- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Safe DNA-extraction Protocol Suitable for Studying Tree-fungus Interactions
Published: Vol 10, Iss 11, Jun 5, 2020 DOI: 10.21769/BioProtoc.3634 Views: 8083
Reviewed by: Amey RedkarDheeraj Singh RathoreAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
We present a safe and low-cost method suitable for DNA extraction from mycelium and tree tissue samples. After sample preparation, the extraction takes about 60 min. Method performance was tested by extracting DNA from various tree tissue samples and from mycelium grown on solid and liquid media. DNA was extracted from juvenile and mature host material (Picea abies, Populus trichocarpa, Pseudotsuga menziesii) infected with different pathogens (Heterobasidion annosum, Heterobasidion parviporum, Leptographium wagenerii, Sphaerulina musiva). Additionally, DNA was extracted from pure cultures of the pathogens and several endophytic fungi. PCR success rate was 100% for young poplar material and fungal samples, and 48-72% for conifer and mature broadleaved plant samples. We recommend using 10-50 mg of fresh sample for the best results. The method offers a safe and low-cost DNA extraction alternative to study tree-fungus interactions, and is a potential resource for teaching purposes.
Keywords: DNA extractionBackground
DNA extraction is a central technique in plant-microbe interaction research and for plant disease diagnostic purposes. The abundance of plant-fungal interactions is reflected by the high numbers of cultivable strains isolated from plant samples (Arnold et al., 2001; Higgins et al., 2007; Terhonen et al., 2011). Processing of large numbers of samples for routine PCR reactions with commercial kits can be costly. Additionally, the requirement for hazardous chemicals, such as 2-mercaptoethanol, chloroform, or phenol can restrict the suitability of the protocols for teaching and training purposes.
Due to high secondary metabolite content, many plant samples, and in particular tree tissues, can pose challenges for nucleic acid extraction. Current protocols for DNA extraction from recalcitrant plant tissue utilize organic solvents (chloroform, 2-mercaptoethanol) or surfactants (e.g., cetyl trimethylammonium bromide) (Porebski et al., 1997; Chiong et al., 2017; Yi et al., 2018). Despite their benefits for DNA extraction, these chemicals pose hazards to user health and the environment. Several versions of low-cost, fast, and low health risk protocols for DNA extraction exist for mycelium (Chi et al., 2009), juvenile plant tissue (Edwards et al., 1991; Lu, 2011), grains (Saini et al., 1999), and dried plant tissues (Chabi Sika et al., 2015). However, these methods have not been applied to study tree-fungus interactions, and many times they have been tested only on limited number of sample types. Our goal was to develop and test a safe and low-cost DNA-extraction protocol that is suitable for extracting DNA from various tree tissues and tree-associated fungal samples. After sample preparation, the extraction takes about 60 min. The extracted DNA is suitable for PCR-based downstream applications, such as DNA-based pathogen detection with species-specific primers. Due to its safety and affordability, the method is also a potential resource for teaching and training purposes.
Materials and Reagents
- Standard materials and reagents
- Sterile microcentrifuge tubes, 1.5 or 2.0 ml
- Micropipette tips: 10, 200, 1,000 µl
- Miracloth (Calbiochem, e.g., VWR, catalog number: 475855-1 )
- Plant or fungal samples
- Purified (RO, DI, MilliQ, Nanopure) and sterilized water
- 100 bp DNA ladder (Jena Bioscience, catalog number: M-214S )
- 1 kb DNA ladder (New England Biolabs, catalog number: N0552 )
- NaCl
- KCl
- EDTA
- SDS
- Tris (pH 7.5)
- Polyvinypyrrolidone (PVP, CAS 900-39-8, FW 40,000, e.g., Caisson Labs, catalog number: P071-100GM )
- Isopropanol
- Ethanol (EtOH)
- Extraction buffer (see Recipes)
- Wash buffer (see Recipes)
- Special materials and reagents for different tissue homogenization optionsB.Special materials and reagents for different tissue homogenization options
- Option 1 for soft leaf tissue and mycelium: No special materials or reagents
- Option 2 for various plant tissue and mycelium, larger than 100 mg: Liquid nitrogen
- Option 3 for various plant tissue and mycelium, smaller than 100 mg: Bead beater tubes (e.g., Lysing Matrix I, MP Biomedicals, catalog number: 116918050-CF )
Note: To reduce plastic waste and save on costs, the bead beater tubes can be washed and re-used (see Notes).
Equipment
- Standard equipment
- Scalpels
- Tweezers
- Spatulas
- Scale (e.g., Metler-Toledo, model: ML54T )
- Micropipettes: 1, 10, 100, 1,000 µl
- Heat block (e.g., VWR, catalog number: 12621-096 )
- Vortex (e.g., VWR, catalog number: 10153-838 )
- Microcentrifuge (e.g., Eppendorf, model: 5424 )
- One of the following to heat up water: Microwave, waterbath (e.g., VWR, model: WB05 ), or hot plate (e.g., VWR, catalog number: NO97042-642 )
- Freezer, -20 °C or -80 °C
- Special equipment for different tissue homogenization options
- Option 1 for soft leaf tissue and mycelium: No special equipment
- Option 2 for various plant tissue and mycelium, more than 100 mg:
Dewar for liquid nitrogen
Ceramic mortars and pestles - Option 3 for various plant tissue and mycelium, less than 100 mg:
Bead beater (e.g., Biospec, model: Mini-Beadbeater 16 , catalog number: 607/607EUR ) - For sampling xylem tissue from mature trees: chisel (e.g., Grainger, catalog number: 2AJA6 ) and mallet (e.g., Grainger, catalog number: 4YR61 ), cutting board
Procedure
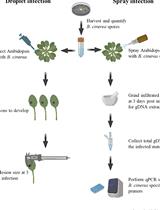
- Sample preparation: Weigh 10-50 mg of sample (see Figure 1).
