- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Live Cell Measurement of the Intracellular pH of Yeast by Flow Cytometry Using a Genetically-Encoded Fluorescent Reporter
Published: Vol 10, Iss 12, Jun 20, 2020 DOI: 10.21769/BioProtoc.3653 Views: 5114
Reviewed by: Steven BoeynaemsIndranil MalikAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2019


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The intracellular pH of yeast is a tightly regulated physiological cue that changes in response to growth state and environmental conditions. Fluorescent reporters, which have altered fluorescence in response to local pH changes, can be used to measure intracellular pH. While microscopy is often used to make such measurements, it is relatively low-throughput such that collecting enough data to fully characterize populations of cells is challenging. Flow cytometry avoids this drawback, and is a powerful tool that allows for rapid, high-throughput measurement of fluorescent readouts in individual cells. When combined with pH-sensitive fluorescent reporters, it can be used to characterize the intracellular pH of large populations of cells at the single-cell level. We adapted microscopy and flow-cytometry based methods to measure the intracellular pH of yeast. Cells can be grown under near-native conditions up until the point of measurement, and the protocol can be adapted to single-point or dynamic (time-resolved) measurements during changing environmental conditions.
Keywords: Flow cytometryBackground
The intracellular pH of yeast is correlated with characteristics like viability and growth rate, and the regulation of intracellular pH consumes a large proportion of cellular energetic resources (Orij et al., 2011). However, intracellular pH can change rapidly and is highly environmentally sensitive, so it is crucial to have a fast, minimally perturbative method of measurement for this important aspect of cell physiology.
Genetically-encoded biosensors that convert local concentrations of a compound of interest into fluorescent readouts have revolutionized our ability to characterize the intracellular environment. For some sensors, the absolute fluorescence intensity is correlated with the readout. This can be a problem when performing in-cell measurements, since the fluorescence depends both on the sensor expression level, which varies cell to cell, and the characteristic of interest. Ratiometric sensors, which depend on the ratio of fluorescence in two different parts of the spectrum in the same fluorophore, do not suffer from this drawback. One such sensor, pHluorin (Miesenböck et al., 1998), is a pH-sensitive fluorescent biosensor based on GFP that can be used to measure intracellular pH; the emission intensity (measured around 520 nm) after excitation in the near-UV and blue (405 and 488 nm typically) varies with pH such that the ratio of emission intensities can be related to pH.
In this protocol, we outline how to use pHluorin to measure intracellular pH in living budding yeast cells using flow cytometry. Flow cytometry is a method that combines microfluidic focusing and optical interrogation to measure the fluorescence of single cells in liquid culture with little to no special sample preparation required. Others have also used this method to measure intracellular pH in yeast cells (Weigert et al., 2009; Valkonen et al., 2013). The advantage of flow cytometry is that it is much higher throughput than microscopy-based methods (Bagar et al., 2009; Orij et al., 2009), while still being able to characterize individual cells and measure the fluorescence of multiple fluorophores. This access to both single-cell measurements and enough data to generate population-level statistics with a great deal of confidence is highly valuable. One potential downside to using flow cytometry to analyze the fluorescence of biosensors is that the spatial distribution of the fluorophore within the cell is not accessible (at least with traditional flow cytometry), as only a single, average fluorescence value in each channel is reported for each event (cell). However, for intracellular pH measurements in particular, the variation in any compartment (here, the cytosol, although pHluorin can be targeted to other organelles) (Orij et al., 2009) is expected to be minimal due to the unique properties of proton exchange in buffered aqueous solutions such as the cellular interior (Boron, 2004).
The outline of a typical experiment is illustrated in Figure 1. Cells expressing pHluorin are suspended in buffer of known pH and an ionophore, in this case nigericin, is added. This addition makes the cell membrane permeable to protons and thus equilibrates the intracellular and extracellular pH. The ratiometric fluorescence of pHluorin is measured for these cells with known intracellular pH, and then these data are used to construct a calibration curve that can be used to convert measured fluorescence ratios in other cells to real intracellular pH values.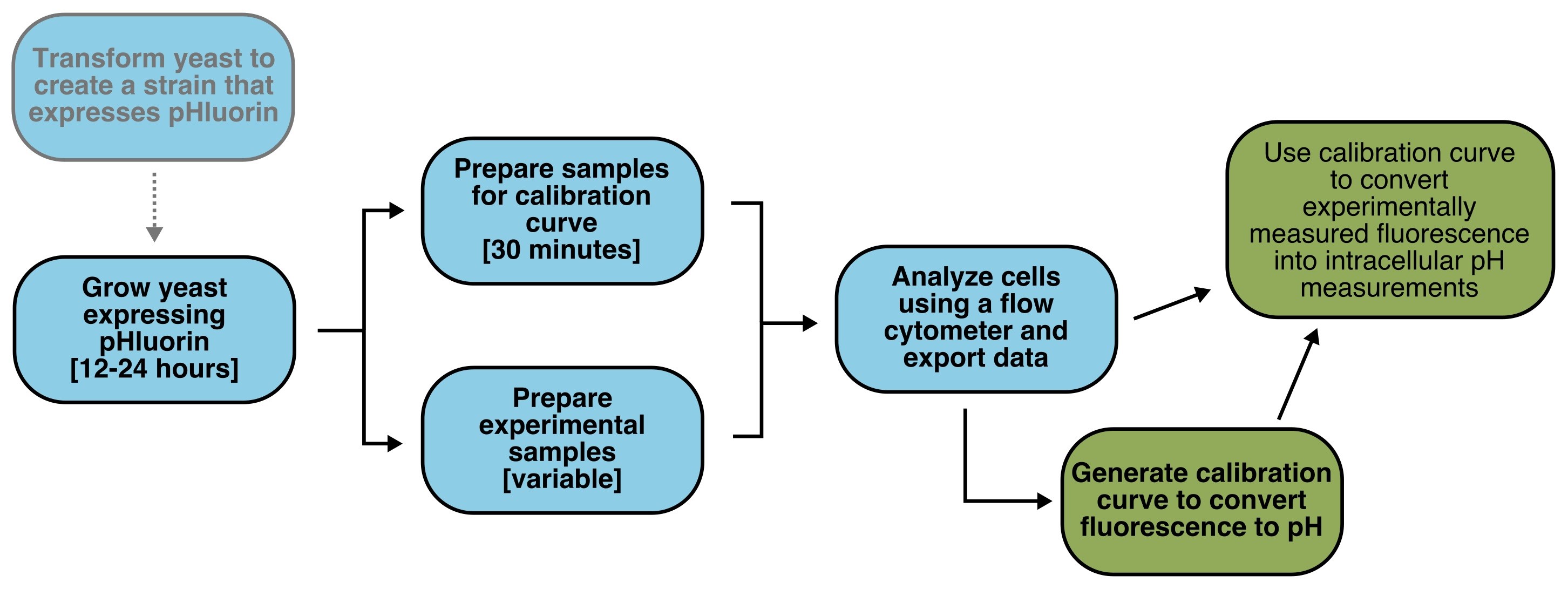
Figure 1. Flowchart of Protocol. Experimental steps are colored blue, analysis steps are in green. Optional steps are in gray text.
