- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RNA Extraction from Ears and Draining Lymph Nodes of Mice Infected with Leishmania amazonensis

Published: Vol 10, Iss 11, Jun 5, 2020 DOI: 10.21769/BioProtoc.3633 Views: 5669
Reviewed by: Alexandros AlexandratosMarieta RusevaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Parasites of the genus Leishmania infect the mammalian hosts, including mice and humans and cause cutaneous or visceral leishmaniasis depending upon the parasite species transmitted by the vector sandfly. Leishmania amazonensis is one of the Leishmania species responsible for the cutaneous form of the disease. We have inoculated with these parasites the ear dermis of mice. RNA preparations were performed from fragmented tissues using a buffer containing guanidin isothiocynate (RLT buffer, RNeasy Mini Kit, Qiagen, SAS, France) and β-mercaptoethanol. Both reagents facilitate the isolation of intact RNA from tissues and the use of the RNeasy Kits present with several advantages that facilitate the isolation of pure non-degraded total RNA: i) This method allows to avoid the presence of phenol in the RNA extraction buffer, commonly used in alternative protocols; ii) Moreover Diethylpyrocarbonate (DEPC) treatment of glassware, to avoid RNAses contamination of the samples, is not required with this protocol; iii) Finally, it is a fast procedure and the isolated total RNA may be concentrated in a small volume thus facilitating its use for downstream experimental procedures.
Keywords: RNA isolationBackground
Pathogens of the genus Leishmania cause vector born zoonotic diseases with a variety of clinical manifestations and a variability in disease severity (Mansueto et al., 2007). The cutaneous form of leishmaniasis is caused by several Leishmania species including Leishmania amazonensis (McMahon-Pratt and Alexander, 2004; Christensen et al., 2019).
The existence of mouse models, with high, intermediate or no susceptibility to the cutaneous form of leishmaniasis have contributed to the development of experimental protocols for parasite dissemination (de Oliveira Cardoso et al., 2010). The parasites are naturally transmitted to the mammalian hosts by the bite of infected phlebotomine sandflies of the genus Lutzomyia (Bates, 2007). Thus transmission protocols use the ear dermis, the dorsal skin or the hind footpad (Schuster et al., 2014). In our protocol the Leishmania amazonensis parasites were inoculated through the ear dermis of the mouse, a method that closely imitates the natural transmission of the parasites by the sandfly (Giraud et al., 2019b). In addition, the ear pinna is appropriate for phenotypic evaluation of the lesion as well as for the isolation of nucleic acids from the inoculation sites, for two reasons: 1) parasites, in their infectious path, induce two distinct phases: the first, is a clinically silent phase characterized by the absence of lesions and the increase of parasite load and the second phase, during which visible lesions develop, and are associated with immune cell infiltration at the site of inoculation (Liu and Uzonna 2012; Giraud et al., 2019a). 2) The immune cells expand in the draining lymph nodes, which are also used for nucleic acids preparation (Cortes et al., 2010). Therefore the immune response of the host to the parasites can be studied on the site of inoculation and on the secondary immune adjacent tissue, the ear draining lymph nodes.
Leishmania amazonensis infection triggers an inflammatory response characterized by the presence of macrophages on the lesion sites. The parasites develop in the resident dermal macrophages (MF) and in Dendritic cells (DC) (Liu and Uzonna, 2012). The mechanisms of the parasite adaptation in the mammalian phagocytes and tissues remain elusive. Macrophages do not prevent the proliferation of the infection but on the contrary are the resident cells for these parasites.
