- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Primed Track: Reliable Volumetric Single-cell Tracking and Lineage Tracing of Living Specimen with Dual-labeling Approaches

Published: Vol 10, Iss 11, Jun 5, 2020 DOI: 10.21769/BioProtoc.3645 Views: 4359
Reviewed by: Samantha E. R. DundonAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Mammalian embryonic development starts with a single fertilized zygote that develops into a blastocyst embryo consisting of three cell types that evolve into either embryonic or extra-embryonic tissues. Lineage tracing of these cells can provide important information about the molecular and cellular dynamics contributing to fate allocation during early development. While global labeling techniques allow for visualization of all cells at the same time, lineage tracing of cells over several divisions can become complicated due to embryo movement and rotation as well as increasing cell densities. Here, we use green-to-red photoconvertible proteins for both global and sparse labeling of cells of interest in the developing murine embryo. We use primed conversion to achieve precise photoconversion of single nuclei in 4-cell stage embryos followed by volumetric live imaging to capture development up to the blastocyst stage. We developed an image analysis pipeline, called primed Track, that uses the dual labeling strategy for both straightforward segmentation and registration of all cells in the embryo as well as correction of rotational and spatial drift. Together, this strategy allows for reliable and fast tracking and lineage tracing of individual cells, even over increased imaging time intervals that result in a major reduction in data volume, all essential conditions for volumetric long-term imaging techniques.
Keywords: Pre-implantation embryosBackground
The first lineages of mammalian development are specified within the first days after fertilization before the embryo adheres to the wall of the uterus during implantation (Welling et al., 2016). Still, how and when these first specialized cell types appear and which molecular mechanisms are responsible for symmetry breaking from an initially homogeneous set of cells, remains largely unknown. Selective plane illumination microscopy (SPIM) provides a unique means for capturing developmental progression of living samples for several consecutive days in a non-invasive manner (Strnad et al., 2016; McDole et al., 2018). However, a large fraction of mammalian pre-implantation embryos shows significant spatial and rotational drift, which makes it difficult to track individual cells over time and often results in the exclusion of these embryos from the analysis (Strnad et al., 2016; Welling et al., 2019). While increased imaging rates could help address this challenge, this approach requires additional data storage space and could result in increased chances of photodamage making it a less attractive solution for sensitive specimen like mammalian embryos.
The introduction of fluorescent proteins (FPs) as genetic labels transformed biological and developmental imaging due to their ease of use in conventional imaging platforms. Compared to previous common cell labelling methods, such as vital dyes (Vogt, 1925) and interspecies transplantations (Honig and Hume, 1989), FPs can be easily introduced into different cell types and can provide specific and indelible labelling when stably expressed. Furthermore, the use of tissue- or cell-specific promoters to drive expression of FPs increases contrast for lineage tracing and can reveal important information about cellular identity (Baier et al., 1996).
A subclass of FPs, called photomodifiable FPs, is of particular interest because they can change their spectral properties (i.e., excitation and emission spectra) upon violet (405 nm) or ultraviolet (UV) light illumination. For example, the photoactivatable GFP, paGFP (Patterson and Lippincott-Schwartz, 2002), can permanently change from a non- or weak fluorescent state to a strongly fluorescent one upon violet or UV light illumination, while the photoconvertible FP Dendra2 can change from a green fluorescent state to a red one (Gurskaya et al., 2006). Photoconvertible FPs offer the advantage of being able to visualize both global and sparse cell populations and therefore provide a powerful resource of cell tracking and lineage tracing of cell populations in living samples (Dempsey et al., 2012). However, until recently labelling using photoconvertible FPs has not been used in live volumetric imaging studies due to the potential phototoxicity of conventional 405nm light and its inherent inability to confine axially photoconversion.
