- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Biochemical Pulldown of mRNAs and Long Noncoding RNAs from Cellular Lysates Coupled with Mass Spectrometry to Identify Protein Binding Partners


(*contributed equally to this work) Published: Vol 10, Iss 11, Jun 5, 2020 DOI: 10.21769/BioProtoc.3639 Views: 7079
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
RNA binding proteins (RBPs) interact with cellular mRNAs, controlling various steps throughout the lifetime of these transcripts, including transcription, cellular transport, subcellular localization, translation and degradation. In addition to binding mRNA transcripts, a growing number of RBPs are shown to bind long noncoding RNAs (lncRNAs), controlling key cellular processes, including gene expression and translation of proteins. Current methodologies aimed at identifying and characterizing protein binding partners of specific RNAs of interest typically rely on tagging of the RNA with affinity aptamers, using in vitro transcribed RNA or immobilized oligonucleotides to capture RNA-protein complexes under native conditions. These assays are coupled with mass spectrometry or Western Blot analysis to identify or/and confirm interacting proteins. Here, we describe an alternative approach to identify protein binding partners of mRNAs and large long noncoding RNAs. This approach relies on biochemical pulldown of specific target RNAs and interacting protein partners from cellular lysates coupled with mass spectrometry to identify novel interacting proteins. By using 24-48 ~20 mer biotinylated DNA probes that hybridize to the target RNA, the method ensures high specificity and minimal off target binding. This approach is reproducible and fast and serves as a base for discovery studies to identify proteins that bind to RNAs of interest.
Keywords: mRNABackground
RNA-binding proteins (RBPs) interact with mRNAs in cells to form ribonucleoprotein (RNP) complexes that regulate the post-transcriptional activity of these transcripts by affecting their processing, subcellular localization, translation and stability (reviewed in Lunde et al., 2007; Glisovic et al., 2008). Recent studies have expanded the number of mRNA binding proteins in mammalian cells (for example Baltz et al., 2012; Castello et al., 2012; Kwon et al., 2013); additionally, a growing number of long noncoding RNAs (lncRNAs) have been shown to bind RBPs (for example Gumireddy et al., 2013; Ng et al., 2013; McHugh et al., 2015; Yoon and Gorospe, 2016), emphasizing the importance of characterizing the roles of RNA-protein interactions. There are several methodologies available to date to identify and characterize protein binding partners of specific RNAs (reviewed in Ramanathan et al., 2019). Shortly, these include tagging of RNAs of interest with affinity aptamers (Slobodin and Gerst, 2010), using in vitro transcribed RNAs (Abdelmohsen et al., 2011; Tominaga et al., 2011; Grammatikakis et al., 2016), RNA affinity pulldown typically using a limited number of RNA or DNA oligonucleotides (Kar et al., 2011; Castello et al., 2012; Yoon et al., 2012; Abdelmohsen et al., 2014; Zhang et al., 2016) coupled with Western blot or mass spectrometry analyses (Simon et al., 2011, McHugh et al., 2015, Di Tomasso et al., 2016, Kar et al., 2017, McHugh and Guttman, 2018), or immobilized oligonucleotides to capture RNA-protein complexes under native conditions (Zeng et al., 2006).
Here, we built upon the Chromatin Isolation by RNA Purification (ChIRP) method (Chu et al., 2011 and 2012), adapted it to maximize the yield of proteins bound to specific target RNAs and coupled it with mass spectrometry analysis for thorough identification and characterization of protein binding partners of these RNA transcripts (Figure 1). Similarly to the ChIRP approach (Chu et al., 2011), to obtain high specificity in isolation of a specific target RNA we used sets of pre-biotinylated single molecule FISH probes. A typical set is comprised of 24-48 ~20 mer DNA probes, complementary to and tiled along the target RNA. As in the ChIRP protocol (Chu et al., 2011), we used glutaraldehyde crosslinking as compared to other crosslinking methods, it yielded the highest signal-to-noise ratio. However, this protocol can be adjusted to use various cross linkers, including ultraviolet light and formaldehyde, to focus on specific interactions of interest.