Materials and Reagents
- 50 ml conical flasks (Olympus Plastics, catalog number: 28-108 )
- 0.22 μm filter (Fisher, catalog number: CLS 431118 )
- 5 ml polystyrene round-bottom tubes, 12 x 75 mm style (Corning Falcon, catalog number: 358058 )
- Strain of Saccharomyces cerevisiae from the S288C background, such as BY4741/2/3
Alternatively, another strain of yeast may be used, as long as both non-fluorescent and pHluorin-expressing strains are available or can be made by the investigator (see optional reagents below). More information on yeast strains and genotypes can be found at https://wiki.yeastgenome.org/index.php/Commonly_used_strains. - 2-Deoxy-D-glucose (Sigma, Sigma Life Sciences, catalog number: D8375-5G )
- Nigericin (Adipogen, Adipogen Life Sciences, catalog number: AG-CN2-0020 ), prepared as a 10 mM stock in 100% ethanol and stored at -20 °C
- Yeast Nitrogen Base (YNB) + Nitrogen (Sunrise Scientific, catalog number: 1501-250 )
- Synthetic Complete (SC) dropout mix (Sunrise Scientific, catalog number: 1300-030 )
- D-(+)-Glucose (Research Products International, catalog number: G32040-5000.0 )
- MES (Fisher, catalog number: BP300-100 )
- HEPES (GoldBio, catalog number: H-400-1 )
- KCl (potassium chloride) (Fisher, catalog number: P217-3 )
- NaCl (sodium chloride) (Fisher, catalog number: BP358-212 )
- 2 M HCl (hydrochloric acid) (Fisher, catalog number: A144-212 )
- 2 M KOH (potassium hydroxide) (Sigma, catalog number: 484016-1KG )
- Ammonium acetate (Sigma, catalog number A1542-250G)
- 2X Calibration Curve Buffer (see Recipes)
- Yeast growth media (see Recipes)
Optional reagents
- pCGT05 or pHluorin expression vector (see Notes and attached plasmid map for more details; if the strain expressing pHluorin does not already exist and must be made by the investigator, then this reagent is not optional)
- PmeI (New England Biolabs, catalog number: R0560S ; comes with CutSmart® Buffer)
- Salmon testes DNA (Millipore Sigma, catalog number: D1626-250MG ) prepared as a 2 mg/ml solution in TE Buffer (see Recipes)
- Molecular Biology grade agarose (Apex Bioresearch Products, catalog number: 20-102GP )
- 10x TBE Buffer (Bio-Rad, catalog number: 161-0733 ), diluted to 1x in ultrapure water
- SCD (2%) plates without leucine, for selection
- Tris-HCl (Fisher, catalog number: BP153-1 )
- Tetrasodium EDTA (Fisher, catalog number: S311-100 ) prepared as a 1 M pH 8.0 stock
- Lithium Acetate (Sigma, catalog number: L6883-1KG )
- Polyethylene glycol (PEG) 3350 (Sigma, catalog number: P4338-500G )
Equipment
- Microcentrifuge capable of spinning 1.5 ml tubes at 3,000 x g
- BD Biosciences LSR Fortessa flow cytometer (see note for instrument settings), or any flow cytometer capable of exciting between 380 and 410 nm and between 470-490 nm, and measuring emission (for both excitations) between 500 and 550 nm (see Notes). Other laser configurations could also be used; see Miesenböck et al. 1998 for full characterization of pHluorin
- Electronic pH meter such as the Mettler Toledo SevenCompact pH meter S220 (Mettler Toledo, model: 30019032 )
- (Optional) For yeast transformation
- Heat block/dry bath/water bath
- Mold for casting agarose gels
- Standard electrophoresis power supply and setup for running agarose gels
- Heat block/dry bath/water bath
Software
- Software that can read FCS files. In this protocol we use R (CRAN, https://www.r-project.org/) with the flowCore package (Bioconductor, https://bioconductor.org/packages/release/bioc/html/flowCore.html); see example script for data analysis pipeline
Procedure
- (Optional) Create pHluorin-expressing yeast strain
- To integrate at the leu2 locus (S288C backgrounds), cut vector pCGT05 with PmeI by mixing the following reagents and incubating at 37 °C for 60 min.
Reagent (Concentration) Amount (μl) pCGT05 (~200 ng/μl) 5 Cutsmart Buffer (5x) 5 PmeI (10,000 units/ml) 1 Ultrapure water 39 - Alternatively, a plasmid with a selectable marker may be used rather than integration; see Notes. Use this as a template in the transformation in Step A3 and plate on appropriate selection media.