In 2015, we (Dempsey et al., 2015) reported a new photoconversion method, called primed conversion. We showed that Dendra2 can be successfully photoconverted from green to red by simultaneous illumination with 488 nm and 730 nm light. In contrast to conventional 405 nm photoconversion, primed conversion allows for precise conversion of small volumes in 3D space; conversion occurs only in the defined volume in which the two beams (488 nm and 730 nm) intersect, enabling labelling of single cells and/or subcellular compartments in very dense 3D environments (Dempsey et al., 2015; Mohr et al., 2016; Mohr and Pantazis, 2016). As primed conversion uses lower energy light than 405 nm photoconversion, it is less phototoxic. Recently, studies have demonstrated that primed conversion can be also achieved using conventional red lasers (630-690 nm) found in most commercial fluorescence microscopes (Klementieva et al., 2016; Mohr et al., 2017). This work also revealed the basic principle of the primed conversion mechanism (Mohr et al., 2017); the acquired knowledge was used to render most known green-to-red photoconvertible proteins primed convertible (pr-FPs) (Mohr et al., 2017; Turkowyd et al., 2017) (see https://www.addgene.org/Periklis_Pantazis/). Out of these, we have selected pr-mEosFP with high brightness and photostability as the optimal photoconvertible protein for our imaging experiments in living mouse embryos (Welling et al., 2019).
Here we show that confined primed conversion of green-to-red photoconvertible proteins in combination with volumetric long term imaging of murine pre-implantation embryos allows for tracking of individual cells with high fidelity during development up to the blastocyst stage (Welling et al., 2019). Taking advantage of both the global green and sparse red labels, we have developed an image analysis pipeline, called primed Track, for registration and computational correction of observed spatial and rotational drift during development, facilitating segmentation and reliable reconstruction of lineage trees with minimal tracking uncertainties (Welling et al., 2019). In addition, the dual labeling approach combined with primed Track allows for accurate tracking of specimen imaged with larger time intervals, which reduces photodamage in sensitive samples and decreases the amount of data produced. Finally, primed Track is not limited to pre-implantation embryos with photoconvertible labels but can also be generally used to facilitate tracking and lineage tracing in other types of dual-labelled living samples, such as organoids.
Materials and Reagents
- Materials
- Tissue culture dishes 35 mm (Falcon, catalog number: 351008 )
- Glass bottom culture dishes (35-mm Petri dish, no. 1.0; MatTek, catalog number: P35G-1.0-14C )
- Plasmid template for mRNA synthesis for injection into mouse embryos: pCS2+-H2B-pr-mEosFP (Addgene Plasmid #141101). The plasmid containing pr-mEosFP can be found on Addgene (Addgene Plasmid #99213)
- Reagents
- EmbryoMax M2 medium (Millipore, catalog number: MR-015-D )
- EmbryoMax KSOM medium (Millipore, catalog number: MR-020P-5F )
- Mineral oil (Sigma, catalog number: M8410 )
Equipment
- CO2 incubator (Binder, model: CB160 )
- Confocal microscope (Zeiss LSM 780) with Nonlinear Optics (NLO) (Ti:Sapphire) laser in Continuous Wave (CW) mode (Coherent Chameleon Ultra II)
- Dissection microscope (Leica M205C)
- Primed conversion filter plate (see Mohr et al., 2016) for detailed setup. Individual parts can be ordered at AHF Analysentechnik, Germany
- LS1 live light-sheet microscope system (Viventis Microscopy Sarl, Switzerland)
Software
- ZEN software (Zeiss)
- Imaris version 8.4 (Bitplane)
- MATLAB (MathWorks)
Procedure
- Confined primed conversion of single nuclei in mouse pre-implantation embryos
- Prepare KSOM medium according to the manufacturer’s instructions one day in advance and let it equilibrate overnight in a CO2 incubator at 37 °C.
- Prepare glass bottom imaging dishes with ~2 μl drops of KSOM covered with mineral oil and leave it in a CO2 incubator until use.
- Prepare a sample holder for the LS1 light-sheet microscope with ~10-15 μl KSOM covered with ~300 μl mineral oil and leave it in a CO2 incubator until use.