- Plant tissue: Use tweezers and scalpel to cut the sample to approximately 5 × 5 × 1 mm pieces. Smaller and thinner pieces will result in better sample quality.
- Mycelium: Use scalpel/spatula to scrape mycelium from the surface of Petri plates, or use spatula to collect mycelium from liquid culture.
- Xylem tissue from mature trees: Place wood sample on cutting board. Use chisel and mallet to harvest pieces of xylem tissue. Cut to 5 × 5 × 1 mm pieces with scalpel and spatula.

Figure 1. Examples of sample sizes for DNA extraction. A. Fresh phloem from 8-week-old Populus trichocarpa trees. Weight 20 mg. B. Fresh phloem from mature P. trichocarpa trees. Weight 15 mg. C. Fresh Leptographium wagnerii mycelium harvested from liquid malt extract cultures. Weight 20 mg. Interval between vertical lines = 1 mm. - Homogenize sample and add extraction buffer
Three options are available depending on sample type. Complete homogenization is not necessary.- Option 1: Mycelium and soft leaf tissue
- Place sample in a 1.5 or 2.0 ml microcentrifuge tube.
- Add 1 ml extraction buffer.
- Vortex rigorously for 20 s.
- Proceed to Procedure C.
- Place sample in a 1.5 or 2.0 ml microcentrifuge tube.
- Option 2: Various plant tissue and mycelium ≥ 50 mg:
- Grind sample in mortar with pestle and liquid nitrogen.
- Transfer 10-50 mg of homogenized sample with a spatula or by decanting to a 1.5/2.0 ml microcentrifuge tube.
- Add 1 ml extraction buffer.
- Vortex rigorously for 20 s.
- Proceed to Procedure C.
- Grind sample in mortar with pestle and liquid nitrogen.
- Option 3: Various plant tissue and mycelium ≤ 50 mg:
- Transfer the sample into a 2.0 ml beat beater tube.
- Add 1 ml extraction buffer.
- Process for 20 s in a bead beater.
- Proceed to Procedure C.
- Transfer the sample into a 2.0 ml beat beater tube.
- Option 1: Mycelium and soft leaf tissue
- DNA extraction
- Heat the wash buffer in a water bath or in a beaker with warm water (approximately 65 °C) to dissolve any precipitants. Temperature is not critical, as long as no precipitants remain. Mix by inversion.
- Lysis and debris elimination: Incubate the samples in extraction buffer at 65 °C for 15 min. Vortex once during incubation. Centrifuge at 6,000 x g for 10 min.
- Eliminate debris: After centrifugation, transfer ca. 0.5 volume of the supernatant into a new tube. Add 1 volume of pre-heated wash buffer, and vortex samples for 20 s. Centrifuge at 21,000 x g for 10 min.
Note: Complete debris elimination is not critical when pipetting the supernatant. - Precipitate: Transfer ca. 0.7 volume of supernatant into a new tube, add 0.85 volume of isopropanol (room temperature), and mix by inversion for 20 s. Centrifuge at 21,000 x g for 10 min.
Note: Minimize transferring any debris while pipetting the supernatant. - Wash the pellet: Pour out the supernatant, and remove remaining supernatant by tapping the tubes upside down on a paper towel. Add 200 µl of 70% ethanol, and centrifuge at 21,000 x g for 5 min.
Note: Consider local regulations for correct handling of isopropanol waste. - Dry the pellet: Pipet out the ethanol. Leave the caps open, and dry pellets in a heat block at 65 °C for 5 min.
Note: For faster drying, remove as much of the ethanol as possible. - Resuspension: Dissolve the pellet in 20-50 µl of TE buffer or nuclease free water. Vortex to dissolve if needed, and centrifuge briefly to collect any droplets to the bottom of the tube. Store DNA samples at -20 °C or -80 °C until used.
Note: If the DNA pellet is not colorless or white post 70% ethanol wash, add 20-50 µl of TE buffer or nuclease-free water to resuspend the DNA without disturbing the pellet. Gently pipet the liquid a few times in the tube and collect the supernatant as DNA for downstream processes. Keep the pellet until DNA is quantified. Use the DNA for PCR-based detection, or store in freezer until used.