Typically, we grow cells in desired conditions, cross link them and freeze cell pellets for future use or use them immediately. Cell pellets are lysed and pre-cleared cell lysates are incubated with a set of 24-48 biotinylated DNA probes, followed by additional incubation with streptavidin beads (Figure 1). Eluted proteins are separated using SDS-PAGE with each lane cut into 8 equal sections and in-gel digested as per Shevchenko et al., 2006 (Figure 1). Extracted peptides are LC-MSMS analyzed (Figure 1). This method allows for the identification and characterization of protein binding partners of specific RNAs of interest.
Materials and Reagents
- 150 mm tissue culture plate (TPP, catalogue number: 93150 )
- Cell scrapers (TPP, catalog number: 99003 )
- Falcon tubes (TPP catalog number: 91015 )
- Surgical Blade (Swann-Morton, catalog number: 0 103 )
- 96 Deepwell Plate (Thermo, catalog number: 95040450 )
- 2 ml Protein LoBind Tubes (Eppendorf, catalog number: 00 30108132 )
- 1.5 ml Protein LoBind Tubes (Eppendorf, catalog number: Z666505 )
- 0.5 ml Protein LoBind Tubes (Eppendorf, catalog number: 00 30108094 )
- 12 Channel Pipette, 30-300 μl (Eppendorf, catalog number: 3122000060 )
- 10% NP-40 (Thermo Fischer Scientific, catalog number: 85124 )
- PBS (Gibco, catalog number: 10010-015 )
- Glutaraldehyde (Merk, catalog number: 111-30-8 )
- Glycine (Fluka, catalog number: 15527H )
- Protease inhibitors cOmplete, Mini, EDTA-free (Roche, catalog number: 0 4693159001 )
- DTT (Merck, catalog number: 3483-12-3)
- Superase-In (Ambion, catalog number: AM2694 )
- Stellaris biotinylated probes (custom made by Biosearch as per gene of interest)
- MagReSyn® Streptavidin Affinity binding/capture of biotinylated biomolecules (AEC-AMERSHAM SOC Ltd, catalog number: MR-STV002 )
- Precision Plus Protein Dual Color Standards (Bio-Rad, catalog number: 161-0374 )
- 4x Laemmli sample buffer (Bio-Rad, catalog number: 1610747 )
- Coomassie Brilliant Blue R-250 (Bio-Rad, catalog number: 1610436 )
- Bovine Serum Albumin Fraction V (Roche, catalog number: 10735078001 )
- LC-MS Water (Honeywell, catalog number: LC365 )
- Ammonium Bicarbonate (Fluka, catalog number: 0 9830 )
- Methanol (Honeywell, catalog number: LC230 )
- Acetonitrile (Honeywell, catalog number: LC015 )
- Iodoacetamide (Sigma-Aldrich, catalog number: I1149 )
- Trypsin (Promega, catalog number: V511A )
- Formic Acid (Thermo, catalog number: 28905 )
- Tris-HCl (Sigma-Aldrich, catalog number: T1503-250G )
- KCl (Sigma-Aldrich, catalog number: P9333-1KG )
- Triton X100 (Sigma-Aldrich, catalog number: X-100-100ML )
- NaCl (Sigma-Aldrich, catalog number: s7653-1KG )
- SDS (Sigma-Aldrich, catalog number: L3771-100G )
- Formamide (Thermo Fischer Scientific, catalog number: 17899 )
- 20x SSC (Sigma-Aldrich, catalog number: S6639-1L )
- RNase A (Sigma-Aldrich, catalog number: 10109142001 )
- RNase H (Sigma-Aldrich, catalog number: 10786357001 )
- DNase 1 (Invitrogen, catalog number: EN0521 )
- Lysis Buffer (see Recipes)
- Hybridization Buffer (see Recipes)
- Washing Buffer (see Recipes)
- DNase Buffer (see Recipes)
- Elution Buffer (see Recipes)
- Destaining Buffer (see Recipes)
- Dehydration Buffer 1 (see Recipes)
- Reduction Buffer (see Recipes)
- Alkylation Buffer (see Recipes)
- Dehydration Buffer 2 (see Recipes)
- Digestion Buffer (see Recipes)
- Trypsin Solution (see Recipes)
- Extraction Buffer (see Recipes)
- Loading Solvent (see Recipes)
- Gradient solvent A (see Recipes)
- Gradient solvent B (see Recipes)
Equipment
- Refrigerated microcentrifuge (Beckman Coulter, model: 22R )
- Shaker (TCS Scientific Corp Nutator)
- Rotator (Intelli-mixer RM-2L)
- Magnetic separator (Resyn Biosciences)
- Deepwell Plate Thermal Mixer (Eppendorf, catalog number: 5355 )
- Deepwell Plate Vacuum Drying System (Gel plugs dried in vacuum desiccator jar trough Agilent’s DS 102 dual stage rotary vane pump, catalog number: 9499315 )
- Vacuum Concentrator (Labconco, catalog number: 7310035 )
- Water Bath Sonicator (Bandelin, catalog number: 301 )
- Dionex Ultimate 3000 RSLC System (Thermo, catalog number: IQLAAAGABHFAPBMBEZ )
- Desalting Column (Thermo, catalog number: 164567 )
- Separation Column (Thermo, catalog number: 164534 )
Software
- Protein Pilot (Sciex, https://sciex.com/products/software/proteinpilot-software)
Procedure
- Crosslinking of cells
- Grow ~1 x 107 cells in desired conditions in 150 mm plates (in a volume of 30 ml medium).
