in blocking solution (1:600, see Materials and Reagents) for 2 h at room temperature. Also add Phalloidin-Alexa488 (1:200, Life Technologies) as an independent marker.
- Rinse four times with 200 µl blocking solution and once with 200 µl filtered PBS.
- Keep the samples submerged in filtered PBS at 4 °C in a Petri dish or a 6-well plate, protected from light until dSTORM imaging.
Notes:
- Apply prolonged washing steps if high background is detected during imaging.
- Use the Phalloidin Alexa488 channel to unequivocally distinguish the H-zone and I-band during data processing.
- Achieving ideal labeling density is crucial. Multiple primary antibody dilutions must be tested at constant secondary antibody concentration. The starting dilution must be inspected with confocal microscopy before STORM imaging.
- Depending on the primary antibody, some samples must be processed immediately, while others can be stored for weeks prior to imaging without a significant drop in quality.
E. Sample preparation for dSTORM
- Prepare dSTORM imaging buffer freshly (see Recipe 6).
- Put 40 µl dSTORM imaging buffer on the slide, cover it with the coverslip containing your sample, and seal it with twinsil speed silicone.
- Clean and dry the outer surface of the coverslip if necessary.
- Place the sample on the microscope stage.
F. dSTORM acquisition
- Activate the focus stabilizing system.
- If the sample is labeled only with Alexa Fluor 647 the ‘Single channel filter set’ must be used, otherwise select the ‘Quad channel filter set’.
- Use the 647 nm excitation laser at low power (30 mW, 0.06% AOTF and < 10 W/cm2) to select undamaged, straight, and isolated myofibrils (Video 1).
- Adjust the focus and image the central part of the myofibril.
- Set ROI size to include only the excited region of the sample.
- Capture and save a snapshot image (Figures 1A and 1C).
- Close the shutter of the camera.
- Increase the 647 nm laser power (300 mW 100% AOTF and ≈2.5-3 kW/cm2) to trigger the photo-switching state of the fluorescent dyes. Wait 5-10 seconds and open the shutter to assess blinking of the fluorophores (Video 2).
Notes:- If there are overlapping molecules because of too dense labeling, bleach the sample for 1 minute by using simultaneously the 647 nm excitation laser at full power (300 mW 100% AOTF and ≈2.5-3 kW/cm2) and the 405 nm laser at moderate power (10 mW, 40% AOTF and ≈20 W/cm2), Repeat this bleaching procedure for multiple times. If blinking is still too dense, a new sample must be prepared with lower labeling density.
- If there are overlapping molecules because of insufficient excitation power density (and the laser power cannot be further increased), then stop the measurement, reduce the excitation spot size and restart the measurement. If you cannot reduce the beam size then prepare a new sample with lower labeling density.
- If high background is detected (e.g., IR background when the ‘Single channel filter set’ is used or anti-Stokes fluorescence from the second dye (Gajdos et al., 2020) when the ‘Quad channel filter set’ is used) use the 690/70 emission filter in addition to the LP 647 filter.
- For more details on the microscope configuration see Szikora et al., 2020.
 Video 2. Examples for ideal and suboptimal labeling density and fluorophore blinking behavior. In an ideal case blinking of the fluorophores should be bright and clearly separated from each other. In contrast to this, sparse labeling results in too few signals, whereas in case of too dense labeling the signals are not clearly separated from each other. Scale bar: 5 µm.
Video 2. Examples for ideal and suboptimal labeling density and fluorophore blinking behavior. In an ideal case blinking of the fluorophores should be bright and clearly separated from each other. In contrast to this, sparse labeling results in too few signals, whereas in case of too dense labeling the signals are not clearly separated from each other. Scale bar: 5 µm. - Acquire 20,000-50,000 frames at an exposure time of 20 ms or 30 ms, and with the EM gain set to 100.