- Culture embryos labeled with photoconvertible proteins (by micro-injection of mRNA into zygotes or 2-cell stage embryos as described previously (Plachta et al., 2011), or transgenic lines) ex vivo in a 15-25 μl drop of KSOM covered with mineral oil in a culture dish at 37 °C and 5% CO2 up to desired developmental stage in which you want to convert single nuclei.
- Set up the microscope for primed conversion as described in detail in Mohr et al. (2016). Since the laser power may decay along the light path, the effective laser power for the 488 nm and 730 nm laser at the focal plane needs to be measured using a power meter device prior to starting the conversion experiments.
- Transfer the embryos to a drop of KSOM covered with mineral oil in the already prepared (Step A2) glass-bottom culture dish and place the dish in the microscope.
- Make a z-stack of both the green and red channels prior to photoconversion.
- Insert the primed conversion filter cube into the light path and convert the region of choice (here the H2B-labeled nucleus) by zooming into that particular region: use simultaneous 488 nm light (30-50 μW) and 730 nm light (5-10 mW), ~20 s conversion time and maximum scan speed (pixel dwell time = 0.79 μs).
Note: Depending on the intensity of the fluorescent labeling, the laser power used for primed conversion and conversion time need to be optimized. See note at the end of this manuscript for more details. - Remove the primed conversion filter cube from the light path and check the efficiency of conversion in live mode. The efficiency of conversion can be calculated by measuring fluorescence intensities in both the red and green channel before and after photoconversion followed by calculating the change in intensities for each channel. Test which photoconversion settings give the highest fold change in both the red and green channel for your samples. Repeat primed conversion if necessary (see Figure 1).
- Make a post-conversion z-stack of the whole embryo of both the green and red channels.
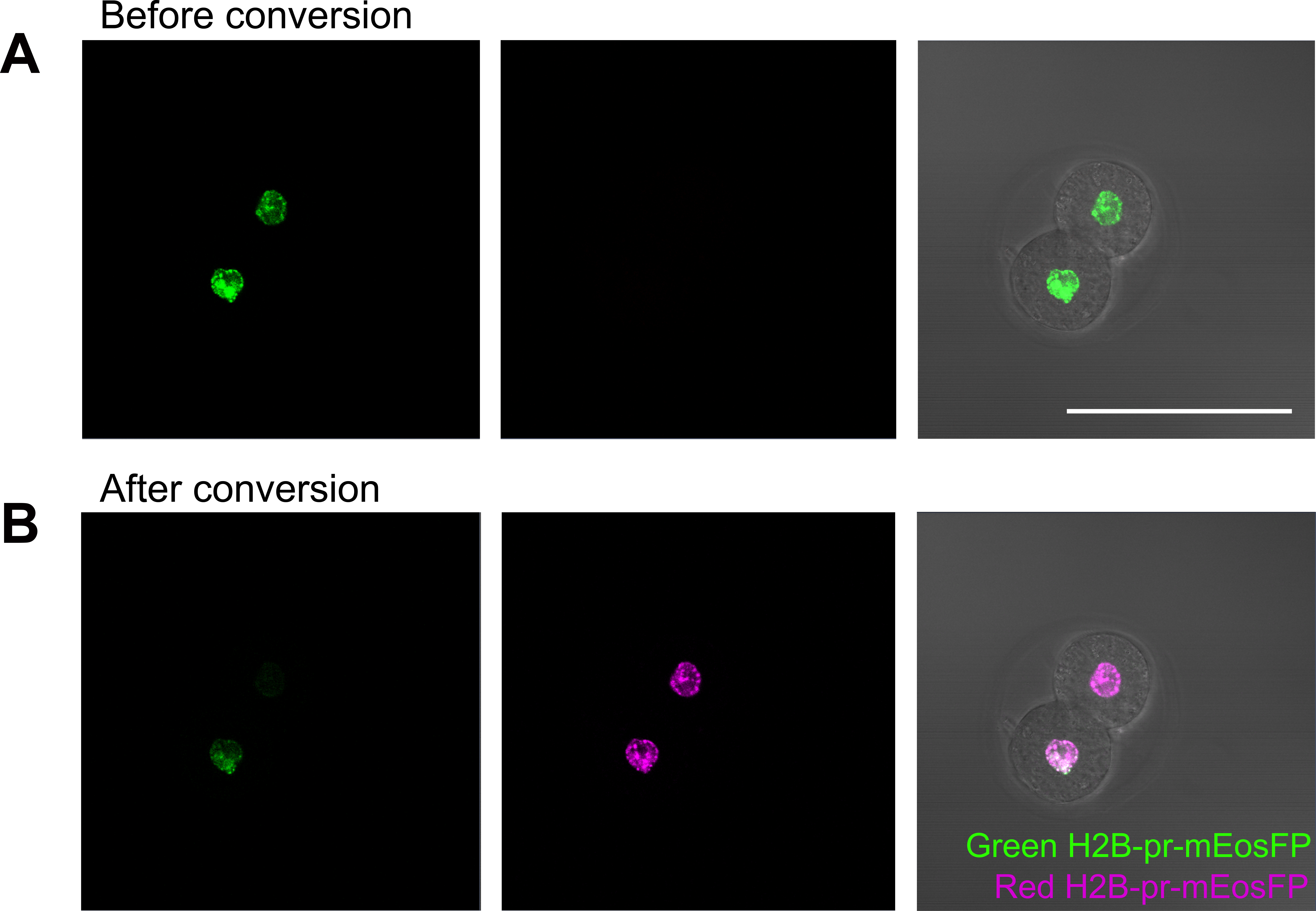