Note: Higher number of cells can be used, we found 1 x 107 cells to be the minimal amount required to obtain data at high resolution. - Wash in 20 ml 1x PBS.
- Crosslink cells in the plate with 20 ml 1% Glutaraldehyde in 1x PBS for 10 min at RT on a shaker.
Note: Alternative cross linkers can be used instead of Glutaraldehyde, including Ultraviolet light and Formaldehyde. The protocol has to be adjusted accordingly. - Quench with 20 ml 0.125 M Glycine for 5 min at RT, on a shaker.
- Wash with 20 ml ice-cold 1x PBS.
- Scrape cells and transfer to 15 ml falcon tubes.
- Centrifuge cells at 2,500 x g, 5 min, at 4 °C and discard supernatant.
- Snap freeze pellet and store at -80 °C or use immediately.
- Grow ~1 x 107 cells in desired conditions in 150 mm plates (in a volume of 30 ml medium).
- Lysis of cells
- Resuspend pellet in 250-300 μl Lysis buffer + 1x protease Inhibitors.
- Transfer the re-suspended cells to an Eppendorf, mix well and incubate at RT for 25 min on a shaker.
- Centrifuge cells at 4 °C for 10 min at 10,000 x g.
- Transfer the supernatant to a new Eppendorf and discard pellet.
- Centrifuge supernatant to pre-clear, at 4 °C for 10 min at 12,000-14,000 x g.
- Collect the supernatant (the pellet is not always visible at this stage).
- Probe hybridization
- Prepare the hybridization reaction:
200 μl lysate
100 μl Hybridization buffer
0.3 μl 1 M DTT (to a final of 0.1 mM)
1x Protease inhibitors
1.5 μl 20 U/μl Superase (to a final of 0.1 U/μl)
Add biotinylated probes to a final concentration of 100 pmol per reaction.
Note: We recommend the use of a set of scrambled biotinylated single molecule probes as a negative control. Alternatively, a set of probes that target the antisense strand can be used. Additionally, we recommend a parallel reaction, were the set of probes are omitted. Data sets of the experiment should be compared with these control datasets to eliminate proteins that tend to aggregate or bind in a non-specific manner. - Incubate hybridization reaction at 37 °C for 4 h, rotating.
- Prepare the hybridization reaction:
- Streptavidin beads preparation (Video 1)
Note: In the video the tube in which the reaction will occur is labeled “Reaction”. Subsequently, there are three tubes, labeled “1x PBS”, “1 mg/ml BSA” and “Hyb” (hybridization buffer).- Wash 40 μl slurry Streptavidin beads (equals to ~10-15 μl solid Streptavidin beads) in ~250 μl 1x PBS x 3.
- Block beads with 250-500 μl 1 mg/ml BSA in 1x PBS for 1 h at RT, shaking.
- Discard the blocking solution and wash beads with Hybridization buffer. Repeat wash three times.
 Video 1. Streptavidin beads preparation
Video 1. Streptavidin beads preparation
- Biotin-Streptavidin reaction
- Add the hybridization reaction to beads and incubate at 37 °C for 30-60 min, rotating.