Note: The exposure time is set based on the blinking properties of the fluorescent dye (camera count and the number of overlapping molecules). Ideally the exposure time equals the ON-state lifetime of a single fluorescence molecule. The number of acquired frames depends on the bleaching ratio and efficiency of the reactivation. - Use the 405 nm reactivation laser at any time if necessary at low power (< 1 mW).
- Repeat the dSTORM acquisition procedure to obtain data for at least 6 regions per sample.

Figure 1. Demonstration of the resolution achievable in the IFM with the combination of dSTORM and structure averaging. A, C. Widefield and B, D. dSTORM images of α-Actinin (MAC 276) immunostained flight muscle myofibrils. E. The reconstruction of α-Actinin distribution after structure averaging of rotationally and translationally registered STORM images.
Data analysis
A. Evaluation of the acquired dSTORM data using rainSTORM
- Start Matlab, change the working directory to the rainSTORM folder and run the “startup; rainSTORM” command.
Note: After a short initialization in the workspace, the main rainSTORM GUI should pop up. If the startup script fails, it will drop an error message in the workspace. Possible sources of errors or warnings are: old Matlab version or missing toolboxes. - Load a TIFF or ND2 stack by clicking ‘Browse’.
- Select the desired method for localization (use the default ‘Least-Squares Gaussian 2D linear Bg’ option first).
Note: Other localization and background subtraction methods are available in rainSTORM. See rainSTORM_2016_user_guide_ver1.pdf on the rainSTORM webpage for more details (available here). - Click ‘Process All’ to complete the localization process for the raw measurement data.
- Wait for a summarized image to show up, which will indicate the end of the process.
Note: A progress can also be tracked in the main Matlab window.
B. Analysis, correction and visualization of localizations using rainSTORM
- Select the ‘Simple Histogram’ reconstruction method and set 16 as the Reconstruction Scale.
Note: In our system this sets the super-pixel size to 10 nm. - Adjust the acceptance filters to the following:
For Thompson precision: ‘Std X’: 0-25 nm and ‘Std Y’: 0-25 nm
For PSF size: ‘Sigma X’: 0.7-1.5 px and ‘Sigma Y’: 0.7-1.5 px - Click ‘Process All Recons’ to do the initial reconstruction of the super-resolved image.
- Click ‘Show SupRes Image’ to display the reconstructed image (Figures 1B and 1D).
- To apply drift correction (if necessary), click the ‘Auto Drift Correction’ button.
- Click ‘Show SupRes Image’ to display the reconstructed image.
- Click ‘Report Current Recon’ to save the current reconstruction.
Note: The localization table is saved in csv file format, while the super-resolved images and the evaluation histograms are in png format.
C. Exporting individual sections
- After the reconstruction click the ‘Export Box Section’ button on the ‘rainSTORM_Image_Viewer’ window.
- A rectangle shaped area around the studied structure (“double line”, “single line” or “gap”) can be selected manually on the super resolved image.
Note: As a result, two files, a .csv and a .mat file are exported into the folder of the raw measurement data file. Their names are generated by attaching "ROI_Xxx_Yyy" to the original filename, where xx and yy are the x and y coordinates of the center of the selected areas.
D. Switching analysis tools
- Clear Matlab workspace and remove the rainSTORM folder from the path.
- Change the working directory to the IFM Analyzer folder and run the “startup; IFMAnalyser” command.
E. Creating localization distribution histograms using IFM Analyzer
- To import the sections click ‘Add Files’ on the ‘Process Sections Data’ tab (Figure 3A).
- Select the most appropriate model: ‘Single Line’, ‘Double Line’, or ‘Gap’.
- Adjust the fitting options, if needed.
Notes:- See the user guide for a more detailed description (available here).
- Use the default values first and change only one parameter at a time.
- Additional information on the geometric parameters of subsarcomeric structures can be found in the methods section of the original research paper (Szikora et al., 2020).
- Click the ‘Process Selected’ button to run the initial analysis on each file. This will predict the symmetry axis of the structure (Figure 2A), fit a second degree polynomial onto the structure (Figure 2B), and merge the localizations to create the localization distribution histogram (Figure 2C).