Figure 1. H2B-pr-mEosFP labeled embryos before and after primed conversion with different conversion settings per nucleus. A. 2-cell stage embryo before photoconversion. B. The same embryo after conversion with different powers used for the 730 nm laser: 5 mW for lower nucleus, 10 mW for upper nucleus. The conversion time was 20 s for both nuclei. Scale bar = 100 μm.
- Live volumetric imaging of mouse pre-implantation embryos
- Right after performing confined primed conversion on single nuclei of mouse embryos, transfer 10-15 embryos to a pre-equilibrated sample holder for the LS1 light-sheet microscope system (Step A3).
- Place the sample holder in the LS1 inverted SPIM setup in an incubator chamber at 37 °C and 5% CO2.
- Acquire a z-stack consisting of 80 planes, 3 μm apart, for each embryo at regular time intervals (10-15 min) in both the green and red channels. Embryos can be imaged up to the blastocyst stage.
Note: Imaging frequency can be extended to 30-40 min without this dramatically influencing tracking and lineage tracing fidelities.
Data analysis
Step-by-step segmentation, tracking and lineage tracing using primed Track
The acquired time-lapse 3D images of photoconverted mouse embryos in two channels can be analyzed using primed Track, in which a custom MATLAB code is used and implemented in Imaris (Bitplane). All codes of primed Track can be downloaded from this code repository:
First segmentation
- Use the Imaris Spot detection tool to detect green and red cells in the embryo. Use a low threshold for quality to segment all cells even at the cost of including spurious spots. Choose a spot radius that fits the volume of the cells that need to be segmented and enable “different spot sizes”.
- In the fourth step of the spot detection algorithm, choose a quality that allows for including the cells that have lower fluorescent signal at later developmental stages.
- In the sixth step of the spot detection algorithm, choose a region threshold to set the size of the spots according to the size of the surface of the cell they are placed on.
- Set a filter to exclude small bright background spots and micronuclei from the segmentation (see Figure 2). A new spot object will be created.