- Discard the unbound reaction.
- Wash beads with 40x solid bead volume (~400-600 μl) of washing buffer.
- Repeat wash three times.
- Resuspend beads in 3x the original volume of solid beads (~30-45 μl) of DNase buffer.
- Add 100 μl Elution buffer and incubate at 37 °C for 30 min, rotating.
- Add Sample Loading Buffer and boil at 95 °C for 5 min.
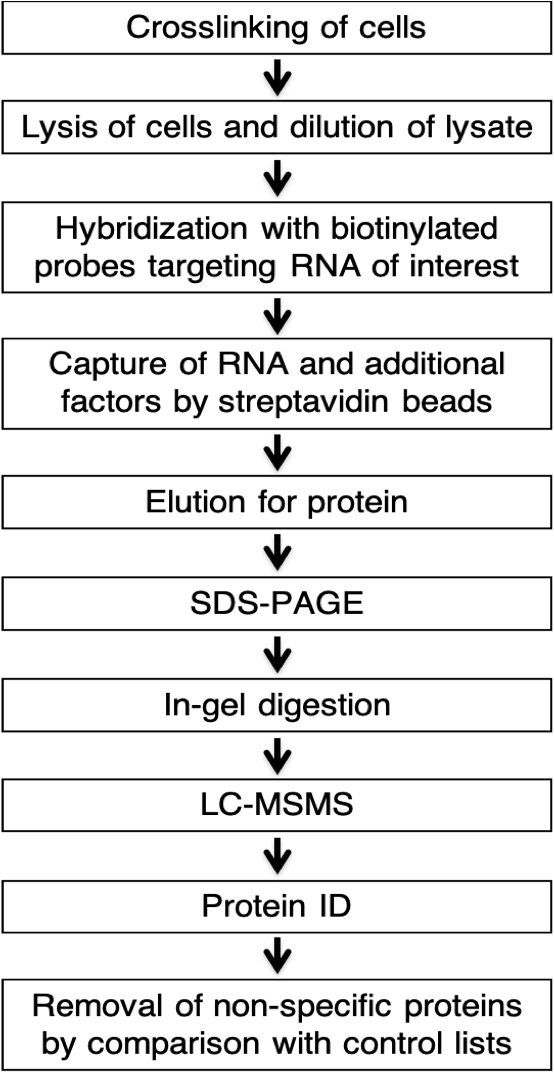
Figure 1. Schematic of biochemical pulldown of RNAs and associated proteins and characterization by LS-MSMS
Elution Option II:- Add ~50 μl 2x Sample Loading Buffer and boil at 95 °C for 5 min.
- Run samples on a SDS-PAGE page, stain with Coomassie and cut bands for mass-spectrometry. A representative SDS-PAGE gel is shown in Figure 2.
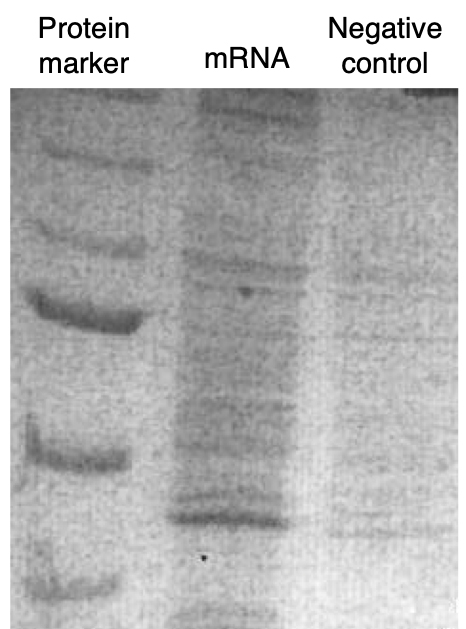
Figure 2. A representative image of an SDS-PAGE stained with Coomassie representing a pulldown experiment in 3T3 mouse fibroblasts: a set of probes against an mRNA of interest were used. The negative control consisted of a set of scrambled, non-specific probes
- In-gel digestion (based on Reference 19)
- Cut relevant gel lane with surgical blade into multiple slices (about 8 horizontal slices along length of lane in this case, large high intensity protein bands were cut out separately to minimize potential masking of low abundance proteins).