Notes:- When the ‘Auto Process’ option is disabled then each initial prediction must be verified manually. This can be done in a popup window after each fit, where each fitting option can be adjusted.
- The output files are saved in four subfolders (initLine, fittedCurve, histFigure, histData).
- After initial fitting, the selected sections can be moved to another tab for further analysis using the ‘Move to Process Histogram’ or the ‘Move to Merge’ buttons.
Note: The results are saved in subfolders.
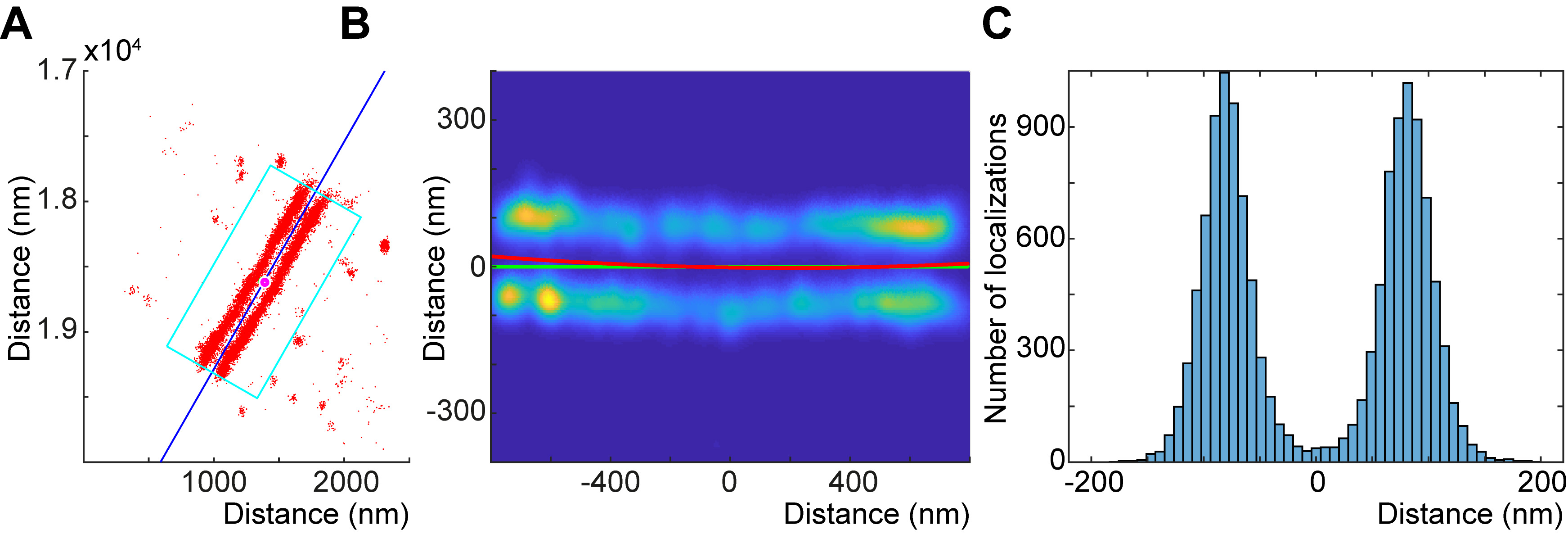
Figure 2. Creating localization distribution histograms using IFM Analyzer. A. Initial prediction of the symmetry axis of the structure (labeled with anti-Projectin (P5) and Alexa Fluor 647). B. A second degree polynomial (red line) is fitted onto the Gaussian blurred structure. C. The localization distribution histogram created using the second degree polynomial as a reference line.
F. Quantification of the localization distribution histograms using IFM Analyzer
- Quantification of the generated histograms (which are located in the histData subfolder) can be done in the ‘Process Histogram Data’ tab (Figure 3B).
- The acceptance range can be narrowed to discard the overlapping and out of focus localizations.
