- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Evaluation of the Efficiency of Genome Editing Tools by a Frameshift Fluorescence Protein Reporter

(*contributed equally to this work) Published: Vol 10, Iss 10, May 20, 2020 DOI: 10.21769/BioProtoc.3622 Views: 5299
Reviewed by: Alba BlesaShyam SolankiChangyi Zhang
Original research article
The authors used this protocol in:

Jul 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
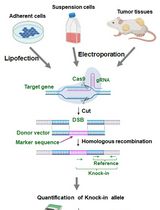
In the last decade, genome editing has been the center of attention as a novel tool for mechanistic investigations and for potential clinical applications. Various genome editing tools like meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector-based nucleases (TALEN), and the clustered regularly interspaced short palindromic repeats (CRISPR)-associated genes (Cas), have been developed in recent years. For the optimal use as well as continued developments of these genome editing tools, the evaluation of their efficiencies and accuracies is vital. Here, we present a protocol for a reporter based on frameshift fluorescence protein which we recently developed to evaluate the efficiency and accuracy of genome editing tools. In this method, a ~20 bp target sequence containing frame-shifting is inserted after the start codon of a cerulean fluorescence protein (CFP) to inactivate its fluorescence, and only a new insertion/deletion event in the target sequence will reactivate the CFP fluorescence. To increase the traceability, an internal ribosome entry site and a red fluorescence protein, mCherryFP, are placed downstream of the reporter. The percentage of CFP-positive cells resulted from in/del mediated fluorescence restoration can be quantified by fluorescence measuring devices as the readout for genome editing frequency. As a demonstration, we present the usage for CRISPR-Cas9 technique here with flow cytometer as the readout for fluorescence changes.
Keywords: Insertion-deletionBackground
Genome editing tools are very important for the investigations of biological mechanisms and prevention and/or treatment of genetic diseases (Maeder and Gersbach, 2016). In the last couple of decades, several genome editing tools have been introduced, which include the meganucleases (Epinat et al., 2003), zinc finger nucleases (ZFNs) (Kim et al., 1996), transcription activator-like effector-based nucleases (TALEN) (Christian et al., 2010), and the clustered regularly interspaced short palindromic repeats (CRISPR)-associated genes (Cas) (Jinek et al., 2012; Cong et al., 2013; Sander and Joung, 2014). In general, these tools create DNA double stranded breaks (DSB) to trigger genome editing in vivo (Maeder and Gersbach, 2016). The evaluation of the efficiencies and specificities of genome editing tool is essential for their applications and further developments. In our recent published study, we described a reporter that can generate quantitative readout for genome editing efficiency (Kumar et al., 2019). In this system, a ~20 bp target sequence is placed in a multiple cloning site (MCS) which is right after the start codon of Cerulean fluorescence protein (CFP) to generate a frame-shift of the open reading frame (ORF). This frameshifted-CFP (FsCFP) can be used as a reporter of genome editing because only when there is a successful DNA-double strand break (DSB) event on the target sequence followed with a non-homologous end joining (NHEJ) to generate an in/del event to shift the reading frame to a correct order (by a chance of up to 1/3), the CFP fluorescence will be reactivated as a positive readout. To facilitate the quantification, an internal ribosome entry site (IRES) and a red fluorescence protein, mCherryFP, is placed after the reporter. In principle, this reporter can be applied to any genome editing system as long as a DSB and NHEJ are expected from the editing. This approach can effectively detect low-efficiency editing in a population of cells with very low false negative or false positive. Furthermore, in this method, the positive cells can be conveniently identified and enriched for the examination or validation of the in/del event in the genome. Also, this method can be easily adapted for screening to optimize the genome-editing enzyme or the other components (such as guide-RNA) in the positive cells. Here, we used the CRISPR-Cas9 technique as a demonstration and the flow cytometry as the readout of the fluorescence events.
In this protocol, the target sequence is inserted between restriction sites of NotI and XhoI before CFP reporter together with sequence for the optimal recognition of Cas9 and a premature STOP codon to create a frame shift. The reporter region is then integrated in the nuclear genome of the target cell by the assistance of lentivirus. The target cells expressing the red fluorescence protein are then isolated by fluorescence-activated cell sorting (FACS), before vectors containing the Cas9 and gRNA are introduced into these cells. After incubation, the ratio of the CFP over mCherryFP was measured in flow cytometry to provide quantitative measurement for the efficiency of the genome editing.