Data analysis
Analysis of protocol performance:
- Plant and fungal material used for DNA extractions
To test the suitability of the protocol, we extracted DNA from artificially inoculated trees, naturally infected trees, and mycelium (Table 1). The artificially inoculated samples included 8-week-old Populus trichocarpa plants spray-inoculated with Sphaerulina musiva (LeBoldus et al., 2010; Abraham et al., 2018), and 3-year-old Picea abies plants plug-inoculated with Heterobasidion sp. (Terhonen et al., 2019). The naturally infected plant samples included stem cankers on mature P. trichocarpa trees caused by S. musiva infection, and mature Pseudotsuga menziesii roots infected with Leptographium wageneri. Necrotic or discolored phloem or xylem samples with 10-380 mg of tissue (average 86 mg) were used for DNA extraction. For every sample, 1 ml of extraction buffer was used regardless of sample weight. Fungal DNA was extracted from mycelium harvested from pure cultures (Table 1). For solid medium, either malt extract agar (2% malt extract, 2% agar) or KV8 agar (18% V8 juice, 0.2% CaCO3, 2% agar) were used. Cultures on solid medium were grown in ambient room temperature. For liquid medium, either malt extract medium (2% malt extract) or KV8 medium (18% V8 juice, 0.2% CaCO3) was used (Table 1). Cultures in liquid medium were grown in ambient room temperature on a rotary shaker (100-150 rpm). The mycelium was harvested from liquid medium by filtering through Miracloth (Calbiochem) and rinsed with DI water. - Assessment of DNA sample quality
DNA concentrations were measured with Nanodrop, Nanophotometer, or Qubit. PCR, quantitative real-time PCR (qPCR), agarose gel electrophoresis (Figure 2), and fungal ITS sequencing were used to evaluate sample quality. For PCR and qPCR, no-template negative controls and positive template controls were included into each run to evaluate detection reliability.
For the P. trichocarpa samples that were inoculated or naturally infected with S. musiva, we used a host-pathogen specific assay (Abraham et al., 2018). For detection of Heterobasidion species from inoculated wood samples, species-specific primers for H. annosum and H. parviporum (Hantula and Vainio, 2003) were used (Terhonen et al., 2019). For detection of L. wageneri from P. pseudotsuga roots and fungal cultures, we used Leptographium-specific primers (Schweigkofler et al., 2005). Primers for P. menziesii (Winton et al., 2002) were used to distinguish PCR-inhibition from negative samples, and to amplify DNA extracted from Douglas-fir needles.
DNA samples from Diplodia sapinea cultures were amplified with primers targeting the nuclear large subunit, elongation factor and calmodulin regions (Vilgalys and Hester, 1990; Carbone and Kohn, 1999; Grünig et al., 2007; Nelsen et al., 2011). Additionally, DNA from D. sapinea and fungal endophytes was amplified with primers ITS1-F and ITS4 for the fungal ribosomal internal transcribed spacer region (White et al., 1990; Gardes and Bruns, 1993). The PCR conditions are specified in Table 2 and primer sequences in Table 3. All PCR amplicons were visualized under UV light on 1.5% agarose gels with StainINTM RED or GelRedTM nucleic acid stains. For fungal species used for sequencing, the PCR products were purified and sequenced using the respective primers (Table 1) at Microsynth SEQLAB (Göttingen, Germany).
The effect of potential PCR inhibitors in the DNA samples on target detection was evaluated with a multiplex Taqman qPCR protocol (Abraham et al., 2018). DNA extracted with a commercial kit (DNeasy Plant Mini, Qiagen) from comparable tissue samples was used as a reference for low-inhibitor samples. Seven-point dilution series were prepared for P. trichocarpa (10-fold dilution series, 60-6 × 10-4 ng/µl) and S. musiva DNA samples (5-fold dilution series, 50-3.2 × 10-3 ng/µl) (Abraham et al., 2018) extracted with the developed method and with the commercial kit. The quantification cycle (Cq) values for the samples from the two extraction methods were compared, to evaluate the impact of potential PCR inhibitors on target detection.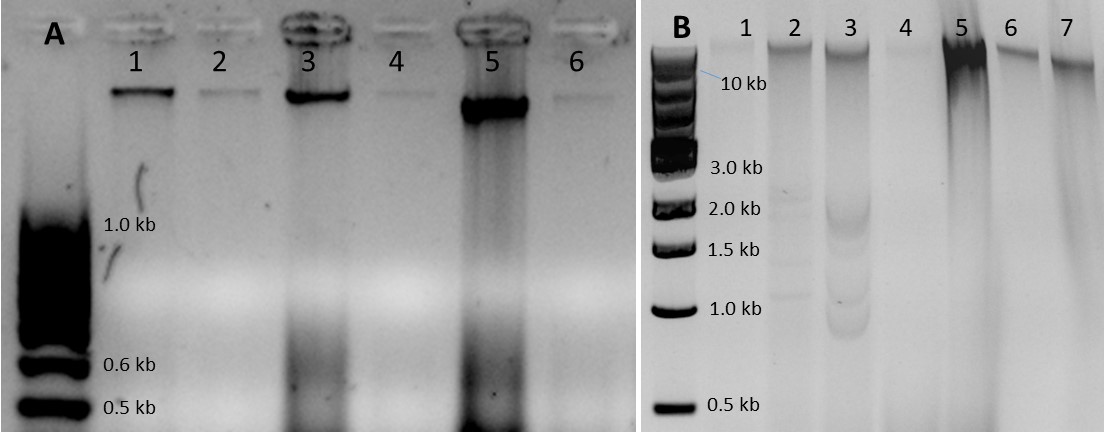
Figure 2. Examples of DNA samples extracted with the developed protocol. A. Three Diplodia sapinea DNA samples extracted from mycelium grown on malt extract agar (MEA). Lanes 1, 3, and 5: 50-500 ng of DNA. Lanes 2, 4, and 6: 10-fold dilutions of samples in lanes 1, 3, and 5. Gel: 2.0% agarose in 1× TAE, 100 V, 35 min. Ladder: 100 bp DNA ladder. B. DNA extracted from Leptographium wagnerii mycelium from liquid malt extract (lane 1), Sphaerulina musiva mycelium grown on KV8 agar (lane 2), S. musiva grown in liquid KV8 medium (3), fresh poplar phloem (lane 4), fresh poplar leaves (lane 5), mature Douglas-fir xylem (lane 6), and fresh Douglas-fir needles (lane 7). Lanes 1-4 and 6-7: 20-100 ng DNA. Lane 5: 500 ng DNA. Gel: 1% agarose in 1× TAE, 120 V, 70 min. Ladder: 1 kb DNA ladder.