Note: For our microscope system the optimal PSF size was 0.9 (CCD pixel), so the acceptance range was set to 0.8-1.0. - Set the appropriate model function: single and double line structures are best described by the ‘Gaussian’ option. There is a special ‘Gap’ model for the gap structure.
- ‘Sample Type’ should be set based on the structure.
Note: Additional fitting options can be set in the ‘Fitting Options’ tab, see the user guide for a detailed description. - Click ‘Process Selected’ to start the quantification process.
Note: Visual representation of the fit can be checked in the ‘fittedWithImage’ subfolder, while the fitted parameters for each file are stored in the ‘fittedParams/allfittedparams.csv’ file.
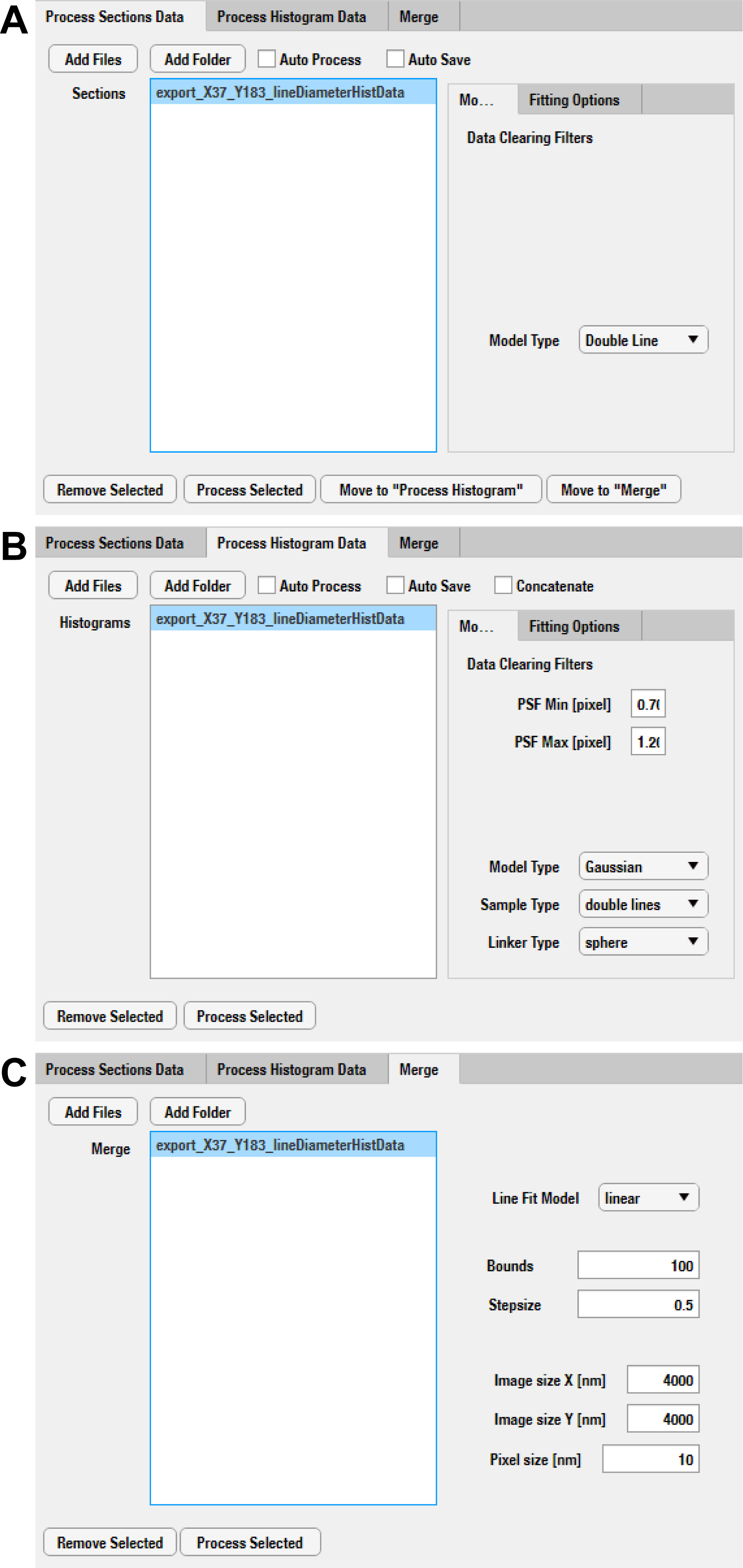
Figure 3. Steps of localization data processing with IFM Analyzer. A. Initial fitting, improved fitting and histogram generation are performed in the Process Sections Data tab. B. Histogram fitting and measurements are performed in the Process Histogram tab. C. The summed super-resolution image of the localization data is generated in the Merge tab.