- Dice each gel slice into small pieces (1 x 1 mm) and place into deepwell plate.
- Add ~200 μl (or enough to cover) of Destaining buffer and vortex (800 rpm max.) in a thermomixer for 20 min and discard the supernatant.
- Repeat Step F3.
- Add 100 μl (or enough to cover) of Dehydration buffer 1, vortex in a thermomixer for 20 min (800 rpm max).
- Remove and discard the supernatant.
- Vacuum the gel pieces to complete dryness (10-20 min).
- Add 25 μl (or enough to cover gel pieces) Reduction buffer to dried gels.
- Vortex and spin briefly (800 rpm max).
- Allow reaction to proceed at 60 °C for 1 h.
- Chill tubes to room temp (22-23 °C).
- Add 500 μl Dehydration buffer 1.
- Incubate for 10 min (22-23 °C).
- Remove supernatant and discard.
- Add 25 μl (or enough to cover gel pieces) Alkylation buffer to the gel pieces.
- Vortex and spin briefly (800 rpm max).
- Allow reaction to proceed in the dark for 20 min at room temperature (22-23 °C).
- Remove supernatant and discard.
- Wash gels with 100 μl Digestion buffer, vortex 10 min, spin.
- Remove supernatant and discard.
- Dehydrate gels with 100 μl (or enough to cover gel pieces) of Dehydration buffer 2, vortex 5 min, spin, remove supernatant and discard (800 rpm max).
- Repeat Step F21.
- Vacuum the gel pieces to complete dryness (~20 min, keep time to minimum).
- Place the deepwell plate on ice.
- Add freshly prepared trypsin solution to just barely cover the gel pieces (~5-50 μl).
- After 30 min check if all trypsin solution has absorbed and add more if necessary. (Gel pieces should be covered completely with trypsin).
- Rehydrate the gel pieces on ice or at 4 °C for further 1 h.
- Add Digestion buffer as needed to cover the gel pieces.
- Incubate at 37 °C for 18 h.
- Transfer the digest solution (aqueous extraction) into 2 ml Protein Lo-Bind tubes.
- To the gel pieces, add 30 μl (enough to cover) of Extraction buffer.
- Vortex 20-30 min.
- Sonicate in ultrasonic bath for 5 min.
- Add aqueous extraction to solution obtained at Step F30.
- Repeat Steps F31-F33.
- Speed Vac the digest solution to dryness.
- LC-MSMS
- Add 20 μl loading solvent to dried peptide digests and vortex for 1 min.
- Transfer 18 μl to glass HPLC vial or 0.5 ml Eppendorf Protein LoBind tube and place in autosampler.
- Inject 2-5 μl on the trap column (PepMap 75 μm x 2 cm) at 5 μl/min using Loading solvent and after 5 min switch inline with analytical column (PepMap 75 μm x 15 cm).
- Elute peptides using a 15 min linear gradient 6-35% Gradient solvent B at 300 nl/min.
- Apply an electrospray voltage of 2.0-2.5 kV to nanospary emitter.
- Acquire data in Data Dependant Acquisition (DDA) mode with precursor ions acquired using a single 250 ms scan in the range 400-1,500 m/z and 50 product ion scans at 50 ms scan time in the range 100-1,800 m/z that are automatically trigger.
Data analysis
Data was processed using Protein pilot v5.0.1 using Paragon search engine (Sciex) using a Uniprot Swissprot Mouse database supplemented with sequences of common contaminating proteins. A 1% FDR cut-off was applied on PSM, peptide and proteins levels. Only proteins that were not present in the control sample lists were selected for further analysis.