We have studied the immune response of the host to Leishmania amazonensis infection in mice (Giraud et al., 2019b). Parasites transmitted through the ear dermis induce the immune response of the adjacent lymph nodes. RNAs prepared from the ear lesions and the lymph nodes were used to address the immune-related gene expression prior and after infection with the parasites. Several methods exist for preparation of ribonucleic acids. The classical protocols use guanidium isothiocyanate, a chaotropic agent that disintegrates the cellular structures and dissociates rapidly the nucleoproteins from the nucleic acids (Reid, 1991). RNases are also inactive in the presence of 4 M guanidium thiocyanate. Beta-mercaptoethanol (β-ME) is added to reduce RNA degradation by RNases. These protocols require disposable sterile glassware and plastic ware for the preparation of non-degraded RNA. Often pretreatment with diethylpyrocarbonate (DEPC) (0.1%) is used to treat glassware for 12 h at 37 °C and then heating at 100 °C for 15 min to remove any traces of diethylpyrocarbonate. The cellular proteins are removed from the nucleic acids preparations by extractions with phenol/chloroform. Ethanol precipitation of the isolated RNAs is then required and the nucleic acids are obtained by precipitation in the presence of 0.3 M sodium acetate. Our method benefits from the existence of a simplified protocol provided by the Qiagen kits. The methodological advantages of this protocol are as follows: 1) the method is simplified and quick, 2) there is no need to use DEPC, 3) phenol extraction of the nucleic acids from the proteins is not required, 4) the silica columns for separation of the nucleic acids from the cells or tissues constituents are very efficient, 5) RNAs are at a correct concentration and ethanol precipitation is not necessary and finally this method yields a high quality of RNAs. Whilst accompanied protocols are provided by the Qiagen kits, the additional details in the description of our method, applied to the mouse ear and lymph nodes, completes the methodology of the corresponding published article (Giraud et al., 2019b), presents with an interest for the isolation of pure non-degraded RNAs and may be useful to other investigators.
Materials and Reagents
- 60 x 15 mm, Cell culture-treated Permanox Petri Dish sterile (Thermo Fisher Scientific, NuncTM, catalog number: 150340 )
- Pipette Barrier (Filter) tips (Certified Free for RNases, DNases, sterile) (Thermo Fisher Scientific, Invitrogen, catalog numbers: AM126-45 (10 μl), AM126-35 (20 μl), AM126-50 (200 μl), AM126-60 (1,000 μl)
- 2 ml and 1.5 ml tubes (Eppendorf, Fisher Scientific, catalog number: 3810X )
- 2 ml hard tissue homogenizing tubes, containing 2.8 mm ceramic (zirconium oxide) beads CK28 (Bertin Instruments, catalog number: P000911-LYSK0-A , for Precellys tissue homogenizer, see Equipment 1)
- Glass Pasteur Pipettes (Merck, catalog number: CLS7095D5X )
- Adult C57BL/6J and B6(Cg)-Spp1tm1Blh/J mice (8-12 weeks of age)
- Phosphate buffered saline (PBS BioPerformance Certified, pH 7.4, Sigma-Aldrich, catalog number: P5368-10PAK , storage at room temperature)
- β-Mercaptoethanol (β-ME), Molecular Biology Grade (Millipore, Merck, Calbiochem, catalog number: 444203-250ML or alternatively dithiothreitol (DTT) may replace β-ME [Mommaerts et al., 2015])
- RLT (RNA lysis buffer, containing guanidine thiocyanate) (Qiagen, catalog number: 79216 )
- RW1 (RNA wash buffer containing ethanol) (Qiagen, catalog number: 74134 )
- RPE (RNA wash buffer) (Qiagen, catalog number: 74134 )
- RNeasyR Plus Mini Kit (Qiagen, catalog number: 74134 )
- Alternatively the Qiagen RNeasy Kit not including the gDNA separation columns may be used (Qiagen, catalog number: 75154 )
- RNase-free DNase Set (Qiagen, catalog number: 79254 )
- Buffers and separation columns (RNeasy Plus Mini Kit) (Qiagen, catalog number: 74134 )
- Ethanol (CH3CH2OH) (Merck, catalog number: 02851-1L )
- Ethanol 70% in H2O (see Recipes)
- RLT + β-ME or instead RLT-DTT buffer (see Recipes)
Equipment
- Adjustable Pipettes (Sartorius Biohit Proline France), or Micropipettes PIPETMAN L (GILSON, catalog number: 070053-070058 )
- Reagent Vessel (Sartorius, catalog number: 783500 )
- Surgical instruments (including a pair of scissors, two pair of fine scissors, and two tweezers) (Figure 1i-iv)
- Mice dissecting table and dissecting glass plate (Figure 1v).
- Precellys 24 System tissue homogenizer (Bertin Technologies, catalog number: P000669-PR240-A.0 )
- Precellys® 2 ml Hard Tissue homogenizing ceramic beads kit (CK28) (Bertin Instruments, catalog number: 10011151 , 230 Rockville, MD 20850 USA)
- 4 °C fridge and -20 °C freezer
- Centrifuge Eppendorf 5415R or 5424R (Eppendorf, catalog number: 5404000410 , Radius 8.3 cm)
- NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, catalog number: ND-1000 ) (Schroeder et al., 2006)

Figure 1. Instruments used for tissues dissection and lymph node isolation. i. Scissors; ii. Curved scissors; iii. Tweezers; iv. Fine scissors; v. Tissue dissection glass plate.