G. Merge localizations from sections using IFM Analyzer
- To create a summed super-resolution image of the localization data, the analyzed sections must be moved from the ‘Process Section Data’ tab to the ‘Merge’ tab (Figure 3C).
Note: The files can be directly loaded from the histData subfolder. - Select all elements in the list.
Note: Adjust options to change the fitting model, image size or pixel size. - Click ‘Process Selected’ to generate a summed super-resolved image (Figure 1E).
Note: The final merged image is generated in the ‘mergeData’ subfolder in png format.
Notes
- Always repeat the staining experiment independently at least three times per labeled structure to achieve representative and reliable measurements.
- Although all the presented measurements were acquired with our custom-made dSTORM microscope system, and the collected data were processed with our rainSTORM localization code, we believe that other SMLM systems (Leica GSDIM, ONI Nanoimager etc.) and localization codes (ThunderSTORM, RapidSTORM etc.) can also be used to generate high quality input data files for the IMF Analyzer.
- Instead of the GLOX switching buffer the Everspark switching buffer (Provost et al., 2019) can also be used. Our preliminary tests using this buffer produced similar results without the time constrain of the oxygen scavenging system.
- We primarily used conventional immunofluorescent labeling, however we also tested some other approaches. For example, proteins can be localized through tags, such as GFP or FLAG. These studies are clearly appealing, as high quality primary antibodies are often missing, and in addition, in the tag based localization approaches position of the epitope is precisely known. Provided that functional equivalence of the tagged and the native proteins has been confirmed with an appropriate test (e.g., rescue experiment), this can be a very powerful approach. In our experience the commercially available GFP antibodies (e.g., Thermo Fisher, A6455 ) and nanobodies (e.g., Chromotek, gb2AF647 ) are ideal for dSTORM imaging, and the FLAG antibodies (e.g., Sigma Aldrich, F7425 ) are also usable.
- One potential disadvantage of conventional immunofluorescent labeling of individual myofibrils is that antibody penetration/epitope accessibility can be limited in the compact IFM due to the size of the antibodies. In this regard, due to their small size, nanobodies are expected to offer an improvement; in addition, they may increase localization precision as they reduce the epitope to fluorophore distance. Our preliminary data suggest that indeed, nanobodies are able to improve accessibility, however, our approach needs to be further optimized before making a strong conclusion about resolution.
- So far the Drosophila IFM was our major model system, however, our first set of preliminary data revealed that this approach is relatively easily adaptable to individual myofibers isolated from mice, and most likely to other non-drosophila myofibrils as well.
Recipes
- 2x Relaxing solution
40 mM phosphate buffer pH 7.0
10 mM MgCl2
10 mM EGTA
10 mM ATP
10 mM DTT
2 mM PMSF
Store at -20 °C and use within 6 months - 1x Relaxing solution
Mix the 2x relaxing solution with ddH2O 1:1 (v/v)
Prepare freshly and keep on ice during sample preparation - Dissection solution
Mix the 2x relaxing solution with glycerol 1:1 (v/v)
Prepare freshly and keep on ice during sample preparation - Fixative solution
Mix the 2x relaxing solution with 16% paraformaldehyde and ddH2O 2:1:1 (v/v)
Prepare freshly and keep on ice during sample preparation - Blocking solution
5% (v/v) Goat serum
0.3% (v/v) Trition X-100
in PBS
Store at 4 °C and use within one week - dSTORM imaging buffer (GLOX)
4% (w/v) glucose
2,000 U/ml glucose-oxidase
40,000 U/ml catalase
25 mM NaCl
22 mM tris(hydroxymethyl)aminomethane
4 mM tris(2-carboxyethyl)phosphine (TCEP)
100 mM β-mercaptoethylamine (MEA)
The imaging buffer must be freshly prepared for each experiment, since its lifetime is limited to a few (~3) hours.