Table 1. Sample types extracted with the protocol, PCR success rates (%), and number of sequenced samples
aGardes and Bruns 1993, White et al., 1990
bGrünig et al., 2007
cCarbone and Kohn, 1999
dVilgalys and Hester, 1990, Nelsen et al., 2011
Table 2. PCR conditions and primers used to test sample quality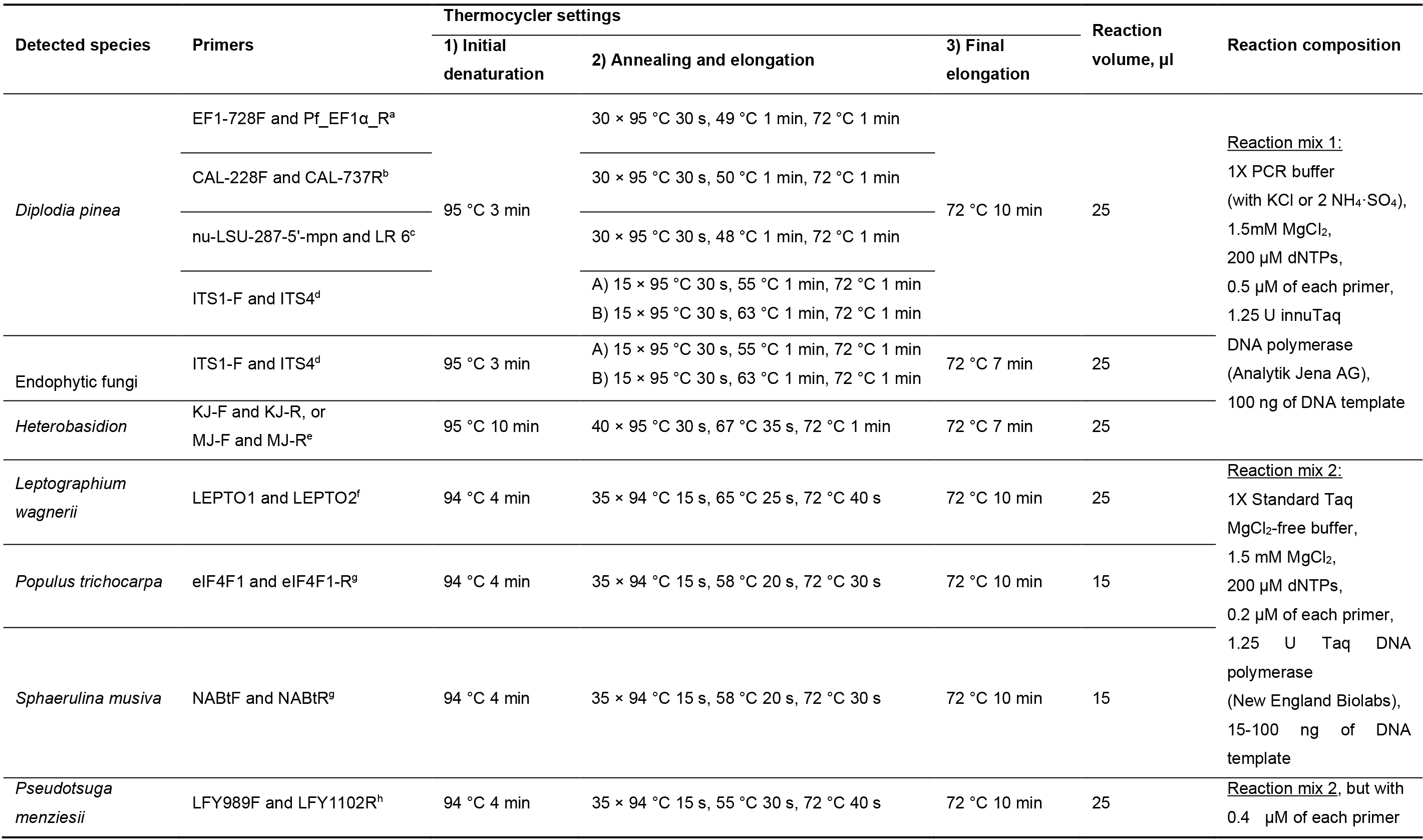
aCarbone and Kohn 1999, Grünig et al., 2007
bCarbone and Kohn, 1999
cVilgalys and Hester, 1990, Nelsen et al., 2011
dGardes and Brunns, 1993; White et al., 1990
eHantula and Vainio, 2003
fSchweigkofler et al., 2005
gAbraham et al., 2018
hWinton et al., 2002Table 3. Primer sequences used in the PCR reactions.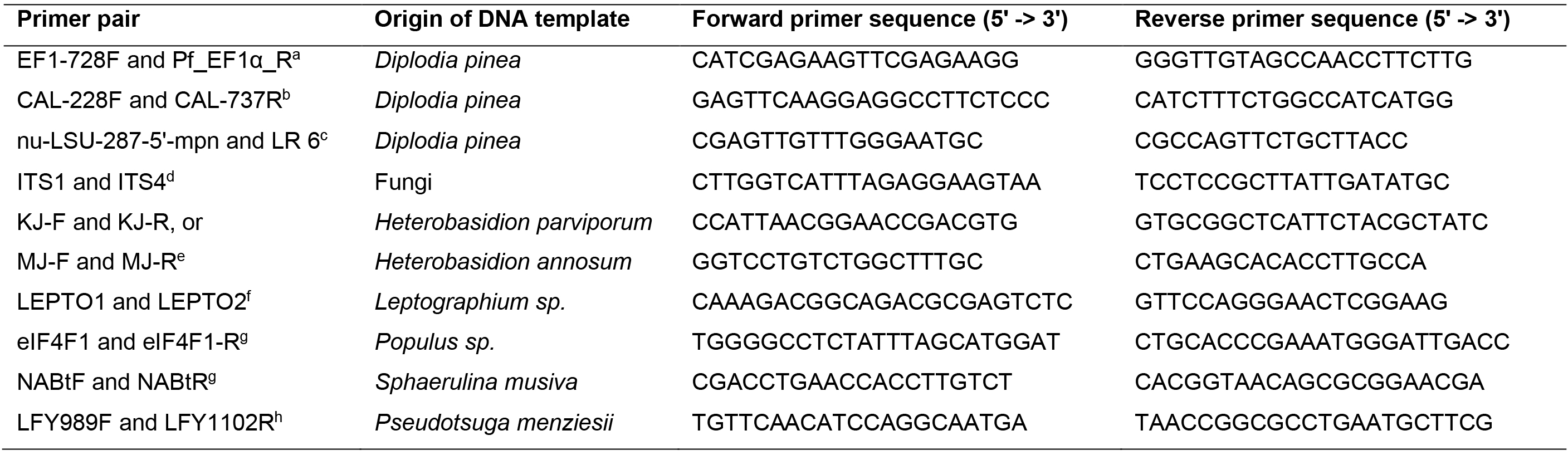
aCarbone and Kohn, 1999, Grünig et al., 2007
bCarbone and Kohn, 1999
cVilgalys and Hester, 1990, Nelsen et al., 2011
dGardes and Brunns, 1993, White et al., 1990
eHantula and Vainio, 2003
fSchweigkofler et al., 2005
gAbraham et al., 2018
hWinton et al., 2002 - Data analysis
Data analysis was conducted in R version 3.6.1. The effect of PCR inhibitors on quantification cycle (Cq) values was estimated using ANOVA followed by Tukey’s HSD tests (Figure 3A). We visualized the contribution of sample weight, DNA concentration, and sample purity (A260/280 and A260/230) on PCR success by plotting the results from principal component analysis (PCA) for the plant and fungal samples (Figures 3B-3D). The PCA results were visualized with the R package factoextra (Kassambara and Mundt 2017). For P. abies samples, necrosis length was also included in the model. Separate PCA’s were computed for P. trichocarpa samples (n = 85), P. abies samples (n = 48), and fungal samples (n = 129). Differences in template properties between failed and successful PCR reactions within the same sample type were compared by two-sample t-tests. If necessary, data were normalized with log-transformations. - Protocol performance
All the DNA samples extracted from 8-week-old P. trichocarpa phloem inoculated with S. musiva were PCR-positive for the pathogen (Table 1). PCR amplification worked for all fungal DNA samples, and the PCR products were suitable for fungal ITS sequencing. In comparison, PCR success rate was lower for DNA samples extracted from naturally infected mature P. trichocarpa samples (72%), inoculated 3-year-old P. abies samples (48%), and naturally infected mature P. menziesii roots (60%) (Table 1). The extraction protocol yields total DNA with the majority of the fragments larger than 10 kb (Figure 2). The samples are stable at least for 2 years in -20 °C. Partial DNA fragmentation (Figure 2B) did not affect PCR performance.