Procedure
Special safety conditions should be applied when working with Leishmania spp. parasites in the laboratory. The main route of exposure is through the use of needles for infecting the ears of the animals. Therefore protective measures should be taken in consideration, i.e., wearing gloves, needle precautions and working under a hood in a P2 plus laboratory. All instruments used for dissection of the tissues should be placed in alcohol 70% recipients and then washed in the presence of chlorine and sterilized.
- Preparation
- Prepare 2 ml hard tissue homogenizing tubes containing the ceramic beads and label with the samples identification.
- Prepare RLT-β-ME buffer (see Recipes).
- Complement RPE wash buffer with 4 volumes of ethanol (100%).
- Prepare a dilution of 70% Ethanol using sterile nuclei acids-free water.
- Label the tubes to be used for the next steps of RNA isolation from the fragmented tissues.
- Prepare cell culture Petri dish and add 3 ml PBS.
- Dissection of tissues
- Adult C57BL/6J and B6(Cg)-Spp1tm1Blh/J mice at various ages selected for the experiments are anesthetized with CO2.
- After washing with Ethanol and subsequently with PBS the areas from which the tissues will be isolated, cut the infected with the parasites ear (left ear) and discard the surrounding tissue of the infected area. Isolate a similar area from the right ear (non-infected) to be used for control (Figure 2).
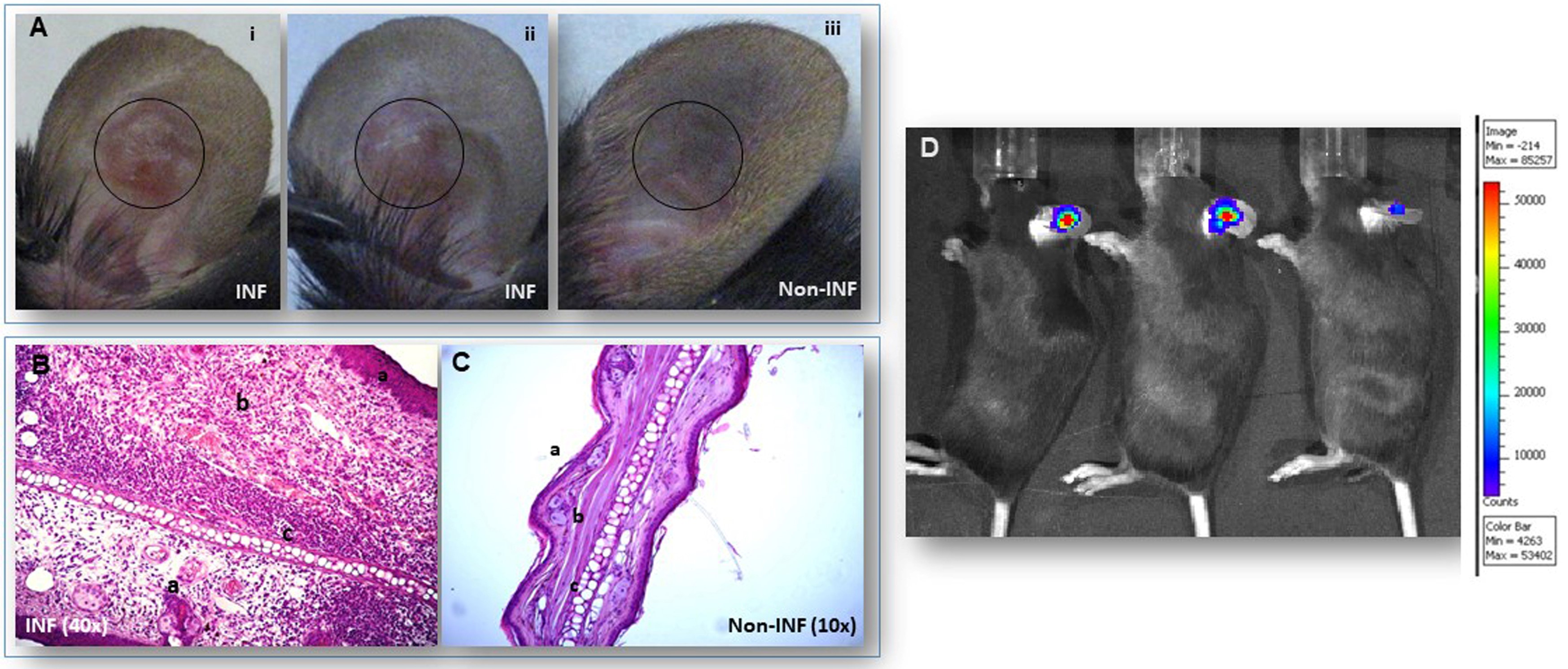
Figure 2. Sites of inoculation with Leishmania amazonensis parasites of the ear dermis of C57BL/6J mice. A. Ear lesions are shown on the infected (INF) ear pinna, (i and ii). Control non-infected ear pinna, (iii). Circles show the ear areas isolated for total RNA preparation. B-C. Hematoxylin and Eosin (H&E) staining of histology sections of ear pinna from C57BL/6J mice. B. Infected with Leishmania amazonensis (INF): a: epidermis with acanthosis; b: inflamed dermis; c: cartilage. C. Non infected (Non INF) ears: a: epidermis; b: dermis; c: cartilage. D. Bioluminescence imaging of luciferase expressing Leishmania amazonensis parasites in infected ear dermis of mice. The color bar indicates the density of the luciferase emission delineating the number of parasites present in the infected area of the ear pinna. - Wash rapidly each tissue fragment with PBS and place it in separate homogenizing tubes.