Acknowledgments
The protocol described here in more details has been published in Szikora et al., 2020. This work was supported by the Hungarian Science Foundation (OTKA) (K109330 and K132782 to J.M.), the Hungarian Brain Research Program (KTIA_NAP_13-2-2014-0007 and 2017-1.2.1-NKP-2017-00002 to M.E. and J.M.), the National Research, Development and Innovation Office (GINOP-2.3.2-15-2016-00001, GINOP-2.3.2-15-2016-00032 to J.M. and GINOP-2.3.2-15-2016-00036 to M.E.), and an OTKA Postdoctoral Fellowship (PD 128623 to S.S.). We would like to thank the Idylle Labs for sending a test kit of the Everspark by Eternity buffer.
Competing interests
The authors declare no competing financial interests.
References
- Burkart, C., Qiu, F., Brendel, S., Benes, V., Hååg, P., Labeit, S., Leonard, K. and Bullard, B. (2007). Modular proteins from the Drosophila sallimus (sls) gene and their expression in muscles with different extensibility. J Mol Biol 367(4): 953-969.
- Gajdos, T., Hopp, B. and Erdelyi, M. (2020). Hot-Band Anti-Stokes Fluorescence Properties of Alexa Fluor 568. J Fluoresc. doi.org/10.1007/s10895-020-02496-0.
- Provost, A., Rousset, C., Bourdon, L., Mezhoud, S., Reungoat, E., Fourneaux, C., Bresson, T., Pauly, M., Beard, N., Possi-Tchouanlong, L., Grigorov, B., Bouvet, P., Diaz, J. J., Chamot, C., Pecheur, E. I., Ladaviere, C., Charreyre, M. T., Favier, A., Place, C. and Monier, K. (2019). Innovative particle standards and long-lived imaging for 2D and 3D dSTORM. Sci Rep 9(1): 17967.
- Sigal, Y. M., Zhou, R. and Zhuang, X. (2018). Visualizing and discovering cellular structures with super-resolution microscopy. Science 361(6405): 880-887.
- Szikora, S., Gajdos, T., Novák, T., Farkas, D., Földi, I., Lenart, P., Erdélyi, M. and Mihály, J. (2020). Nanoscopy reveals the layered organization of the sarcomeric H-zone and I-band complexes. J Cell Biol 219(1). DOI: 10.1083/jcb.201907026.
- Weitkunat, M. and Schnorrer, F. (2014). A guide to study Drosophila muscle biology. Methods 68(1): 2-14.
- Xiao, Y. S., Schöck, F. and González-Morales, N. (2017). Rapid IFM Dissection for Visualizing Fluorescently Tagged Sarcomeric Proteins. Bio-protocol 7(22): e2606.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Szikora, S., Novák, T., Gajdos, T., Erdélyi, M. and Mihály, J. (2020). Superresolution Microscopy of Drosophila Indirect Flight Muscle Sarcomeres. Bio-protocol 10(12): e3654. DOI: 10.21769/BioProtoc.3654.
- Szikora, S., Gajdos, T., Novák, T., Farkas, D., Földi, I., Lenart, P., Erdélyi, M. and Mihály, J. (2020). Nanoscopy reveals the layered organization of the sarcomeric H-zone and I-band complexes. J Cell Biol 219(1). DOI: 10.1083/jcb.201907026.
Category
Cell Biology > Cell imaging > Super resolution imaging
Molecular Biology > Protein > Detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.