Based on the comparison of Cq values for DNA samples extracted with a commercial kit, the extracted DNA samples may contain inhibitors that can affect the accuracy of target quantification by qPCR (Figure 3A). The Cq values were lower for the three highest dilutions (P < 0.040). After 1,000-fold dilution, the extraction method had no effect on quantification. The standard curves prepared from the DNA samples extracted with the protocol had lower amplification efficiencies compared to the commercial kit (Figure 1A). It is possible that the qPCR protocol in Abraham et al. (2018) is not optimal for the samples extracted with the developed DNA extraction protocol. However, the extraction method did not affect target detection.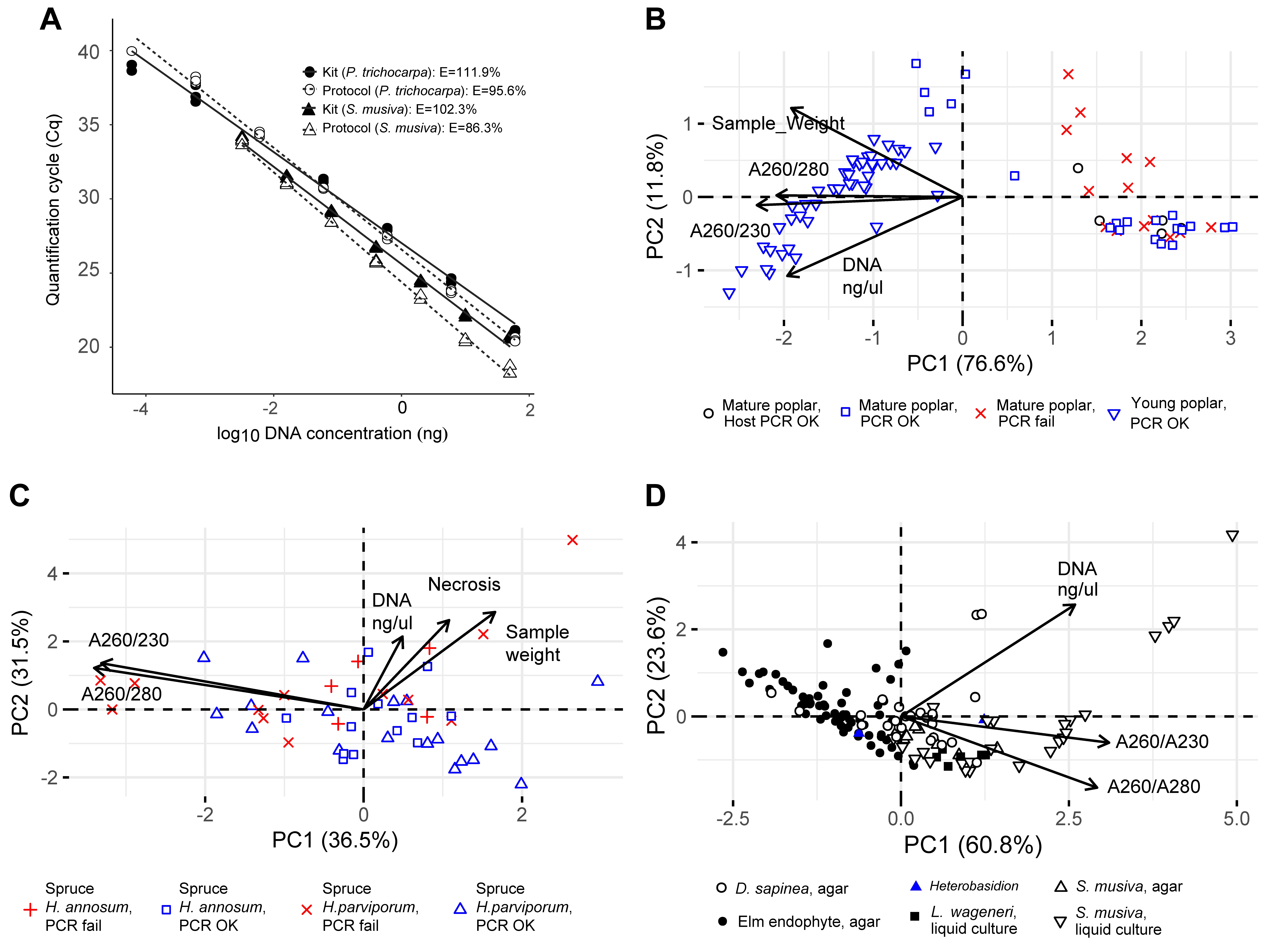
Figure 3. Comparative qPCR analysis to estimate the presence of sample inhibitors (A), and principal component analysis to visualize sample properties in Populus trichocarpa samples (B), young Picea abies samples (C), and fungal samples (D)
We explored the association of DNA sample properties with PCR success by visualizing the results from principal component analysis (PCA). The two first principal components (PC) explained 68-87% of the total variation. The A260/280 and A260/230 values explained majority of variation along PC1 for P. trichocarpa, P. abies, and fungal DNA samples (55%, 85% and 75% contribution to PC1 variation, respectively) (Figures 1B-1D). For P. trichocarpa, the samples split into two groups on both sides of the vertical PC2 axis (Figure 3B). For the samples on the left side of the PC2 axis, PCR success rate was 100% and sample purity was relatively high (Figure 3B).
Sample weight explained 55% and 40% of the variation along the PC2 in P. trichocarpa and P. abies, respectively. Larger initial sample weights were associated with lower PCR success rate for mature poplar and young Norway spruce DNA samples (Table 4). Among the 34 mature poplar samples that clustered on the right side of PC2 axis (Figure 3B), initial sample weights and DNA concentrations were higher for the failed PCR reactions (t = 2.882, P = 0.007 and t = 2.195, P = 0.035, respectively). Similarly, initial sample weights and DNA concentrations were higher for P. abies samples that failed PCR amplification (t = 1.9511, P = 0.057 and t = 2.060, P = 0.045, respectively). For the mature poplar samples, PCR failure both for host and pathogen amplification despite higher DNA concentration is probably associated with low template quality combined with high polyphenolic content in the mature bark tissue samples. For Norway spruce, PCR failure for pathogen amplification despite high DNA concentration and sufficient template quality is probably explained by low amount of pathogen DNA in less colonized samples. Alternatively, high phenolic content in heavily colonized samples may have inhibited pathogen detection, as increasing necrosis length was associated with lower PCR success in Norway spruce samples. This indicates that DNA samples extracted from tree tissue samples with high amounts of lignin or other polyphenolic compounds may have lower PCR success rates.
The fungal DNA samples were highly variable based on A260/230 and A260/280 values (75% of variation on PC1, Figure 3D). The DNA samples extracted from mycelium grown in liquid cultures typically had higher measures of purity compared to fungal samples with agar (Table 4). Based on this, we recommend minimizing the amount of agar in the fungal samples used for DNA extractions. Despite high variation in sample purity, PCR amplification was successful for all the fungal DNA samples.