- After shaving the hair, cut through the skin at the area on the side of each ear and isolate the ear draining lymph nodes from both sides (left side infected and right side to be used as control). Clean the lymph nodes from the surrounding tissue using the glass dissection dish (Figure 1vi).
- Place the lymph node in a new homogenizing tube.
- The dissection procedure of the tissues is described on Figure 3.
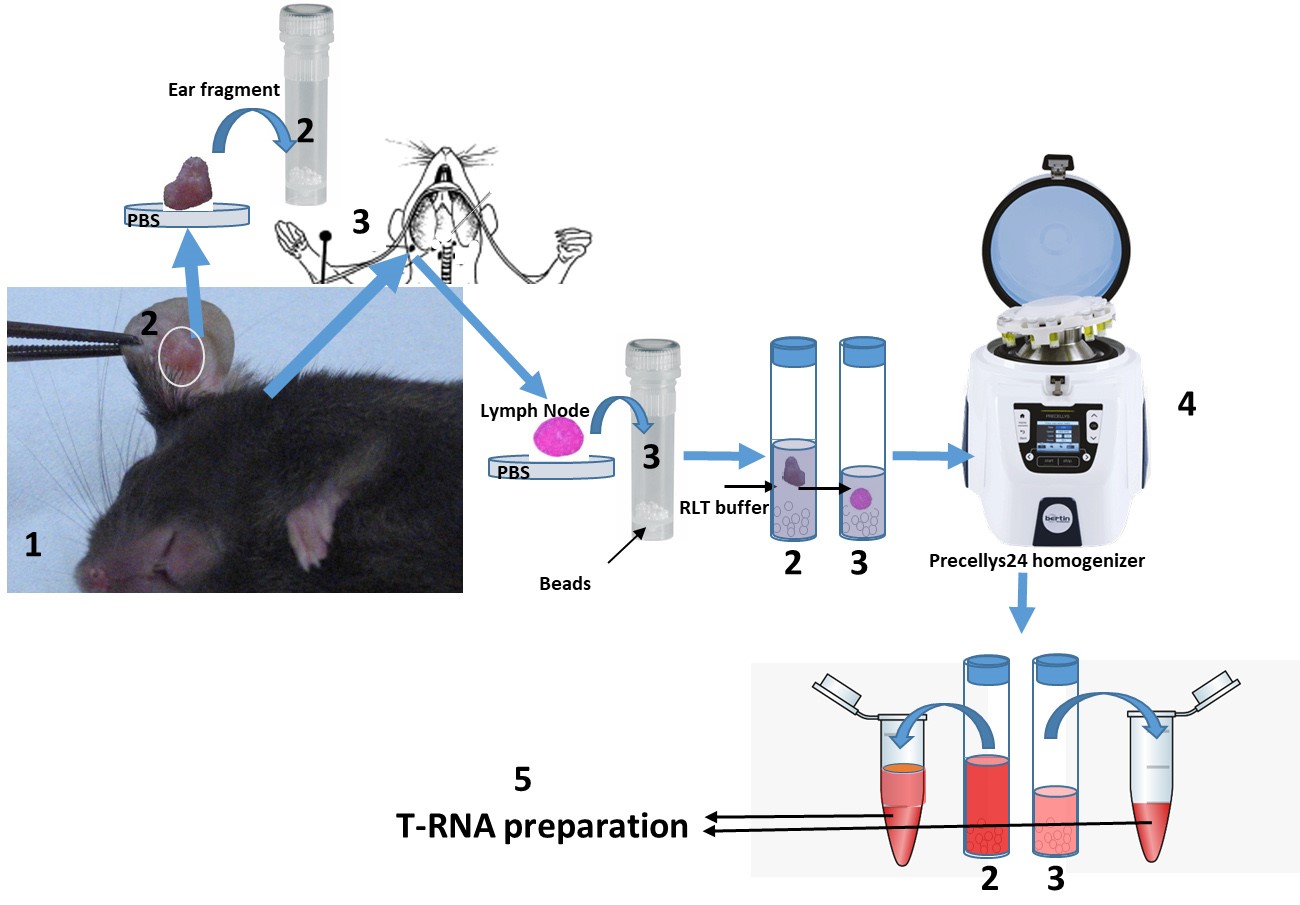
Figure 3. Step by step procedure for isolation of tissues from mice. Mice, after various times of infection with Leishmania amazonensis parasites at the ear pinna, were sacrificed (1). The infected area of the ear tissue is excised and after a brief wash with PBS in a Petri dish plate, the ear fragment is placed in a Precellys 24 tube containing the beads and the RLT buffer (2). The auricular, ear draining lymph node is surgically removed and placed in a new tube containing the beads and the RLT buffer (3). The lymph node is dense and sinks into the tube liquid. The tissues are homogenized in the Precellys 24 homogenizer (4) and then the homogenate is placed into two new Eppendorf 2 ml tubes for total RNA preparation (5). All procedures involving animals have been approved by the Institutional Committees on animal Welfare under strict accordance with the European guidelines.
- RNA isolation from tissues
Note: The description of our procedure for Total RNA preparation follows the protocol of RNA isolation with the RNeasy Plus Mini Kit as supplied by Qiagen with minor modifications. Our starting material was always one ear fragment or one lymph node per preparation. These tissues yielded approximately 10-50 μg of total RNA, depending on the tissue. 1 μg of total RNA yields 1 μg of cDNA. From these cDNA preparations we used 10 ng per Real time PCR reaction.- Prepare the tubes that will contain the tissues by adding 1 ml of RLT + β-ME buffer for the ear fragment and 400 μl of the same buffer for the lymph nodes.