Materials and Reagents
Materials
- Cell Culture dish 150 x 25 mm (Asi, catalog number: TD0150 )
- Cell Culture dish 90 x 20 mm (Asi, catalog number: TD0100 )
- 5 ml serological pipet (Asi, catalog number: SP205 )
- 10 ml serological pipet (Asi, catalog number: SP210 )
- Cell Culture Flask 75 cm2, filter cap (Asi, catalog number: TV0075 )
- Cell Culture Flask 25 cm2, filter cap (Asi, catalog number: TV0025 )
- Syringe filter, PES 25 mm, 0.45 μm (Asi, catalog number: TE45-5 )
- 10 ml syringes (BD, catalog number: 309604 )
- 15 ml and 50 ml conical tubes (Denville, catalog number: C1062-P )
- 18 G x 1 ½ needles (BD, catalog number: 305196 )
- Bottle top filtration–2 μm PES (VWR, catalog number: 97066-202 )
- 15 ml centrifuge tubes (CellPro, catalog number: CN5600 )
- 50 ml centrifuge tubes (CellPro, catalog number: CN5603 )
- Filter Pipet Tips 1,250 μl (TruPoint, catalog number: FT1250 )
- Filter Pipet Tips 200 μl (TruPoint, catalog number: FT1200 )
- Filter Pipet Tips 20 μl (TruPoint, catalog number: FT1020 )
- Filter Pipet Tips 10 μl (TruPoint, catalog number: FT1010 )
- Falcon FACS tubes with 35 μMcell strainer (BD, catalog number: 352235 )
- Falcon FACS collection tubes (BD, catalog number: 352063 )
- Ice buckets
- T4 DNA ligase (New England Biolabs, catalog number: M0202S )
- Gel extraction kit (EZ, catalog number: M1002-50 )
- One ShotTM TOP10 Chemically Competent E. coli (Invitrogen, catalog number: C404003 )
- ZymoPURETM II Plasmid Midiprep Kit (Genesee Scientific, catalog number: 11-550B )
- QIAprep Spin Miniprep Kit (50) (QIAGEN, catalog number: 27104 )
Cells and Plasmids
- Human embryonic kidney cells (HEK 293T, clone 17) (ATCC, catalog number: CRL-11268 )
- Plasmid pQC-XIG (Addgene, catalog number: w497-1 ), deposited by Dr. Eric Campeau, who is currently at Zenith Epigenetics Ltd, Calgary, Canada
- Plasmid pCMV-Delta R8.2 (Addgene, catalog number: 12263 ), deposited by Dr. Didier Trono at EPFL
- Plasmid pCMV-VSV-G (Addgene, catalog number: 8454 ), deposited by Dr. Robert Weinberg at MIT
- gRNAs custom ordered from Vector builder (https://en.vectorbuilder.com/)
Reagents
- High Glucose DMEM (1x) (Life Technologies, catalog number: 11995-065 )
- F-10 media (1x) (Life Technologies, catalog number: 11550-043 )
- 0.25% Trypsin-EDTA (1x) (Life technologies, catalog number: 25200-072 )
- 10% FBS (Hyclone, catalog number: SH30910.03 )
- Polybrene Transfection Reagent (Millipore, catalog number: TR-1003-G )
- Antibiotic-Antimycotic (100x) (Life Technologies, catalog number: 15240062 )
- Polyethylenimine (PEI) (Sigma-Aldrich, catalog number: 408727 )
- T4-ligase Buffer (New England Biolabs, containing 50 mM Tris-HCl, 10 mM MgCl2, 1 mM ATP, 10 mM DTT, pH 7.5)
- DMEM+F10 culture media (45% DMEM + 45% F-10 + 10% FBS, with 1x Antibiotic-Antimycotic)
- Nucleotide oligos/primers:
Table 1. List of primers used in this protocol
Equipment
- Centrifuge 5424 R (Eppendorf, catalog number: 540400138 )
- Labnet Accublock Digital Dry Bath (Labnet, catalog number: 19-41620 )
- Gene Mote Vortex Mixer (Bioexpress, catalog number: S-3200-1 )
- CO2 incubator MCO-19AIC (UV) (Panasonic, catalog number: 13010002 )
- Centrifuge 5702 (Eppendorf, catalog number: 0 22626205 )
- Fluorescent microscope
- Aria-IIU flow cytometer (BD)
Software
- FCS Express 6 (Denovo software–https://denovosoftware.com/)
Procedure
- Generation of reporter and gRNA constructs
- Construction of the Frameshift(Fs) CFP-mCherryFP reporter
- The nucleotide sequence consisting of CFP, IRES and mCherryFP and flanked with NotI and EcoRV restriction sites was synthesized by using the service of Genscript, NJ. See Figure 1 for the illustration of the vector map and the nucleotide sequence.