- If the leu2 locus is not available in the desired strain, integration at another locus is also possible.
- Alternatively, a plasmid with a selectable marker may be used rather than integration; see Notes. Use this as a template in the transformation in Step A3 and plate on appropriate selection media.
- (Optional) Check digestion efficacy
To check digestion, run 2-5 μl of product on a 1% agarose gel, along with uncut vector and a ladder. The cut product should run at two bands, one at 5.2 kb and the other at 3.5 kb; see Figure 2.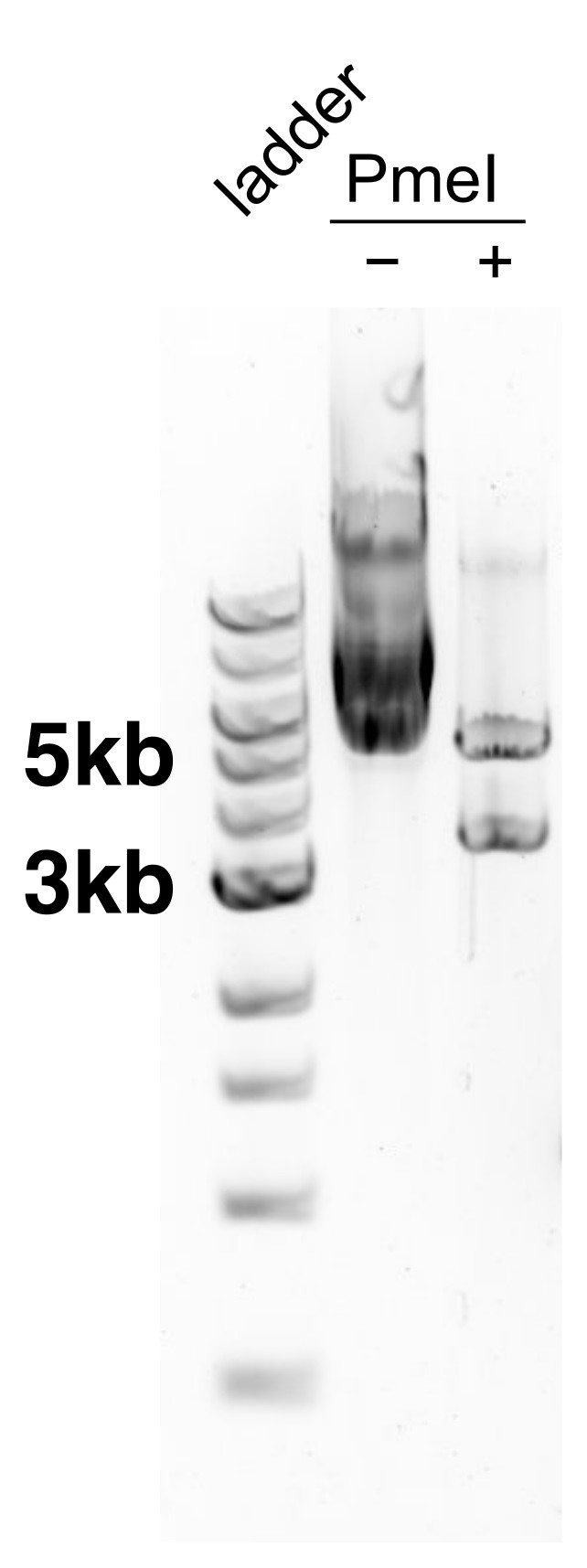
Figure 2. Vector pCGT05 before and after cutting with PmeI. The first lane is a ladder for size reference, the second lane is the vector without incubation with the restriction enzyme PmeI, the third lane is the vector after incubation with PmeI (there are two cut sites such that enzymatic digestion yields two fragments). - Transform into desired strain of yeast
- To integrate into the leu2 locus, follow a standard lithium acetate yeast transformation protocol (Gietz and Schiestl, 1994) using the product of the above digestion reaction as the DNA template.
- Screen colonies on SC plates without leucine (30 °C, allow 2-3 days for growth), and further colony purify on selection plates followed by non-selective plates to ensure that no wild-type cells remain.
- Cells in media may be mixed with glycerol to form a 20% final solution and stored at -80 °C for years.
- To integrate into the leu2 locus, follow a standard lithium acetate yeast transformation protocol (Gietz and Schiestl, 1994) using the product of the above digestion reaction as the DNA template.
- To integrate at the leu2 locus (S288C backgrounds), cut vector pCGT05 with PmeI by mixing the following reagents and incubating at 37 °C for 60 min.
- Grow cells (night before experiment)
- Inoculate starter cultures (at least 5 ml) of each strain of yeast you’d like to measure; include wild-type yeast not expressing pHluorin (for background subtraction) and a sufficient volume of pHluorin-expressing cells to both generate experimental samples and make a calibration curve to convert fluorescence to pH (see below for volume estimates).
- Aim for cells to be at around OD600 0.05-0.1 by the following morning (about 16 h later) so that they can either be analyzed right away, or mildly diluted in order to be analyzed later the same day.
- The calibration curve will require approximately 5 ml of pHluorin-expressing yeast at OD600 0.05-0.2.
- The target OD600 for experimental samples is 0.05 to 0.1, lower than what is typically used for microscopy or biochemistry-based experiments. Calibration curve samples can be slightly higher, up to OD600 0.2.
- Aim for cells to be at around OD600 0.05-0.1 by the following morning (about 16 h later) so that they can either be analyzed right away, or mildly diluted in order to be analyzed later the same day.
- Prepare 2x Calibration Curve Buffer. It can be prepared ahead of time and stored at 4 °C.