- Add the tissues into the tubes and proceed in tissue fragmentation in the Precellys 24 apparatus. Tissues were homogenized at 600 x g for 60 s, in accordance with the manufacturer’s instructions.
- Take out the tubes from the Precellys 24 and centrifuge at 15,000 x g (13,000 rpm), at room temperature for 2 min.
- Place 250 μl from the ear homogenate and the entire lymph node homogenate (400 μl) on separate for each tissue DNA retention columns (gDNA Eliminator Spin Column, Qiagen). Centrifuge at 9,000 x g (10,000 rpm) for 90 s.
- Keep the effluent and discard the DNA separation tubes.
- Add 175 μl of ethanol 70% into the tubes containing the ear homogenate and 280 μl into the tubes containing the lymph nodes. Use a P1000 Pipetman to well homogenize the samples.
- Transfer the samples on the RNA retention column (RNeasy spin column, Qiagen) and centrifuge at 9000 x g (10,000 rpm) for 90 s.
- Eliminate the tube containing the effluent and place the column on a new collection tube (1.5 ml). If DNase treatment is not used continue as in Step C10.
- If DNase digestion is performed (with Qiagen kit for RNA preparation), samples (Step C4) are placed directly on the RNA retention columns and additional Steps D1 to 4 are followed.
- Add on the column 700 μl of RW1 RNA binding buffer (Qiagen) and centrifuge at 9,000 x g (10,000 rpm) for 60 s. Eliminate the effluent but use for the next step the same collector tube.
- Add on the column 500 μl of RPE + ethanol buffer (ensure that it is at room temperature).
- Centrifuge the samples at 9,000 x g (10,000 rpm) for 60 s. Eliminate the effluent and reuse the collector tube.
- Repeat Steps C10 and C11 once more and eliminate the collector tube.
- Place the column onto a new collector tube and centrifuge at 9,000 x g (10,000 rpm) for 2 min to dry out completely the column.
- Repeat the centrifugation step once more at 15,000 x g (13,000 rpm) for 2 min.