Note: The CFP was chosen as the frameshift reporter due to its lack of internal starting codons (ATG) near the 5′-end, which is essential to prevent the generation of smaller proteins that are potentially still fluorescent (such as in the case of mCherryFP). Although GFP also lacks internal ATG near the 5′-end, it is often used as a marker in many vectors containing the genome editing machineries. In addition to fluorescence proteins, proteins that don’t have internal ATG near the 5’-end and can be specifically detected or selected may also be designed as frameshifting reporters. These may include luciferases, β-galactosidase, or aminoglycoside 3′-phosphotransferase.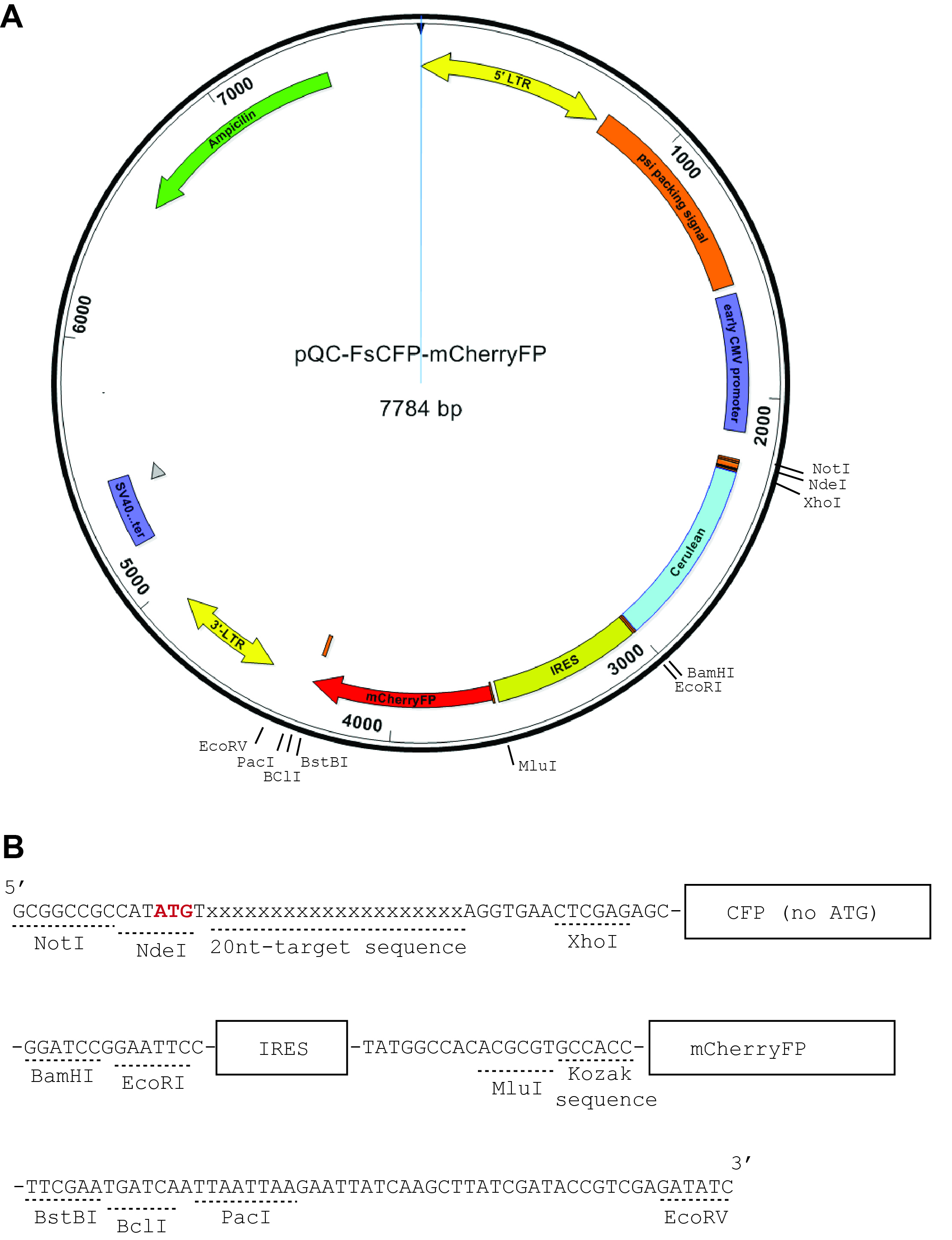
Figure 1. Construction of the reporter plasmids. In (A), there is a schematic representation of the vector map of pQC-FSCFP-mCherryFP, which is modified from pQC-XIG vector (from Addgene) with a synthesized nucleotide sequence as shown in (B). The synthesized sequence (5′-strand shown) is to be cloned into the pQC-XIG vector by restriction sites NotI and EcoRV. This cloning will replace the original IRES and GFP regions. It will also introduce the CFP coding sequence and additional cloning sites. In the demonstrated example here it also included the 20 nt target sequence. - The above-described nucleotide sequence is cloned into a template vector, pQC-XIG, using the restriction sites NotI and EcoRV (5′ end and 3′ end, respectively).