- Inoculate starter cultures (at least 5 ml) of each strain of yeast you’d like to measure; include wild-type yeast not expressing pHluorin (for background subtraction) and a sufficient volume of pHluorin-expressing cells to both generate experimental samples and make a calibration curve to convert fluorescence to pH (see below for volume estimates).
- Prepare calibration curve (morning of experiment)
- Add 5 ml of 2x Calibration curve buffer to eight 50 ml conical tubes. Add 3 ml of ultrapure water (or sufficient volume such that the electrode of the pH meter can be fully submerged in the liquid, keeping in mind that variable amounts of acid and base will need to be added to adjust the pH).
- Using concentrated HCl and KOH, adjust the pH of each tube to half pH units ranging from 5.0 to 8.5 (8 total; this spans the range of pH values to which the fluorophore is sensitive; measurement outside this range is not accurate). The initial pH should be in the range of 5.5-6.5.
Note: Make sure that the buffer is at room temperature before adjusting pH, as the pH can be temperature-sensitive. - Once the pH is in the correct range, add ultrapure water until the final volume is 10 ml (calibration curve buffer is now at 1x). Note the final pH and record the exact value on the outside of the tube.
pH-adjusted 1x calibration curve buffer (without ionophore) can be stored at 4 °C for around a month. When re-using, bring to room temperature, re-measure the pH, and note any changes on the outside of the tube. Small changes (~0.05 pH units) are tolerable, but large changes indicate that the buffer should be discarded and re-made from the 2x stock.
- Prepare the calibration curve (~30 min before experiment)
- Aliquot 1 ml of calibration curve buffer at each pH into labeled 1.5 ml tubes and to each tube add 1 μl of 10 mM nigericin in 100% ethanol to a final concentration of 10 μM. This step should be done as close to the time of measurement as possible, as the ionophore degrades more rapidly at room temperature.
- Label additional 1.5 ml tubes for each pH that will be measured in the calibration curve (for cells), plus one additional tube for unlabeled cells.
- Once cells are between OD600 0.05 and 0.2, aliquot ~500 μl of pHluorin-expressing cells into each labeled tube. Aliquot one additional tube of unlabeled cells, which will be used for background subtraction.
- Spin cells at 3,000 x g for 3 min. Decant supernatant (media) with a pipette, being careful to remove as much of the liquid as possible without disturbing the cell pellet.
- Briefly wash cells by resuspending in 100 μl of deionized water and spinning at 3,000 x g for 3 min. Decant supernatant.
- Resuspend cells in 400 μl of the appropriate pH prepared calibration curve buffer with nigericin.
- If many cells were lost during decanting (for example if the pellet was disturbed by pipetting) or if the concentration is low to begin with, a smaller volume of buffer may be used, but at least 200 μl of volume is needed to make the measurement (this may vary with the model of cytometer used to make the measurements).
- The unlabeled cells may be resuspended in any pH, as the fluorescence background does not vary with the pH of the buffer.
- If many cells were lost during decanting (for example if the pellet was disturbed by pipetting) or if the concentration is low to begin with, a smaller volume of buffer may be used, but at least 200 μl of volume is needed to make the measurement (this may vary with the model of cytometer used to make the measurements).
- Allow cells to incubate in buffer for 15 min to equilibrate pH. After this, proceed to Procedure E as rapidly as possible. It may be possible to hold the cells in buffer for longer without loss of integrity or a change in the fluorescence values, but this should be tested empirically by running full curves multiple times at different time intervals.
- Analyze samples on the flow cytometer
- Briefly vortex tube or mix by inversion before loading each sample.
- Add 200 μl of wild type (non-pHluorin-expressing) cells, in media, into a 5 ml polystyrene flow tube. Place the tube on the flow cytometer and begin acquiring data. While running the sample on the lowest possible flow rate, isolate the events associated with cells by drawing a gate in the forward scatter vs. side scatter plot to collect the vast majority of cell events, see Figure 3 for typical scattering profile.
- Collect at least 10,000, but preferably 20,000+ events for each sample.
- Voltage settings for a BD LSR Fortessa instrument are given in Table 1. Other instruments and settings can be used; see Notes for more details.
- It is essential that the same voltages are used within the same experiment, and preferably should not be changed even between experiments. Data taken with different voltage settings cannot be directly compared.