Table 4. Sample weight, DNA concentrations, and DNA absorbance values for different DNA sample types extracted with the protocol. Median, minimum and maximum values are indicated.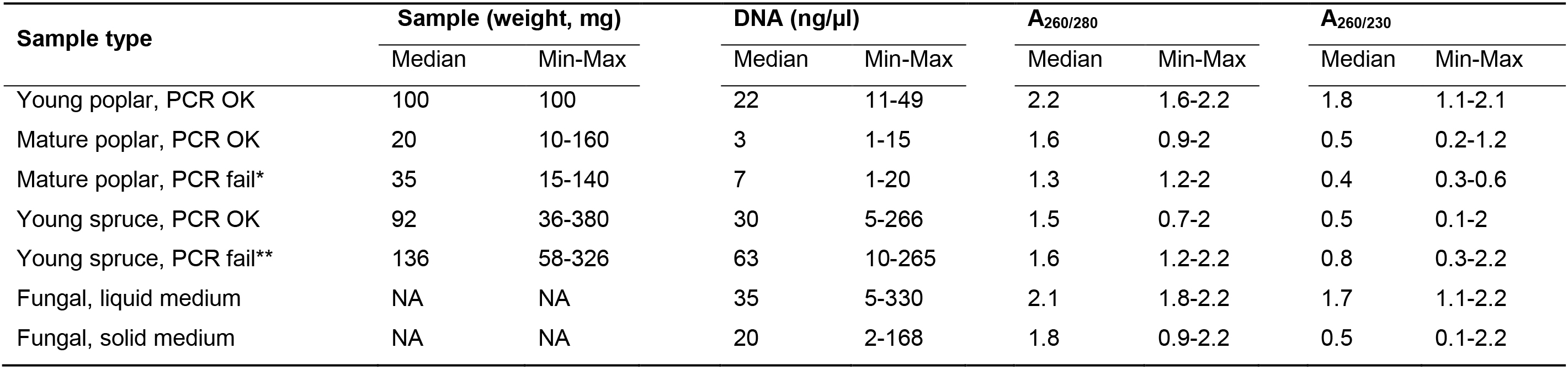
*Both host and pathogen PCR failed
**Only pathogen PCR tested
Notes
- Amount of extraction buffer and sample weight
We recommended to use 1 ml of extraction buffer per 50 mg or less of plant or fungal tissue. Low buffer volume relative to extracted tissue can have a negative effect on PCR success. - Dissolving the pellet
If the DNA pellet is not colorless or white post 70% ethanol wash, add 20-50 μl of TE buffer or nuclease-free water to resuspend the DNA without disturbing the pellet. Gently pipet the liquid a few times in the tube and collect the supernatant as DNA for downstream processes. Keep the pellet until DNA is quantified. Use the DNA for PCR-based detection, or store in freezer until used. - Re-using the bead beater tubes
To reduce costs and plastic waste, the bead beater tubes can be washed, treated with bleach to degrade DNA, autoclaved, and re-used. Separate the beads, caps and tubes in separate containers. Fill the containers with warm soap water, agitate for 5 min, and pour out the soap water. Rinse with tap water until runoff is clear. Shake tubes and caps to remove remaining tap water. Rinse twice with DI-water. To remove any remaining DNA, soak the components 1 h in 3% w/v NaOCl solution (1:1 solution with commercial bleach and DI-water) (Kemp and Smith 2005). Rinse twice with tap water, followed by two DI-water rinses. Let the tube components dry overnight, or dry in an oven. Once the components are dry, compile the tubes and autoclave at 121 °C for 30 min.
Recipes
- Extraction buffer
1 M NaCl
100 mM Tris HCl
10 mM EDTA
2% PVP- Mix all ingredients in a beaker on a stirring hot plate
- Fill to desired volume with sterile purified water
- Heat the solution until PVP is dissolved
- Mix all ingredients in a beaker on a stirring hot plate
- Wash buffer
1% SDS
0.5 M KCl or NaCl- Mix all ingredients in a beaker on a stirring hot plate
- Fill to desired volume with sterile purified water
- Heat the solution until no precipitation is visible
- Heat the wash buffer before use to dissolve any precipitants
- Mix all ingredients in a beaker on a stirring hot plate
Acknowledgments
This research was supported by the DOE Office of Science, Office of Biological and Environmental Research (BER), grant no. DE-SC0018196, and by funding from the Faculty of Forest Sciences and Forest Ecology, University of Göttingen. Jumoke Aduke Babalola and David Robert Rӑscuţoi, University of Göttingen, are highly acknowledged for their contribution to the DNA extraction and PCR experimental set up. Dr. Patrick Bennett is thanked for providing the L. wagenerii strains and naturally infected Douglas-fir root samples, and Dr. Kelsey Søndreli for providing the mature P. trichocarpa samples naturally infected with S. musiva.
Competing interests
No competing interests declared.
References
- Abraham, N. D., Chitrampal, P., Keriö, S. and LeBoldus, J. M. (2018). Multiplex qPCR for detection and quantification of Sphaerulina musiva in Populus stems. Plant Pathol 67(9): 1874-1882.