Note: This replaces the original IRES and GFP sequences in the pQC-XIG vector for the sake of introducing multiple cloning sites flanking the CFP and mCherryFP sequences for future cloning purposes. - Digest the FsCFP-mCherryFP reporter plasmid generated from above with Not1 and Xho1 (5′ end and 3′ end respectively). Gel purify the digested vector using Gel extraction kit (EZ) and store the product at -20 °C.
Note: Do not perform phosphatase treatment during or after the digestion, unless the custom ordered oligos were 3′-phosphorylated.
- The nucleotide sequence consisting of CFP, IRES and mCherryFP and flanked with NotI and EcoRV restriction sites was synthesized by using the service of Genscript, NJ. See Figure 1 for the illustration of the vector map and the nucleotide sequence.
- Introducing the target sequence in FsCFP-mCherryFP reporter (see also Table 1 for the nucleotide sequences of primer/oligos)
- Synthesizing the target sequence–For example, the target sequence for VEGF-A gene in human is 5′-GGGTGGGGGGAGTTTGCTCC-3′. The targeting sequence is placed after the START codon, followed by a protospacer adjacent motif (PAM) site and a premature STOP codon. If necessary, a few (1-5) extra nucleotides can be added to ensure that the CFP is out of frame. In the shown example, the inclusion of a premature STOP codon is to prevent the production of a long-length protein resulting from the frameshifted coding sequence of CFP, which may be harmful to the cell (Figure 2). The nucleotide oligos (both 5’ and 3’ strand) containing the above (target) sequence as well as overhanging nucleotides which would result from NotI (5′) and XhoI (3′) digestion were then synthesized by using the service of Genscript, NJ:
Top-strand target oligo: 5′-ggccgcCATATGTGGGTGGGGGGAGTTTGCTCCAGGTGAAc-3′
Bottom-strand target oligo: 5′-tcgagTTCACCTGGAGCAAACTCCCCCCACCCACATATGGcg-3′
Notes: The two oligos, if annealed, represent the double-digestion product of NotI (5′) and XhoI (3′). (Figure 3)
Figure 2. Schematic representation of the nucleotide sequence flanking the target insert region. The position of the 20-nucleotide (nt) gRNA-matching site, the 3nt-protospacer adjacent motif (PAM) sequence for Cas9 binding is indicated along with the START and STOP codon. The two extra nucleotides in this particular example are included to ensure the out-of-frame CFP. The premature STOP codon is included to prevent the translation of a long-length product from the START codon on the shifted frame of the CFP coding sequence.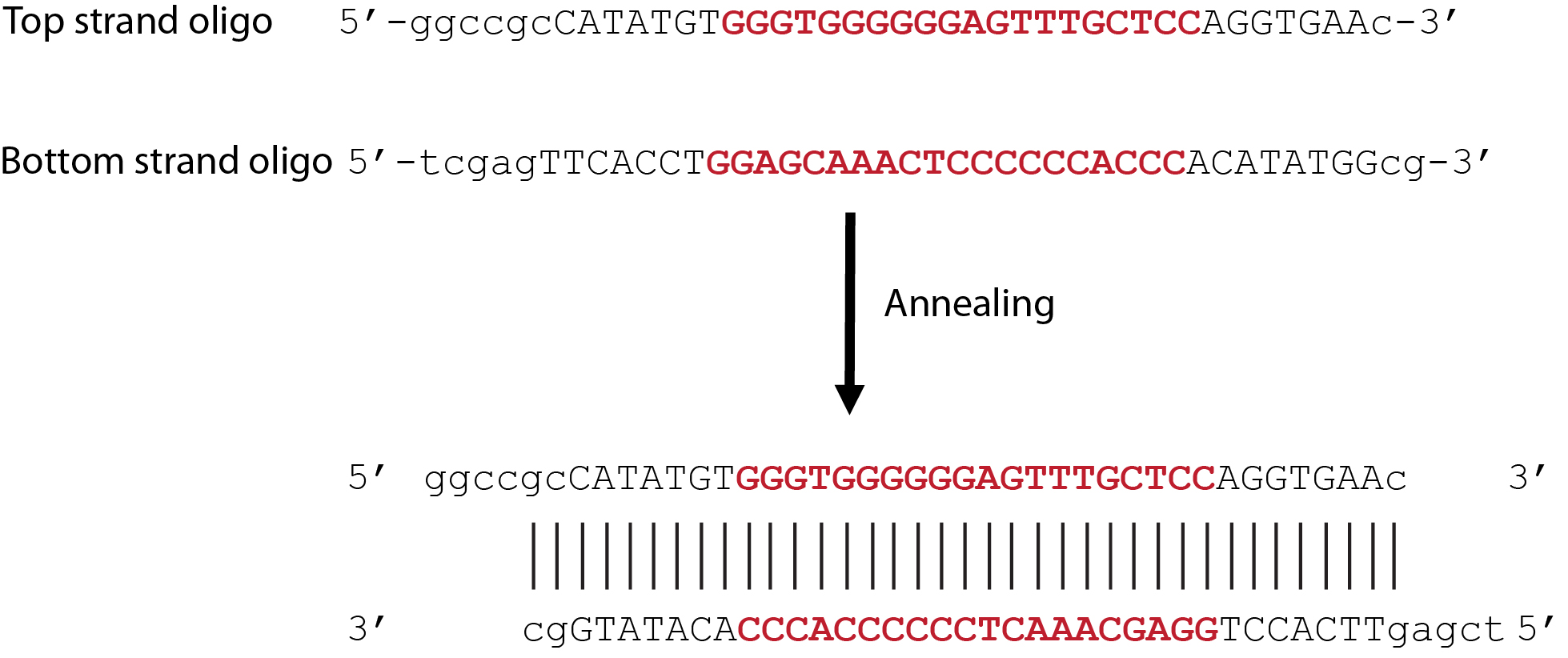