- Replace collector tube with a 1.5 ml Eppendorf new tube and proceed to the RNA elution step by adding 30 μl of RNase/DNase-free sterile water on the center of the column for each sample.
- Centrifuge at 15,000 x g (10,000 rpm) for 60 s.
- Dilute 1.5 μl of each RNA sample in 20 μl of sterile water in a new tube.
- 1 μl of each diluted sample is used for RNA quantification and quality evaluation, by measurements of the Optical Density (OD) in a NanoDrop ND-1000 micro spectrophotometer (Thermo Fisher Scientific) (Figure 4).
- Keep all RNAs at -80 °C until use.
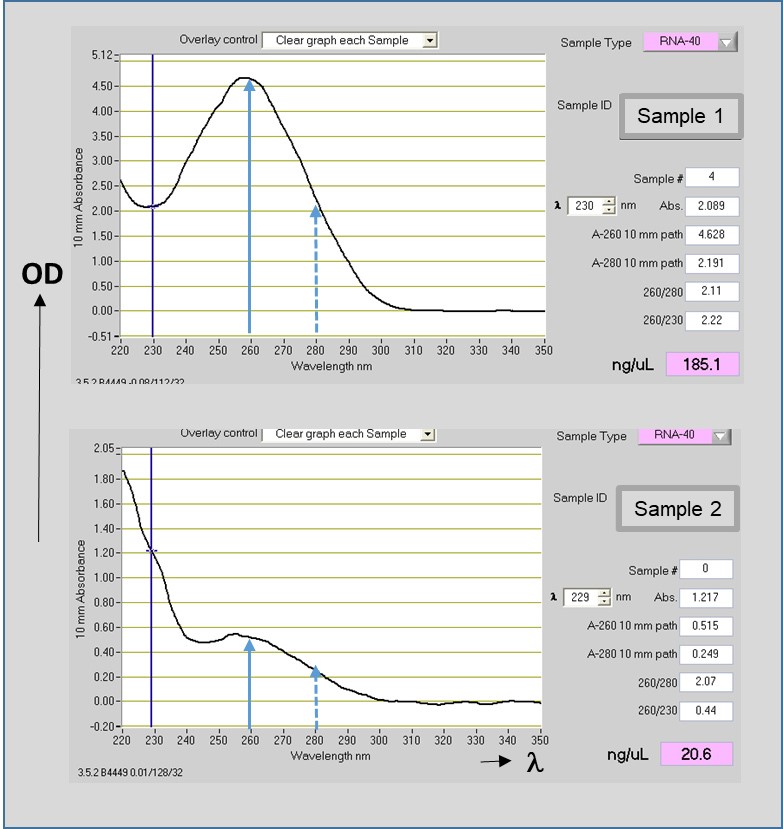
Figure 4. Total RNA concentration and quality evaluation. Representative images of Optical Densities (OD) of RNAs measured by the NanoDrop ND-1000 micro spectrophotometer. Non-degraded RNA preparation (Sample 1, upper panel) and poor quality (degraded) RNA preparation (Sample 2, lower panel) are shown. The wavelengths (λ) of 260 nm for nucleic acid (solid blue lines) and 280 nm for protein (dashed blue lines) are indicated. The ratios of absorbance A260/280 (~2.0) and 260/230 (≥ 2.0) indicate the purity of the RNA (ex. absence of organic compounds and protein contaminants respectively). RNAs concentrations are given in ng/μl, taking in consideration 1 OD A260 unit of ssRNA = 40 μg/ml. Low A260/280 concentrations (lower panel) indicate residual contamination of reagents in the preparation and absence of a clear pick of absorbance at A260 indicates a poor quality of RNA (see Note 2).
- DNase treatment of the samples (Qiagen kit cat. N° 79254 )
- Add to the RNeasy spin column 350 μl Buffer RW1, close the lid gently and centrifuge at 8,000 x g, for 15 s to wash the spin column membrane. Discard the flow-through.
- Prepare 70 μl buffer RDD (supplied with the RNase-free DNase Set) in a tube and add 10 μl DNase I stock solution. Mix gently as DNase is sensitive to physical denaturation. Do not vortex.
- Add 80 μl of the DNase I incubation mix directly into the membrane of the RNeasy spin column and leave on the benchtop for 15 min (20-30 °C).
- Add 350 μl buffer RW1 to the RNeasy spin column, close the lid gently and centrifuge at 8,000 x g for 15 s. Discard the flow-through and continue with the first RPE buffer wash (Step C11).