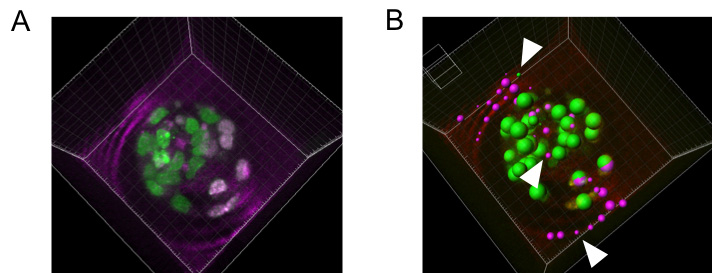
Figure 2. H2B-pr-mEosFP labeled embryo at the blastocyst stage with photoconverted (magenta) and unconverted (green) cells. A. original image before segmentation. B. Initial segmentation of individual nuclei (green and magenta spots) prior to filtering for spot size. Arrows indicate false positive background spots that can be excluded using a filter for spot size.
First finetuning of segmentation
Use the custom MATLAB script to exclude wrongly segmented spots from segmentation in the menu at: Image processing > BSSE > spot functions > validate cells in embryo:
- First, select the spot object that contains all (globally labeled, green) cells as well as the spot object that contains converted (or partially labeled, red) cells.
- Estimate the diameter of the embryo using the fluorescent channel that labels all cells (green for photoconvertible proteins). The radius of the embryo is roughly estimated as the median of all maximal inter-spot distances. Choose a multiplicative factor that could compensate for radius estimation errors and potentially prevent cells at the outside boundary of the embryo to be falsely discarded.
- To eliminate spots that are wrongly segmented as two spots instead of one single spot, identify spots that occur within a user defined distance from another spot in the same channel. Wrongly segmented double spots will be replaced by one new spot (see Figure 3).
- Since autofluorescence in the red channel is higher than in the green channel, there is a higher likelihood of background signal being segmented as a cell in the red channel. Photoconverted red cells will always retain some of the green fluorescence and segmented red spots should therefore overlap with green spots. Delete all wrongly segmented red spots that do not overlap with a green spot (see Figure 3).
- Low green fluorescent signal can lead to failure of detection for segmentation and therefore also false deletion of red spots in the previous step. To compensate for this, the user has to option to recover these red spots if a valid red spot was present at the previous time point within a user-defined search radius.
- Three new spot objects will be created: 1) processed spots 1 [containing all cells (green)], 2) processed spots 2 [only partially labeled spots (red)], and 3) processed spots 1–processed spots 2 (containing green cells that do not overlap with red). The third spot object will be required for facilitating the calculation of lineage trees later.
- Check if the new spot segmentation is satisfying. Adjust segmentation filter settings if necessary.

Figure 3. Examples of segmentation errors. A. Morula stage embryo with one nucleus falsely segmented as 2 spots (in white). B. Autofluorescent background is falsely segmented in the red channel. The image analysis algorithm will allow you to only include segmented red spots that overlap with green spots to overcome segmentation of background signals.
Correction for spatial and rotational drift of the embryo and cropping
- Use the custom MATLAB script to register cells in both green and red channels of the embryo/specimen in the Imaris menu at: Image processing > BSSE > Reference frame functions > set reference frames. MATLAB is used to create Imaris Reference Frame Objects for each time point. The center of mass of the green spots is defined as the origin and their orientation is given by the vector. The position change of the green spots between subsequent time points is compared and determines the rotation angle of the reference frame so that cell drift between time point is minimized. The resampling is performed in Imaris.
- Select spot for estimating embryo center point: choose the processed green spots.
- Select spots for estimating the orientation of the embryo: choose the processed red spot.
- Select the newly created reference frame object and “resample image to align reference frame”. Select "correct image and all objects", "rotation and translational drift" and "crop largest common region".
- Choose how to crop the data to a smaller volume.
- Re-align embryo with the next function in the Imaris menu: Image processing > BSSE > Reference frame functions > correct rotation around reference frame axis
- Select spot for estimating embryo center point: choose the processed green spots.
- Select the newly created reference frame object and “resample image to align reference frame”. Select "correct image and all objects", "rotation and translational drift" and "crop largest common region".