Figure 3. Annealing of 5′-strand and 3′-strand oligos. The oligo sequences containing an example with 20-nt target sequence (in bold red font) derived from human VEGF gene, the START codon, PAM site, and premature STOP codon, as well as overhanging nucleotides which would result from NotI (5′) and XhoI (3′) digestion (shown in lower case). After annealing, the 5′ and 3′ strands together represent the digested product of Not1 and Xho1. - Annealing–In an Eppendorf tube, mix the two oligos from above (Step A2a) in 1:1 ratio to a final concentration of 20 μM (each) in 1x T4 ligase buffer. Place the tube in the boiling water (100 °C) for 10-20 min and allow the water to cool down at room temperature for 8-12 h for the annealing process. The product can then be used freshly or stored at -20 °C.
- Ligation–Mix the annealed oligos with the digested vector from Step A1c. The oligo would be 3-10 times (5 time is recommended) in excess compared to the vector in molecules. Ligation is carried out by the addition of T4 DNA ligase (NEB) in the presence of 1x T4 ligase buffer (NEB). For each 20 μl reaction which typically contains 1 μg of digested vector and 0.05 μg of annealed oligos, 1 μl of ligase was added. The ligation reaction is performed at 16 °C for 2 h followed by 8 °C overnight
- Bacterial transformation–The ligated product from above is introduced into Top10 cells (Invitrogen), according to the manufacturer’s instructions. In general, 2~3 μl of the ligation product was used to transform 50 μl of Bacteria. The transformed bacteria is plated on to LB + Ampicillin (50 μg/ ml) plates for incubation at 37 °C overnight. Grown colonies are picked from the plate to grow in 5 ml liquid LB + Ampicillin (50 μg/ ml) overnight. Plasmid is then isolated using Qiagen QiaPrep Spin Miniprep kit and then submitted for Sanger sequencing with primers specifically targeting the CMV promoter, the CFP region (without cross-reacting with the mCherryFP region), and the IRES region of the plasmid (Figure 1A).
Primers used in this protocol are listed in Table 1.
Forward primer targeting CMV promoter:
5′-AGAGCTCGTTTAGTGAACCGTC-3′
Reverse primer targeting IRES:
5′-GACGGCAATATGGTGGAAAATAACATATAGACAAACGCACACCGG-3′
Forward primer targeting the border between the CMV promoter and the cloning sites:
5′-GAGCTCGTTTAGTGAACCGTCAGATCGCCTGGAGACGCCATCCACG-3′
Reverse primer specifically targeting CFP:
5′-TAGTTGCCGTCGTCCTTGAAGAAGATGGTGCGCTCCTGGACGTAGCC-3′ - Colonies carrying the plasmid containing the expected insertion sequence in the FsCFP-mCherryFP reporter is then grown in 100 ml liquid LB + Ampicillin (50 μg/ml) overnight. Plasmid is then isolated with the Midiprep Kit (ZymoPURETM II Plasmid Midiprep Kit). This product can be stored at 20 °C.