Recipes
- Lysis Buffer
50 mM Tris-HCl pH 7.5
150 mM KCl
1 mM EDTA
0.5 % Triton X100 - Hybridization Buffer
500 mM NaCl
1% SDS
100 mM Tris-HCl, pH 7.0
10 mM EDTA
15 % Formamide
Add fresh before use: 1 mM DTT, Protease Inhibitors, U/μl Superase - Washing Buffer
2x SSC
0.5% SDS
Add fresh 1 mM DTT - DNase Buffer
10 mM NaCl
0.1% NP-40 - Elution Buffer
100 μg/ml RNase A
0.1 U/μl RNase H
100 U/ml DNase 1
All In-Gel Digestion Buffers below are prepared with LC-MS Water - Destaining Buffer
50% Methanol
50 mM Ammonium Bicarbonate - Dehydration Buffer 1
100% Acetonitrile - Reduction Buffer
10 mM Dithiothreitol
25 mM Ammonium Bicarbonate - Alkylation Buffer
55 mM Iodoacetamide
25 mM Ammonium Bicarbonate - Dehydration Buffer 2
50% Acetonitrile
25 mM Ammonium Bicarbonate - Digestion Buffer
25 mM Ammonium Bicarbonate - Trypsin Solution
10 ng/μl Trypsin
25 mM Ammonium Bicarbonate - Extraction Buffer
50% Acetonitrile
5% Formic Acid - Loading Solvent
2% Acetonitrile
0.2% Formic Acid - Gradient solvent A
0.1% Formic acid - Gradient solvent B
80% Acetonitrile
0.1% Formic acid
Acknowledgments
We thank Dr. Sam Barichievy and Dr. Stephanie Fanucchi for stimulating and fruitful discussions. We also thank Dr. Nicolas Beaume for his assistance with the making of the video. This research has been supported by the following grants: V2YGE81 to A.F.S., PG-V2KYPO7, TA 2011 011 from the Council for Industrial and Scientific Research (CSIR, South Africa) to M.M.M and by a grant from the Emerging Research Area Program of The Department of Science and Technology (DST, South Africa) Department of Science & Technology Centre of Competence Grant, SA Medical Research Council SHIP grant, CSIR Parliamentary Grant.
Competing interests
The authors declare no competing interests.
References
- Abdelmohsen, K., Panda, A. C., Kang, M. J., Guo, R., Kim, J., Grammatikakis, I., Yoon, J. H., Dudekula, D. B., Noh, J. H., Yang, X., Martindale, J. L. and Gorospe, M. (2014). 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res 42(15): 10099-10111.
- Abdelmohsen, K., Tominaga, K., Lee, E. K., Srikantan, S., Kang, M. J., Kim, M. M., Selimyan, R., Martindale, J. L., Yang, X., Carrier, F., Zhan, M., Becker, K. G. and Gorospe, M. (2011). Enhanced translation by Nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res 39(19): 8513-8530.
- Baltz, A. G., Munschauer, M., Schwänhausser, B., Vasile, A., Murakawa, Y., Schueler, M., Youngs, N., Penfold-Brown, D., Drew, K., Milek, M., Wyler, E., Bonneau, R., Selbach, M., Dieterich, C. and Landthaler, M. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46(5): 674-690.
- Castello, A., Fischer, B., Eichelbaum, K., Horos, R., Beckmann, B. M., Strein, C., Davey, N. E., Humphreys, D. T., Preiss, T., Steinmetz, L. M., Krijgsveld, J. and Hentze, M. W. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149(6): 1393-1406.
- Chu, C., Quinn, J. and Chang, H. Y. (2012). Chromatin isolation by RNA purification (ChIRP). J Vis Exp(61).
- Chu, C., Qu, K., Zhong, F. L., Artandi, S. E. and Chang, H. Y. (2011). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44(4): 667-678.
- Di Tomasso, G., Miller Jenkins, L. M. and Legault, P. (2016). ARiBo pull-down for riboproteomic studies based on label-free quantitative mass spectrometry. RNA 22(11): 1760-1770.
- Glisovic, T., Bachorik, J. L., Yong, J. and Dreyfuss, G. (2008). RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 582(14): 1977-1986.
- Grammatikakis, I., Zhang, P., Panda, A. C., Kim, J., Maudsley, S., Abdelmohsen, K., Yang, X., Martindale, J. L., Motino, O., Hutchison, E. R., Mattson, M. P. and Gorospe, M. (2016). Alternative splicing of neuronal differentiation factor TRF2 regulated by HNRNPH1/H2. Cell Rep 15(5): 926-934.
- Gumireddy, K., Li, A., Yan, J., Setoyama, T., Johannes, G. J., Orom, U. A., Tchou, J., Liu, Q., Zhang, L., Speicher, D. W., Calin, G. A. and Huang, Q. (2013). Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. EMBO J 32(20): 2672-2684.