Second finetuning of segmentation
- The new registered dataset can contain red cells that were previously not segmented because they were outside of the user defined radius (First finetuning of segmentation step 2). Re-run the validation of the segmentation on the registered embryo (First finetuning of segmentation steps 1-7) to recover red cells that were not segmented previously.
- Three new spot objects will be created: 1) resampled spots 1 [containing all cells (green)], 2) resampled spots 2 [only partially labeled spots (red)], and 3) resampled spots 1–resampled spots 2 (containing green cells that do not overlap with red).
Tracking and lineage tree reconstruction
- Use Imaris’ Lineage module to track the cells over time and reconstruct their lineage tree. The creation of a separate segmentation for the cells occur in the green channel only and having another one that contains photoconverted red cells, reduces the complexity of the lineage calculation by breaking it down into two simpler, computationally less expansive, unrelated sets of objects.
- Optional: Manually correct false connections in the lineage trees.
Notes
The efficiency of primed conversion of photoconvertible proteins is dependent on the levels of fluorescent signal present in the cells. Therefore, different primed conversion parameters need to be tested in order to define the optimal settings for your cells of interest; i.e., the intensities of the priming beam and conversion beam, and the duration of conversion. In our experience, the laser intensities used for the priming beam (488 nm) has the greatest influence on the conversion efficiency. We recommend to keep the 488 nm laser intensity as low as possible for primed conversion to still take place in order to minimize loss of fluorescent signal due to bleaching. This is of particular importance for samples exhibiting low fluorescence intensities prior to photoconversion. In our hands a conversion duration of ~20 s results in a good tradeoff between conversion efficiency and limited bleaching. For samples that exhibit high levels of expression of photoconvertible proteins, a longer conversion time may be required to obtain better photoconversion efficiencies.
Acknowledgments
MW is supported by a Rubicon postdoctoral fellowship from the Dutch Research Council (NWO) and a postdoctoral fellowship from the Peter und Traudl Engelhorn Stiftung. PP holds the Wolfson Research Merit Award from the Royal Society. The protocol is based on work performed in Welling et al. (2016).
Competing interests
PP is an inventor on a patent application filed by ETH Zurich and Caltech that describes primed conversion (WO 2014/147211 and EP 2014/055669). PP is an inventor on a provisional patent application filed by HHMI and ETH Zurich that describes pr-mEosFP (Application Serial No. 62/446,023).
Ethics
All animal experiments were carried out by trained personnel following standard protocols according to our institutional regulatory guidelines. All our experiments were approved by the veterinary authority of the canton Basel Stadt, Switzerland (Permit Number: 2561).
References
- Baier, H., Klostermann, S., Trowe, T., Karlstrom, R. O., Nusslein-Volhard, C. and Bonhoeffer, F. (1996). Genetic dissection of the retinotectal projection. Development 123: 415-425.
- Dempsey, W. P., Fraser, S. E. and Pantazis, P. (2012). PhOTO zebrafish: a transgenic resource for in vivo lineage tracing during development and regeneration. PLoS One 7(3): e32888.
- Dempsey, W. P., Georgieva, L., Helbling, P. M., Sonay, A. Y., Truong, T. V., Haffner, M. and Pantazis, P. (2015). In vivo single-cell labeling by confined primed conversion. Nat Methods 12(7): 645-648.
- Gurskaya, N. G., Verkhusha, V. V., Shcheglov, A. S., Staroverov, D. B., Chepurnykh, T. V., Fradkov, A. F., Lukyanov, S. and Lukyanov, K. A. (2006). Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol 24(4): 461-465.
- Honig, M. G. and Hume, R. I. (1989). Dil and diO: versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends Neurosci 12(9): 333-335, 340-331.
- Klementieva, N. V., Lukyanov, K. A., Markina, N. M., Lukyanov, S. A., Zagaynova, E. V. and Mishin, A. S. (2016). Green-to-red primed conversion of Dendra2 using blue and red lasers. Chem Commun (Camb) 52(89): 13144-13146.