- Synthesizing the target sequence–For example, the target sequence for VEGF-A gene in human is 5′-GGGTGGGGGGAGTTTGCTCC-3′. The targeting sequence is placed after the START codon, followed by a protospacer adjacent motif (PAM) site and a premature STOP codon. If necessary, a few (1-5) extra nucleotides can be added to ensure that the CFP is out of frame. In the shown example, the inclusion of a premature STOP codon is to prevent the production of a long-length protein resulting from the frameshifted coding sequence of CFP, which may be harmful to the cell (Figure 2). The nucleotide oligos (both 5’ and 3’ strand) containing the above (target) sequence as well as overhanging nucleotides which would result from NotI (5′) and XhoI (3′) digestion were then synthesized by using the service of Genscript, NJ:
- Guide RNA (gRNA) construction
The plasmids for gRNA and Cas9 can be designed and purchased from Vector Builder INC (https://en.vectorbuilder.com/), with GFP as a selection marker. For example, the gRNA sequence for VEGF will be –GGGTGGGGGGAGTTTGCTCC. As a control, scramble gRNA should also be ordered.
- Construction of the Frameshift(Fs) CFP-mCherryFP reporter
- Creation of cell lines with FsCFP-mCherryFP reporter and gRNA
Preparation of cell lines with reporter and gRNA is a 2-step sequential process.
Step B1: Cell line of interest + FsCFP-mCherryFP (reporter plasmid) → sort for mCherry positive cells
Step B2: Cell line with FsCFP-mCherryFP + (target) gRNA-GFP → sort for mCherry and GFP double positive cells.
Step B1: Generation of cell line with FsCFP-mCherryFP reporter- Production of lentiviral particles in HEK 293T cell line
To begin with, seed the HEK 293T cells so that they will reach ~75% confluency in a T-25 culture flask, which can be estimated by microscopic observations. Change the DMEM+ F10 culture just before the transfection steps below.- In a 1.5 ml Eppendorf tube (tube 1), add 1 μg of VSV-G, 6 μg of Delta R8.2, and 6.5 μg of reporter plasmid containing the targeting sequence (from Step A2e) to 0.5 ml of DMEM + F10 media. Vortex for 20-30 s for mixing and spin down briefly (less than 10 s) in mini-spinner or in a common desktop centrifuge at 2,000 x g.
- Add 0.5 ml of DMEM to another Eppendorf tube (tube 2), then add 30 μl of activated Polyethylenimine reagent (Sigma) or other types of transfection reagents such as Lipofectamine (Thermo-Fisher) or JetPrime (Polyplus-Transfection). Vortex immediately for 30 s. Spin down the components briefly as above. Add the contents from tube 1 to tube 2. Vortex the tube immediately for 20-30 s and then spin down briefly again. Incubate at room temperature (RT) for 20-30 min.
- After the incubation, add the transfection mix drop by drop to the flask containing the HEK 293T cells and the fresh media. Mix gently and place the flask at 37 °C, 5% CO2 incubator for the production of viral particles in the next three days. The majority of these cells (> 50%) are expected to express mCherryFP (excitation 587 nm, emission 610 nm) after 24 h, which can be visually verified by the presence of red fluorescence under a fluorescence microscope.
- Harvest the media from the virus-producing HEK 293T cells after 24 h, 48 h and 72 h (if necessary). Replace with fresh culture media.
- Place the harvested media in a sterile 50 ml conical tube. Add polybrene to the final concentration of 30 μg/ml. Vortex briefly to mix.
- [Optional] Spin down at a swing-bucket rotor at 1,000 x g for 2 min to remove cell debris.
- Filter the supernatant using syringe and a 0.45-micrometer syringe filter. The supernatant should be used immediately on the target cell, although it can also be stored at 4 °C for up to three days. If a longer storage is required, the supernatant should be distributed into aliquots and snap frozen in liquid nitrogen and then stored at -80 °C.
- Transduction of target cells
As a demonstration, HEK293T cells are used as target cells to receive the lentivirus produced from above (Procedure B, Step B1-1).- Grow HEK 293T cells in T-75 flask. The density of the cells should be under 30% confluent before the 1st viral transduction, as visually estimated under an optical microscope.
- Dilute the virus-containing supernatant harvested from the T-25 of cells at Step B1-1 with 2x volume of fresh culture media. Replace the media in the T-75 flask containing the target cells with this mix containing the virus and polybrene (final concentration 10 μg/ml).
- Repeat the above step for one more time in the next day.
- Check the mCherryFP fluorescence under fluorescence microscope to ensure that the fluorescence positive cells are less than 20%, in order to avoid over-infection.
Note: It is important to keep the target cells less than 75% confluence at any time during the viral induction stage.
- Sorting of target cells carrying the reporter plasmid
- Trypsinize and collect the target cells by flushing with fresh culture media (at least 5 volume of the trypsin solution used). Transfer the mix to a sterile 50 ml conical tube. Collect the cells by centrifugation in a swing bucket rotor at 1,500 x g for 3 min at room temperature.