Data analysis
According to our protocol we have isolated RNAs from animals infected and non-infected with the Leishmania amazonensis parasites. Our method allowed for the preparation of good quantity, non-degraded total RNA sufficient for RT-PCR experiments of several genes (Giraud et al., 2019b).
The data obtained from these preparations are described on Figure 6d and Figure S8 in the original published article (Giraud et al., 2019b).
As mentioned above on section C (RNA isolation from tissues), we have taken in consideration that 1 μg of RNA yields 1 μg of cDNA. For the cDNA preparation we used random hexamers (Roche Diagnostics) and MMLTV-RT reverse transcriptase (Invitrogen, Life technologies). 10 ng of cDNA was used per RT-PCR reaction (with QuantiTect SYBR Green Kit, Qiagen) as described in the Methods section of the original article (Giraud et al., 2019b). Raw Crossing Point (Cp) values were used as input for the qBase software program (https://www.qbaseplus.com/). Calculation of relative quantification values are corrected by appropriate normalization using the qPCR quantification values of the house keeping genes. This normalizations were performed for each experimental tissue. qBase software can calculate the normalized and rescaled quantities in a logarithmic scale. This is an automated process supplied by the integrated algorithm of the qBase program as described by Hellemans et al. (2007).
In our experiments described on Figure 6 and Figure S8 of the manuscript (Giraud et al., 2019b), the relative expressions (quantities) of the opn gene and the genes encoding for the inflammasome multicomplex were calculated on log-transformed ratios by comparison of the fold differences in expression of the Leishmania infected tissues in the osteopontin mutant mice (C57BL/6-/-) versus the wild type osteopontin positive (C57BL/6+/+) mice.
Notes
- The following formula:
g Force (RCF) = (rpm)2 x 1.118 x 10-5 x r is used for the conversion of rpm (revolutions per minute) to g Force or Relative Centrifugal Force (RCF). RCF or g is the acceleration applied to a sample during centrifugation (see: details).
This conversion is dependent on the radius r of the rotor. - RNA concentration and quality are important for the optimum performance of reactions using this nucleic acid. The average concentration of RNA and its purity is measured by spectrophotometric quantification. In the NanoDrop spectrophotometer is included a preset program for RNA concentrations measurements, that automatically reads the OD at 230, 260 and 280 nm and calculates the above discussed ratios (Figure 4). In addition only 1 to 2 μl of sample are required. A Blank is used with the same solution in which the RNA is diluted prior to RNA measurements with the spectrophotometer. RNA has a maximum absorbance (Optical Density) at 260 nm and the absorbance unit used as conversion factor for RNA concentration is A260 ssRNA = 40 μg/ml. For the evaluation of RNA purity OD260 and OD230 measurements also should be taken in consideration. A260/280 ratio of ~2 is accepted for pure RNA. Low 260/280 ratios usually indicates the presence of protein contaminants. A high pick at A230 absorbance indicates the presence of other than proteins contaminants such as residual phenol, guanidine or other reagents. A260/230 ratio values for pure RNA are often higher that the respective A260/280 values. Commonly 260/230 ratios are in the range of 2.0-2.2. Lower than 1.5 ratios indicate the presence of contaminants. RNA integrity can be checked by a run of the RNA on a 1% agarose gel and assess for the integrity of the ribosomal RNA bands. For non-degraded RNA the intensity of the 28S band (upper rRNA band) in eukaryotic cells should be twice the intensity of the lower rRNA band (18S). Both bands should be clear and tight, otherwise the presence of a smear indicates RNA degradation. The presence of high molecular weight bands shows contamination by the DNA. Another possible method for checking RNA integrity is by using the Agilent Bioanalyzer (See Application Notes). This analyzer measures the sizes of the rRNA bands and determines by comparison a RNA integrity number (RIN).
Recipes
- Ethanol 70%
Add 30 ml of sterile (RNase/DNase-free) water in 70 ml of Ethanol (CH3CH2OH) (Merck) in a 200 ml glass bottle. Mix - RLT + β-ME buffer
Note: RNeasy Lysis Buffer (Qiagen RLT or supplied with the Qiagen RNeasy Kits) contains guanidine isothiocyanate (GITC). In order to reduce RNases activity, β-mercaptoethanol (β-ME) is added to the RLT buffer before use.- Add 20 μl of β-ME per 1 ml of RLT buffer. This β-ME containing RLT buffer is stable for up to one month
Warning: β-ME is a toxic chemical and should be dispensed in a fume hood and appropriate protective clothing should be worn. - Alternatively, β-ME may be replaced by 20 μl of Dithiothreitol 2M (DTT) per ml of RLT buffer
- RLT complemented buffer with β-ME or DTT, as described, is added in the homogenization tubes that will contain the ear pinna (1 ml/tube) and the lymph node (400 μl/tube) (Mommaerts et al., 2015)
- Add 20 μl of β-ME per 1 ml of RLT buffer. This β-ME containing RLT buffer is stable for up to one month
Acknowledgments
This work was supported by institutional funding from the Institut Pasteur. We are grateful to the members of the Immunophysiology and Parasitism Unit for their recommendations for these experimental procedures and their participation to the original work described in the published paper from where this protocol is derived (Giraud et al., 2019b).