- McDole, K., Guignard, L., Amat, F., Berger, A., Malandain, G., Royer, L. A., Turaga, S. C., Branson, K. and Keller, P. J. (2018). In Toto Imaging and Reconstruction of Post-Implantation Mouse Development at the Single-Cell Level. Cell 175(3): 859-876 e833.
- Mohr, M. A., Argast, P. and Pantazis, P. (2016). Labeling cellular structures in vivo using confined primed conversion of photoconvertible fluorescent proteins. Nat Protoc 11(12): 2419-2431.
- Mohr, M. A., Kobitski, A. Y., Sabater, L. R., Nienhaus, K., Obara, C. J., Lippincott-Schwartz, J., Nienhaus, G. U. and Pantazis, P. (2017). Rational engineering of photoconvertible fluorescent proteins for dual-color fluorescence nanoscopy enabled by a Triplet-state mechanism of primed conversion. Angew Chem Int Ed Engl 56(38): 11628-11633.
- Mohr, M. A. and Pantazis, P. (2016). Single neuron morphology in vivo with confined primed conversion. Methods Cell Biol 133: 125-138.
- Patterson, G. H. and Lippincott-Schwartz, J. (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297(5588): 1873-1877.
- Plachta, N., Bollenbach, T., Pease, S., Fraser, S. E. and Pantazis, P. (2011). Oct4 kinetics predict cell lineage patterning in the early mammalian embryo. Nature cell biology 13(2): 117-123.
- Strnad, P., Gunther, S., Reichmann, J., Krzic, U., Balazs, B., de Medeiros, G., Norlin, N., Hiiragi, T., Hufnagel, L. and Ellenberg, J. (2016). Inverted light-sheet microscope for imaging mouse pre-implantation development. Nat Methods 13(2): 139-142.
- Turkowyd, B., Balinovic, A., Virant, D., Carnero, H. G. G., Caldana, F., Endesfelder, M., Bourgeois, D. and Endesfelder, U. (2017). A general mechanism of photoconversion of green-to-red fluorescent proteins based on blue and infrared light reduces phototoxicity in live-cell single-molecule imaging. Angew Chem Int Ed Engl 56(38): 11634-11639.
- Vogt, W. (1925). Gestaltungsanalyse am Amphibienkeim mit ortlicher Vitalfarbung. Vorwarts uber Wege und Ziele. I. Methodik und Wirkungsweise der Vitalfarbung mit Agar als Farbtrager.
- Welling, M., Mohr, M. A., Ponti, A., Rullan Sabater, L., Boni, A., Kawamura, Y. K., Liberali, P., Peters, A. H., Pelczar, P. and Pantazis, P. (2019). Primed Track, high-fidelity lineage tracing in mouse pre-implantation embryos using primed conversion of photoconvertible proteins. Elife 8: 44491.
- Welling, M., Ponti, A. and Pantazis, P. (2016). Symmetry breaking in the early mammalian embryo: the case for quantitative single-cell imaging analysis. Mol Hum Reprod 22(3): 172-181.
Article Information
Copyright
![]() Welling et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Welling et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Welling, M., Kalyviotis, K. and Pantazis, P. P. (2020). Primed Track: Reliable Volumetric Single-cell Tracking and Lineage Tracing of Living Specimen with Dual-labeling Approaches. Bio-protocol 10(11): e3645. DOI: 10.21769/BioProtoc.3645.
- Welling, M., Mohr, M. A., Ponti, A., Rullan Sabater, L., Boni, A., Kawamura, Y. K., Liberali, P., Peters, A. H., Pelczar, P. and Pantazis, P. (2019). Primed Track, high-fidelity lineage tracing in mouse pre-implantation embryos using primed conversion of photoconvertible proteins. Elife 8: 44491.
Category
Developmental Biology > Cell growth and fate > Blastocyst
Developmental Biology > Cell growth and fate > Differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.