Note: Create a backup of these unsorted cells by freezing them in -80 °C. - Resuspend the cells in 1 ml of sorting medium (DMEM + antibiotic-antimycotic), which should generate a density of ~10 million cells/ml.
- Pass the cells through the FACS tubes with 35-micrometer cell strainer.
- Add 1 ml of sorting medium in the FACS collection tubes.
- Use target cells without any mCherry plasmid as negative control for gating purposes in flow cytometry.
- Sort the cells for mCherry positive using FACS–BD Aria-Ilu flow cytometer.
Note: For success of subsequent culture, we recommend a minimum of 20,000 cells should be acquired for each sample. - Culture the mCherry positive cells in T-25 flask with 5 ml fresh media. Expand the culture to bigger flasks (such as T-75) to generate cells carrying the reporter for the next step and keep aliquots for storages at -80 °C.
Note: The quality of the cells expressing the reporter should be periodically inspected, which can be verified by the presence of red fluorescence in at least 90% of the cells under fluorescence microscope.
- Trypsinize and collect the target cells by flushing with fresh culture media (at least 5 volume of the trypsin solution used). Transfer the mix to a sterile 50 ml conical tube. Collect the cells by centrifugation in a swing bucket rotor at 1,500 x g for 3 min at room temperature.
Step B2: Generation of cell line co-expressing FsCFP-mCherryFP and gRNA-GFP- Packaging of lentiviral particles in HEK 293T cell line
This step is similar to the one mentioned above (Step B1), except that the gRNA-GFP plasmid (the VEGF-gRNA-GFP or the scrambled-sequence gRNA-GFP) will be used with these plasmids VSV-G (coding for viral envelop protein) and DeltaR8.2 (coding for reverse transcriptase HIV1-pol and packaging factor HIV1-gag suitable for lentivirus). - Transduction of target cells expressing reporter plasmid with the lentivirus containing gRNA-GFP.
Before proceeding, do a quick visual verification under the fluorescence microscope for the target cells expressing reporter plasmid to make sure more than 90% of cells are mCherry-positive.
Note: The target cells (expressing reporter plasmid) grown in the T-25 flask should be around 30% confluent right before the 1st viral induction (24 h).
The target cells can be repeatedly induced for 2-3 days. In the end of the induction, the GFP-producing cells should be less than 50% of the population to avoid over-induction. - Sorting of target cells carrying both the reporter and gRNA
After 72 h, check the transfection efficiency of target cells under fluorescence microscope for the presence of both mCherry and GFP fluorescence.
Note: Create a backup of these unsorted cells by freezing them in -80 °C.- Process the cells for sorting as described above (Steps B3a-B3d)
- Use appropriate positive controls for gating purposes while sorting. Cells don’t express any fluorescence proteins, or expressing GFP-only, mCherryFP-only, or GFP plus mCherryFP should be used as negative controls to set the gates for CFP-positive events. The detection threshold in general can be set at two folds above the maximum signal generated by the negative controls. If necessary, cells expressing functional GFP, mCherryFP, and CFP can also be used as the positive control (although this is usually not necessary).
- Acquire a minimum of 100,000 events for the double-positive cells to ensure reliable detection of any triple-positive cells.
- Sort the target cells for mCherry and GFP double-positive and any mCherryFP, GFP, and CFP triple-positive populations using FACS–BD Aria-Ilu flow cytometer. The setting of the sorting gates for CFP-positive events should be adjusted so that the collection window is at least by 2-folds higher than the edge of the main population at the CFP channel from any of the three negative controls. Conversely, the collected double-positive cells should not contain CFP signal higher than that of the negative controls.
- Grow and expand the double-positive and triple-positive cells for further analysis.
- FACS data analysis
- Analyze the FACS data by using either FCS Express 6 or FlowJo. The percentage of CFP-positive cells among the double-positive cells can be quantified using FCS Express 6 and MS-Excel.
- Forward and side scattering should be used to exclude cell fragments/debris or cell clumps/clusters. The threshold for detection of the GFP and mCherryFP signals should be established with parental cells that were either not treated, or stably transduced with GFP-only or mCherryFP-only expression vectors. The gating threshold for the double-positive cells (GFP and mCherryFP) should be at least one fold away from the individual positives (GFP-only and mCherry-only) (Figure 4).