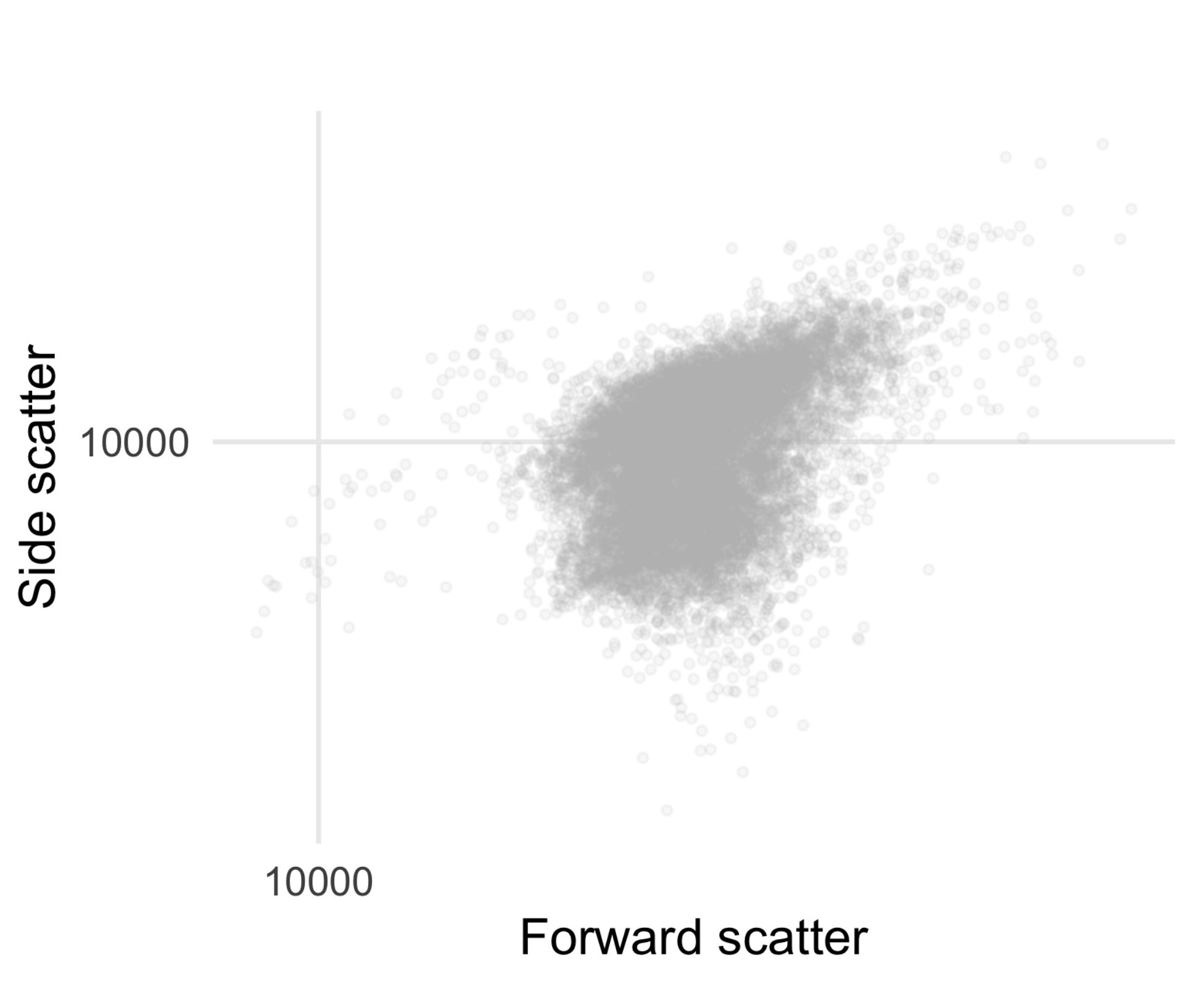
Figure 3. Typical scattering profile of Saccharomyces cerevisiae cells. Data from a BD Biosciences LSR Fortessa with voltages as listed in Table 1.
Table 1. Instrument Settings (BD LSR Fortessa)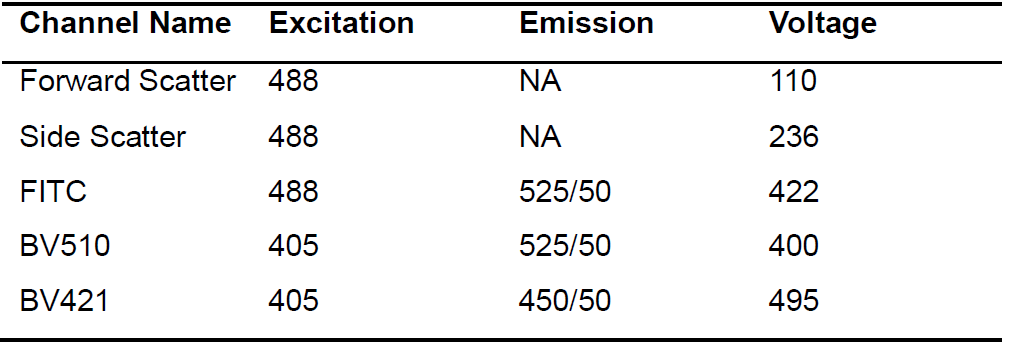
- Collect at least 10,000, but preferably 20,000+ events for each sample.
- Using the same settings and volumes, run and collect data for pHluorin-expressing cells in media, and wild type cells in calibration curve buffer. All of these samples are essential for background subtraction.
- Using the same settings and volumes, run and collect data for all the samples in calibration curve buffer prepared in Procedure C.
- Run any additional experimental samples using the same settings.
Note: In the case of experimental samples in different medium, a separate sample of unlabeled cells in the corresponding medium must also be analyzed in order to do proper background subtraction; see Data analysis section below.
- Export Data
Export all data. The standard format for flow cytometry data is the .fcs format; data analysis below assumes data in this format.
Data analysis
The goal of the analysis is to first use data from cells in buffer to construct a calibration curve, which can then be used to convert the fluorescence readings of cells in media to intracellular pH. In the example script, the two green channels used are called ‘FITC’, corresponding to excitation at 488 nm and emission at 525/50 nm, and ‘BV510’, corresponding to excitation at 405 nm and emission at 525/50 nm. Below, they will be referred to below as F405 for emission intensity at 525 after excitation at 405, and F488 for emission intensity at 525 after excitation at 488. Superscripts will refer to the strain and background.
The following is a general description of the data analysis pipeline. An example script executing this analysis using the R programming language is attached.
- Load the data, contained in an FCS file, and convert it to human-readable data in table format
In the attached example script, this is accomplished by reading the file using the flowCore package for the R programming language, and then converting the resulting object into an R data frame. - Calculate background readings
The median fluorescence of unlabeled cells (in both the FITC and the BV510 channels) will be subtracted as background. Unlabeled cells in buffer will be used to subtract background from labeled cells in buffer, and likewise for samples consisting of cells in media. - Make calibration curve
- For each cell (event), calculate the background-subtracted ratio of fluorescence that varies with pH.