Competing interests
The authors declare that there is none financial or non-financial competing interests associated with this work.
Ethics
All the animals used in our experiments were kept under SPF conditions at the animal facility of the Institut Pasteur, raised under a 12/12 hour dark-light cycle with ad libitum access to food and water. All experimental protocols were approved by the Institutional Committees on animal Welfare under strict accordance with the European guidelines (Directive 2010/63/EU) for animal care.
References
- Bates, P. A. (2007). Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol 37(10): 1097-1106.
- Christensen, S. M., Belew, A. T., El-Sayed, N. M., Tafuri, W. L., Silveira, F. T. and Mosser, D. M. (2019). Host and parasite responses in human diffuse cutaneous leishmaniasis caused by L. amazonensis. PLoS Negl Trop Dis 13(3): e0007152.
- Cortes, D. F., Carneiro, M. B., Santos, L. M., Souza, T. C., Maioli, T. U., Duz, A. L., Ramos-Jorge, M. L., Afonso, L. C., Carneiro, C. and Vieira, L. Q. (2010). Low and high-dose intradermal infection with Leishmania major and Leishmania amazonensis in C57BL/6 mice. Mem Inst Oswaldo Cruz 105(6): 736-745.
- de Oliveira Cardoso, F., de Souza Cda, S., Mendes, V. G., Abreu-Silva, A. L., Goncalves da Costa, S. C. and Calabrese, K. S. (2010). Immunopathological studies of Leishmania amazonensis infection in resistant and in susceptible mice. J Infect Dis 201(12): 1933-1940.
- Giraud, E., Martin, O., Yakob, L. and Rogers, M. (2019a). Quantifying Leishmania metacyclic promastigotes from individual sandfly bites reveals the efficiency of vector transmission. Commun Biol 2: 84.
- Giraud, E., Rouault, E., Fiette, L., Colle, J. H., Smirlis, D. and Melanitou, E. (2019b). Osteopontin in the host response to Leishmania amazonensis. BMC Microbiol 19(1): 32.
- Hellemans, J., Mortier, G., De Paepe, A., Speleman, F. and Vandesompele, J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8(2): R19.
- Liu, D. and Uzonna, J. E. (2012). The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front Cell Infect Microbiol 2: 83.
- Mansueto, P., Vitale, G., Di Lorenzo, G., Rini, G. B., Mansueto, S. and Cillari, E. (2007). Immunopathology of leishmaniasis: an update. Int J Immunopathol Pharmacol 20(3): 435-445.
- McMahon-Pratt, D. and Alexander, J. (2004). Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev 201: 206-224.
- Mommaerts, K., Sanchez, I., Betsou, F. and Mathieson, W. (2015). Replacing beta-mercaptoethanol in RNA extractions. Anal Biochem 479: 51-53.
- Reid, G. A. (1991). A vade-mecum for molecular biologists. Trends in Biotechnology 9(1): 213-214.
- Schroeder, A., Mueller, O., Stocker, S., Salowsky, R., Leiber, M., Gassmann, M., Lightfoot, S., Menzel, W., Granzow, M. and Ragg, T. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7: 3.
- Schuster, S., Hartley, M. A., Tacchini-Cottier, F. and Ronet, C. (2014). A scoring method to standardize lesion monitoring following intra-dermal infection of Leishmania parasites in the murine ear. Front Cell Infect Microbiol 4: 67.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Giraud, E. and Melanitou, E. (2020). RNA Extraction from Ears and Draining Lymph Nodes of Mice Infected with Leishmania amazonensis. Bio-protocol 10(11): e3633. DOI: 10.21769/BioProtoc.3633.
Category
Immunology > Animal model > Mouse
Microbiology > Microbe-host interactions > In vivo model > Mammal
Molecular Biology > RNA > RNA extraction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.