- Kar, A., Fushimi, K., Zhou, X., Ray, P., Shi, C., Chen, X., Liu, Z., Chen, S. and Wu, J. Y. (2011). RNA helicase p68 (DDX5) regulates tau exon 10 splicing by modulating a stem-loop structure at the 5' splice site. Mol Cell Biol 31(9): 1812-1821.
- Kar, A. N., Vargas, J. N. S., Chen, C. Y., Kowalak, J. A., Gioio, A. E. and Kaplan, B. B. (2017). Molecular determinants of cytochrome C oxidase IV mRNA axonal trafficking. Mol Cell Neurosci 80: 32-43.
- Kwon, S. C., Yi, H., Eichelbaum, K., Föhr, S., Fischer, B., You, K. T., Castello, A., Krijgsveld, J., Hentze, M. W. and Kim, V. N. (2013). The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20(9): 1122-1130.
- Lunde, B. M., Moore, C. and Varani, G. (2007). RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8(6): 479-490.
- McHugh, C. A., Chen, C. K., Chow, A., Surka, C. F., Tran, C., McDonel, P., Pandya-Jones, A., Blanco, M., Burghard, C., Moradian, A., Sweredoski, M. J., Shishkin, A. A., Su, J., Lander, E. S., Hess, S., Plath, K. and Guttman, M. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521(7551): 232-236.
- McHugh, C. A. and Guttman, M. (2018). RAP-MS: a method to identify proteins that interact directly with a specific RNA molecule in cells. Methods Mol Biol 1649: 473-488.
- Ng, S. Y., Bogu, G. K., Soh, B. S. and Stanton, L. W. (2013). The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol Cell 51(3): 349-359.
- Ramanathan, M., Porter, D. F. and Khavari, P. A. (2019). Methods to study RNA-protein interactions. Nat Methods 16(3): 225-234.
- Shevchenko, A., Tomas, H., Havliš, J., Olsen, J. V. and Mann, M. (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1(6): 2856-2860.
- Simon, M. D., Wang, C. I., Kharchenko, P. V., West, J. A., Chapman, B. A., Alekseyenko, A. A., Borowsky, M. L., Kuroda, M. I. and Kingston, R. E. (2011). The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A 108(51): 20497-20502.
- Slobodin, B. and Gerst, J. E. (2010). A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA 16(11): 2277-2290.
- Tominaga, K., Srikantan, S., Lee, E. K., Subaran, S. S., Martindale, J. L., Abdelmohsen, K. and Gorospe, M. (2011). Competitive regulation of nucleolin expression by HuR and miR-494. Mol Cell Biol 31(20): 4219-4231.
- Yoon, J. H., Abdelmohsen, K., Srikantan, S., Yang, X., Martindale, J. L., De, S., Huarte, M., Zhan, M., Becker, K. G. and Gorospe, M. (2012). LincRNA-p21 suppresses target mRNA translation. Mol Cell 47(4): 648-655.
- Yoon, J. H. and Gorospe, M. (2016). Cross-linking immunoprecipitation and qPCR (CLIP-qPCR) analysis to map interactions between long noncoding RNAs and RNA-binding proteins. Methods Mol Biol 1402: 11-17.
- Zeng, F., Peritz, T., Kannanayakal, T. J., Kilk, K., Eiriksdottir, E., Langel, U. and Eberwine, J. (2006). A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat Protoc 1(2): 920-927.
- Zhang, W., Xie, M., Shu, M. D., Steitz, J. A. and DiMaio, D. (2016). A proximity-dependent assay for specific RNA-protein interactions in intact cells. RNA 22(11): 1785-1792.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Savulescu, A. F., Stoychev, S., Mamputha, S. and Mhlanga, M. M. (2020). Biochemical Pulldown of mRNAs and Long Noncoding RNAs from Cellular Lysates Coupled with Mass Spectrometry to Identify Protein Binding Partners. Bio-protocol 10(11): e3639. DOI: 10.21769/BioProtoc.3639.
Category
Biochemistry > Protein > Interaction
Molecular Biology > RNA > RNA-protein interaction
Biochemistry > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.