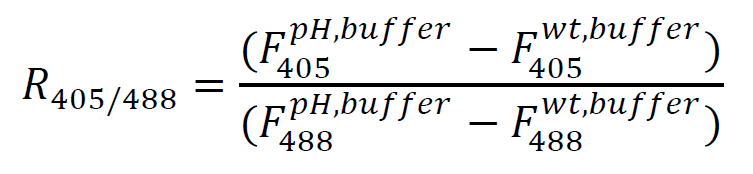
- Bin/group events from each calibration curve sample, corresponding to each pH buffer. Calculate the median fluorescence ratio for each pH. It may be helpful to establish a fluorescence cutoff in order to account for any low, non-expressing, or damaged cells. However, this protocol should retain the majority of the events, and may not be necessary for strains with pHluorin genomically integrated.
- To make the calibration curve, first plot the known pH of each buffer against the median R405/488 value, see Figure 4 for an example calibration curve.
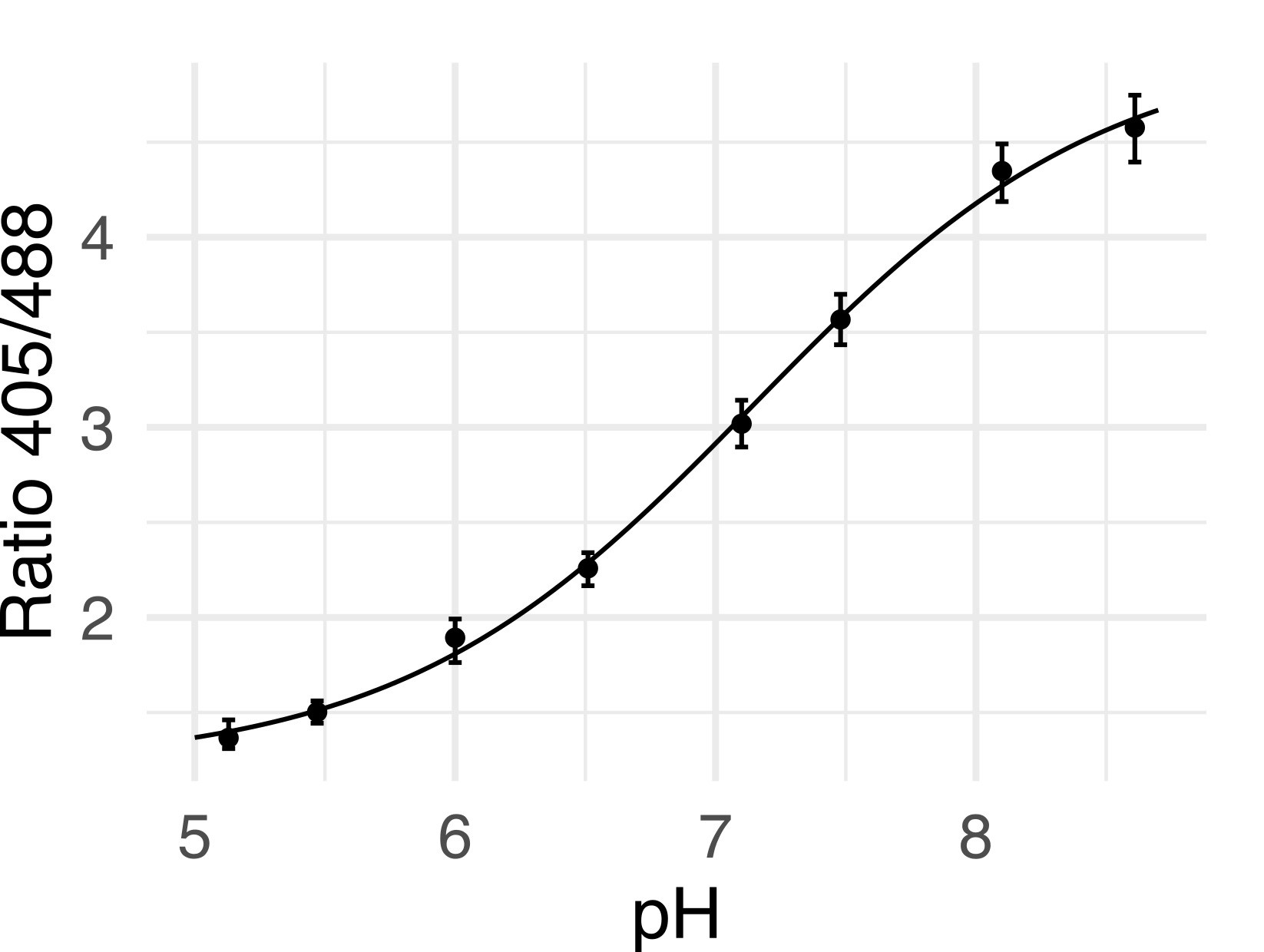
Figure 4. pHluorin calibration curve. Points are medians of a population of at least 10,000 cells, error bars are the 25th and 75th percentile. - Fit the points to a sigmoid of the form:
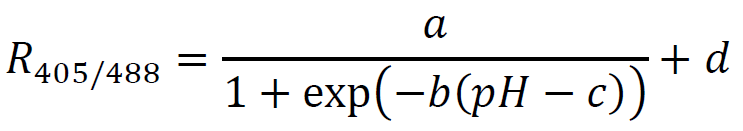
where a, b, c, and d are fitting parameters (a corresponds to the max of the curve, b to the steepness of the rise, c to the midpoint, and d to the baseline). In the example script this is accomplished using the nonlinear least squares method implemented by the nls() function in the R programming language; see documentation at https://stat.ethz.ch/R-manual/R-devel/library/stats/html/nls.html for more information on this method, and Figure 4 for an example calibration curve. Once these parameters have been fit, this function can be used to map between the fluorescence ratio for any individual cell and intracellular pH by calculating the background subtracted R405/488 value, and rearranging the above equation as follows: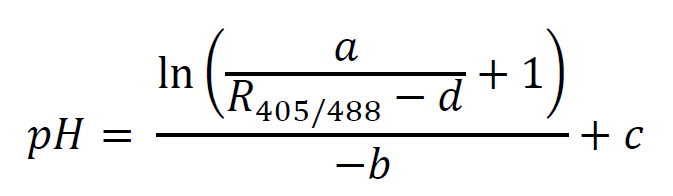
Note: This function can only be used in the range of sensitivity of the fluorophore, pH 5.0 to 8.5 (Miesenböck et al., 1998). In general, a calibration curve must extend to these limits in order to set the baselines correctly, and only ratios that fall within the minimum and maximum pHs used in the calibration curve can be confidently assigned to a pH. Ratios that fall outside this range are ambiguous and should not be analyzed.