- Arnold, A. E., Maynard, Z. and Gilbert, G. S. (2001). Fungal endophytes in dicotyledonous neotropical trees: patterns of abundance and diversity. Mycol Res 105(12): 1502-1507.
- Carbone, I. and Kohn, L. M. (1999). A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91(3): 553-556.
- Chabi Sika, K., Kefela, T., Adoukonou-Sagbadja, H., Ahoton, L., Saidou, A., Baba-Moussa, L., Jno Baptiste, L., Kotconi, S. O. and Gachomo, E. W. (2015). A simple and efficient genomic DNA extraction protocol for large scale genetic analyses of plant biological systems. Plant Gene 1: 43-45.
- Chi, M. H., Park, S. Y. and Lee, Y. H. (2009). A quick and safe method for fungal DNA extraction. Plant Pathol J 25: 108-111.
- Chiong, K. T., Damaj, M. B., Padilla, C. S., Avila, C. A., Pant, S. R., Mandadi, K. K., Ramos, N. R., Carvalho, D. V. and Mirkov, T. E. (2017). Reproducible genomic DNA preparation from diverse crop species for molecular genetic applications. Plant Methods 13(1): 106.
- Edwards, K., Johnstone, C. and Thompson, C. (1991). A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res 19(6): 1349.
- Gardes, M. and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes--application to the identification of mycorrhizae and rusts. Mol Ecol 2(2): 113-118.
- Grünig, C. R., Brunner, P. C., Duo, A. and Sieber, T. N. (2007). Suitability of methods for species recognition in the Phialocephala fortinii-Acephala applanata species complex using DNA analysis. Fungal Genet Biol 44(8): 773-788.
- Hantula, J. and Vainio, E. (2003). Specific primers for the differentiation of Heterobasidion annosum (s.str.) and H. parviporum infected stumps in northern Europe. Silva Fennica 37.
- Higgins, K. L., Arnold, A. E., Miadlikowska, J., Sarvate, S. D. and Lutzoni, F. (2007). Phylogenetic relationships, host affinity, and geographic structure of boreal and arctic endophytes from three major plant lineages. Mol Phylogenet Evol 42(2): 543-555.
- Kassambara, A., and Mundt, F. (2017). factoextra: Extract and visualize the results of multivariate data analyses.
- Kemp, B. M. and Smith, D. G. (2005). Use of bleach to eliminate contaminating DNA from the surface of bones and teeth. Forensic Sci Int 154(1): 53-61.
- LeBoldus, J. M., Blenis, P. V. and Thomas, B. R. (2010). A method to induce stem cankers by inoculating nonwounded Populus clones with Septoria musiva spore suspensions. Plant Dis 94(10): 1238-1242.
- Lu, Y. (2011). Extract genomic DNA from Arabidopsis leaves (can be used for other tissues as well). Bio-protocol 1(13): e90.
- Nelsen, M. P., Lücking, R., Mbatchou, J. S., Andrew, C. J., Spielmann, A. A. and Lumbsch, H. T. (2011). New insights into relationships of lichen-forming Dothideomycetes. Fungal Diversity 51(1): 155-162.
- Porebski, S., Bailey, L. G. and Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15(1): 8-15.
- Saini, H. S., Shepherd, M. and Henry, R. J. (1999). Microwave extraction of total genomic DNA from barley grains for use in PCR. J Inst Brew 105(3): 185-190.
- Schweigkofler, W., W.J, O., S.L, S., D.R, C., Maeda, K., Peay, K. and Garbelotto, M. (2005). Detection and quantification of Leptographium wageneri, the cause of black-stain root disease, from bark beetles (Coleoptera: Scolytidae) in Northern California using regular and real-time PCR. Canadian Journal of Forest Research 35: 1798-1808.
- Terhonen, E., Langer, G. J., Bußkamp, J., Rӑscuţoi, D. R. and Blumenstein, K. (2019). Low water availability increases necrosis in Picea abies after artificial inoculation with fungal root rot pathogens Heterobasidion parviporum and Heterobasidion annosum. Forests 10(1): 55.
- Terhonen, E., Marco, T., Sun, H., Jalkanen, R., Kasanen, R., Vuorinen, M. and Asiegbu, F. (2011). The effect of latitude, season and needle-age on the mycota of Scots pine (Pinus sylvestris) in Finland. Silva Fenn 45
- Vilgalys, R. and Hester, M. (1990). Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J Bacteriol 172(8): 4238-4246.
- White, T., Bruns, T., Lee, S., Taylor, J., Innis, M., Gelfand, D. and Sninsky, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols, Academic Press, Inc., pp. 315-322.
- Winton, L. M., Stone, J. K., Watrud, L. S. and Hansen, E. M. (2002). Simultaneous one-tube quantification of host and pathogen DNA with real-time polymerase chain reaction. Phytopathology 92(1): 112-116.
- Yi, S., Jin, W., Yuan, Y. and Fang, Y. (2018). An optimized CTAB method for genomic DNA extraction from freshly-picked pinnae of fern, Adiantum capillus-veneris L. Bio-protocol 8(13): e2906.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Keriö, S., Terhonen, E. and LeBoldus, J. M. (2020). Safe DNA-extraction Protocol Suitable for Studying Tree-fungus Interactions. Bio-protocol 10(11): e3634. DOI: 10.21769/BioProtoc.3634.
Category
Plant Science > Plant molecular biology > DNA > DNA extraction
Microbiology > Pathogen detection > PCR
Molecular Biology > DNA > DNA extraction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.