- For each cell (event), calculate the background-subtracted ratio of fluorescence that varies with pH.
- Analyze experimental samples
- Subtract background from each experimental sample by first calculating the median fluorescence of unlabeled cells in the same medium as the sample, then calculating the R405/488 as in step C1 above (substituting F405 and F488 values for unlabeled cells in medium rather than buffer).
- Using the relationship established in step C4, convert R405/488 to pH.
Notes
- On yeast strains and strain availability: In the attached example script, data collected from strain yCGT028, a diploid (BY4743 background) yeast strain expressing pHluorin from the leu2 locus, which is empty (leu2Δ0) in the parent strain, and SSA4::mCherry. However, any strain expressing pHluorin, either from the genome or from a plasmid maintained with a selectable marker could be used in this protocol. Because the probe is ratiometric, the expression level variation expected for plasmid expression should not significantly affect the data analysis, however care should be taken that any cells that fail to express the fluorophore are not included; because expressing and non-expressing cells are easily distinguished (see Figure 5), non-expressing cells can be manually gated out on the instrument, or excluded during data analysis.
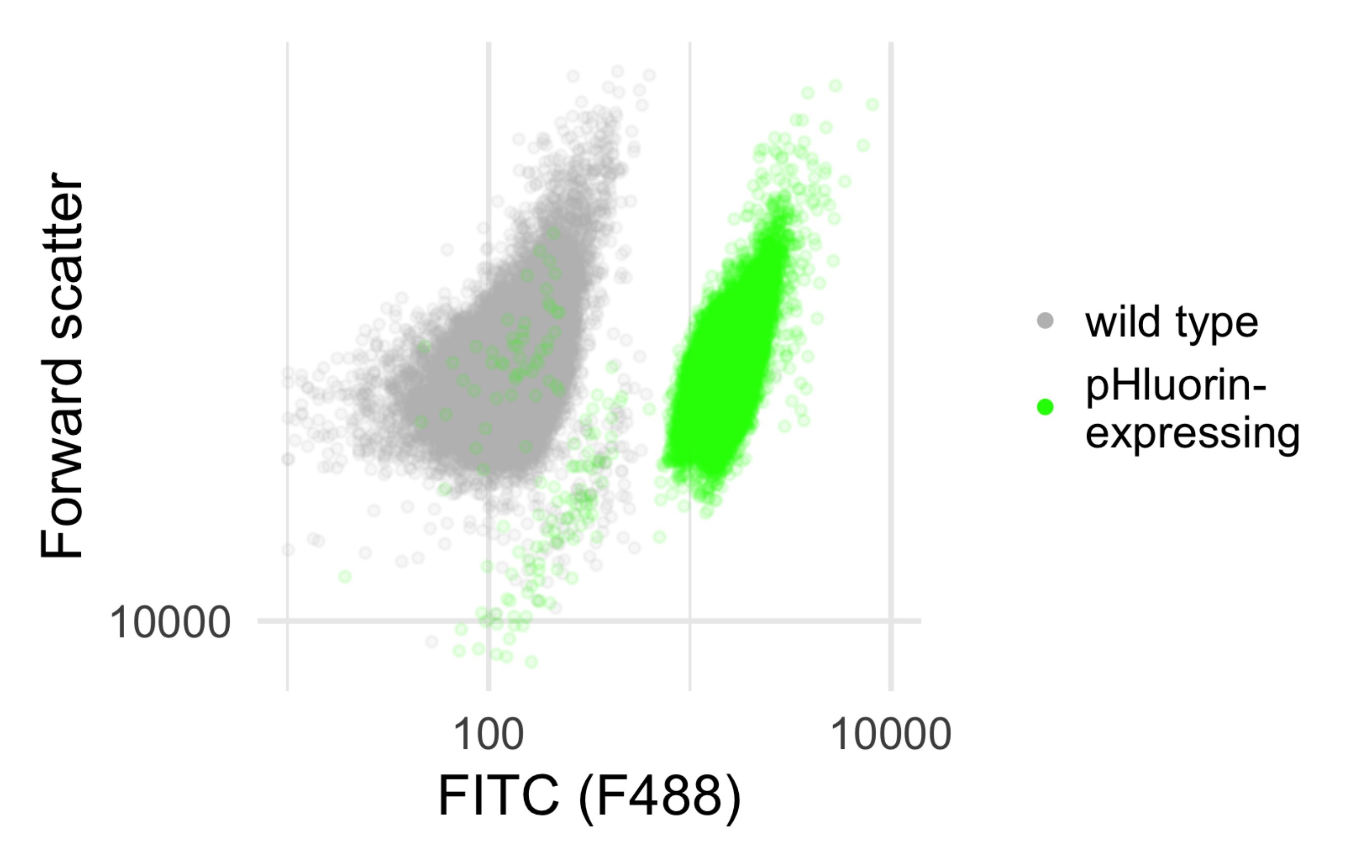
Figure 5. Separation of pHluorin-expressing and wild type populations by fluorescence. Data from a BD Biosciences LSR Fortessa with voltages as listed in Table 1. - On model of flow cytometer: In this protocol we give specific instructions for running samples on a BD Biosciences LSR Fortessa; but any cytometer capable of measuring excitation and emission in the correct range: excitation between 380 and 410 nm and between 470 and 490 nm and emission (for both excitations) between 500 and 550 nm. To set the voltages for these channels, it is helpful to run both cells in pH 5 and pH 8.5 calibration curve buffer, as these represent the maximum and minimum fluorescence intensities expected (Miesenböck et al., 1998). Both samples should be well-resolved in both channels (i.e., not saturating the detector at zero or the max value).
- On fluorescence data: The data exported from the flow cytometer may contain pulse height and width information as well as the total fluorescence (area). Area measurements are used in all aspects of this protocol.
- On compensation: Although compensation (a procedure used to calculate and correct for the overlap between fluorescence channels) is common in multicolor flow cytometry, in this protocol only one fluorophore is being analyzed and it is not necessary to perform compensation. pHluorin may be co-expressed with other fluorophores, such as mCherry, that are not excited in the same range, and analyzed in the same fashion.
- On frequency of measuring calibration curve: Because instrument settings and mechanics can vary day-to-day, it is best practice to measure a new calibration curve every time an experiment is performed. If ratio values seem to be very consistent day-to-day, then only the minimum and maximum pH buffers can be analyzed; if the previously generated calibration curve correctly predicts these samples, then that calibration curve can be used to analyze data taken that day.
- On reproducibility of calibration curves: Although the absolute value of the ratios may change on different days, the midpoint of the curve, which is the apparent pKa of pHluorin, should be consistent. To check whether a calibration curve accurately reproduces the known characteristics of the fluorophore, plot the pH of the buffer for each sample against the following quantity (Bagar et al., 2009):
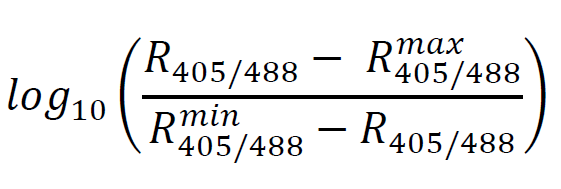
This plot can be fit by linear least squares, and where the resulting line crosses the y = 0 line is the apparent pKa of the fluorophore. The published in vitro pKa is 6.9 (Bagar et al., 2009). See the attached script for an example of this analysis. - On filtering events: In some samples, depending on handling, there is a subpopulation of cells that both lose fluorescence in the pHluorin-associated channels, but also are much more (auto) fluorescent in another channel (ex 405, em 450/50). We interpret these cells as being dead or damaged in some way, and exclude them from analysis. They generally appear in experimental samples and not in calibration curve samples. See analysis script for an example of isolating and filtering out this subpopulation.
Recipes
- 2x Calibration Curve Buffer
100 mM MES
100 mM HEPES
100 mM KCl
100 mM NaCl
400 mM ammonium acetate
20 mM 2-Deoxy-D-glucose - TE Buffer
10 mM Tris-HCl
1 mM Na2EDTA, pH 8.0 - Yeast growth media (1 L)
6.7 g YNB
2.0 g SC Dropout mix
20 g Glucose
Bring to 1 L with ultrapure water and filter through a 0.22 μm filter to sterilize
Acknowledgments
Research reported in this publication was supported by the National Institute of Biomedical Imaging And Bioengineering of the National Institutes of Health (NIH) under Award Number T32EB009412, and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1144082. DAD acknowledges support from the NIH, award numbers R01 GM126547 and R01GM127406, and from the US Army Research Office, award number W911NF-14-1-0411. The authors would also like to thank the University of Chicago Flow Cytometry Core.
Methodology reported in this paper was used in the preprint ‘Transient intracellular acidification regulates the core transcriptional heat shock response’, https://doi.org/10.1101/414706.
Competing interests
The authors declare they do not have any competing interests.
References
- Bagar, T., Altenbach, K., Read, N. D. and Benčina, M. (2009). Live-Cell imaging and measurement of intracellular pH in filamentous fungi using a genetically encoded ratiometric probe. Eukaryot Cell 8(5): 703-712.
- Boron, W. F. (2004). Regulation of intracellular pH. Adv Physiol Educ 28(1-4): 160-179.
- Gietz R. D. and Schiestl R. (1994). Transforming yeast with DNA. Methods Mol Cell Biol 5(5): 255-269.
- Miesenböck, G., De Angelis, D. A. and Rothman, J. E. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394(6689): 192-195.
- Orij, R., Brul, S. and Smits, G. J. (2011). Intracellular pH is a tightly controlled signal in yeast. Biochim Biophys Acta 1810(10): 933-944.
- Orij, R., Postmus, J., Ter Beek, A., Brul, S. and Smits, G. J. (2009). In vivo measurement of cytosolic and mitochondrial pH using a pH-sensitive GFP derivative in Saccharomyces cerevisiae reveals a relation between intracellular pH and growth. Microbiology 155(Pt 1): 268-278.
- Valkonen, M., Mojzita, D., Penttilä, M. and Bencina, M. (2013). Noninvasive high-throughput single-cell analysis of the intracellular pH of Saccharomyces cerevisiae by ratiometric flow cytometry. Appl Environ Microbiol 79(23): 7179-7187.
- Weigert, C., Steffler, F., Kurz, T., Shellhammer, T. H. and Methner, F. J. (2009). Application of a short intracellular pH method to flow cytometry for determining Saccharomyces cerevisiae vitality. Appl Environ Microbiol 75(17): 5615-5620.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Triandafillou, C. G. and Drummond, D. A. (2020). Live Cell Measurement of the Intracellular pH of Yeast by Flow Cytometry Using a Genetically-Encoded Fluorescent Reporter. Bio-protocol 10(12): e3653. DOI: 10.21769/BioProtoc.3653.
Category
Microbiology > Microbial cell biology > Cell-based analysis > Ion analysis
Cell Biology > Cell-based analysis > Flow cytometry
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.