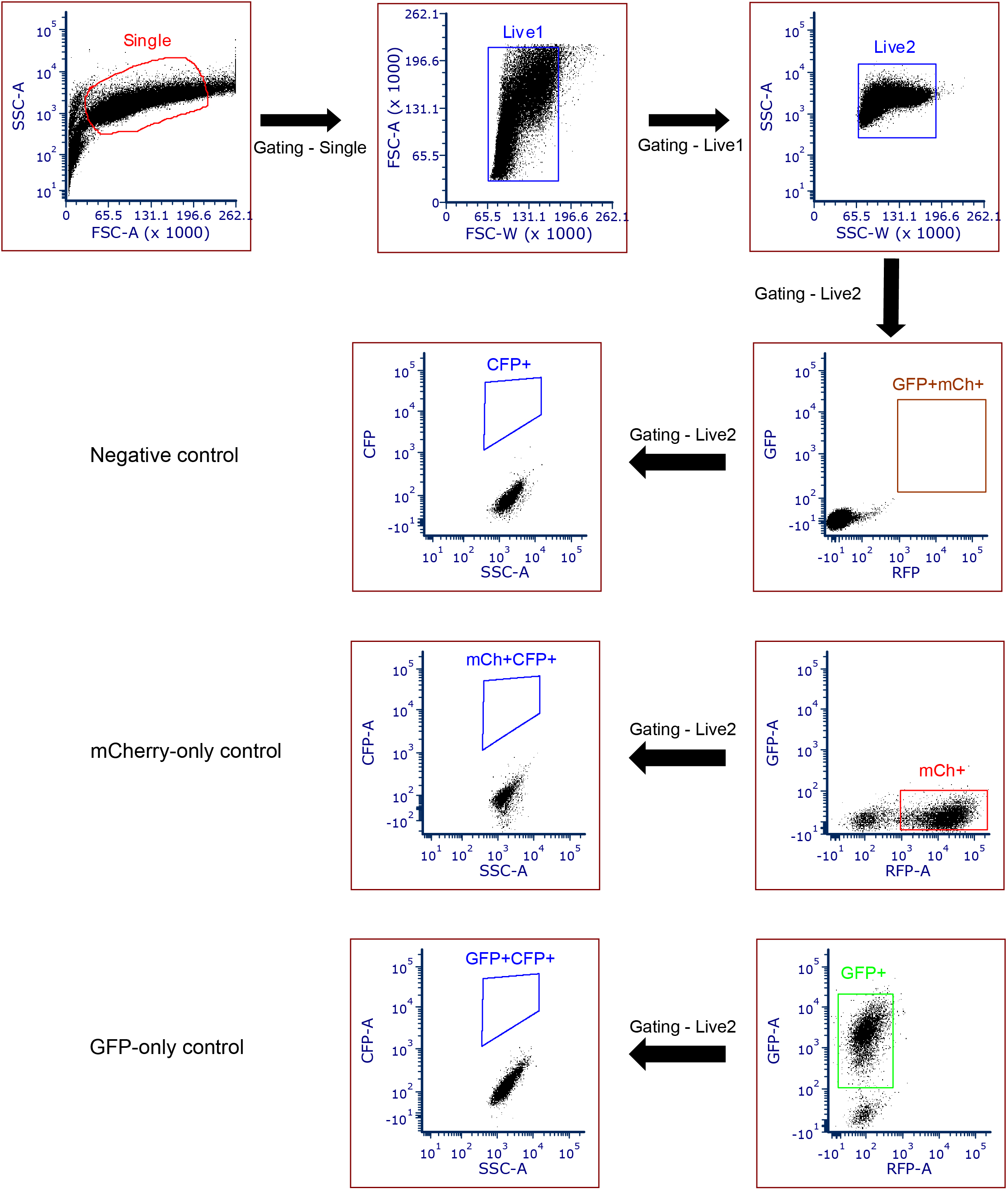
Figure 4. A schematic showing flow cytometry gating strategy. Apply the gating for single and live cells by using forward and side scattering. For the individual fluorescence (GFP and mCherry), gating was applied based on the negative control, GFP-only control, and mCherry-only controls. The gates for selecting GFP and mCherryFP double-positive cells should be at least one fold above the upper edge of the signals from the GFP-only and mCherryFP-only cells. - Apply the gating for GFP and mCherryFP double-positive cells in the sampling group and then look for the CFP positive cells in this population. The gate for CFP should be established by using a CFP-negative control cell expressing GFP plus mCherryFP but not CFP. In our experience, this threshold can also be set by using cells carrying the reporter (with mCherryFP marker) and pQC-XIG (with GFP marker). The gating should be set up so that it is at least two folds higher than the border of the main population of the CFP signal generated from the CFP-negative cells.
- The same set of gatings should be universally applied to all sample groups. The presence of CFP fluorescence indicates the occurrence of in/del events and thereby successful genome editing (See examples in Figure 5). The non-specific (ns) scramble-sequence gRNA-GFP (Figure 5) reflects the background signals of spontaneously generated CFP events by non-specific gene editing.
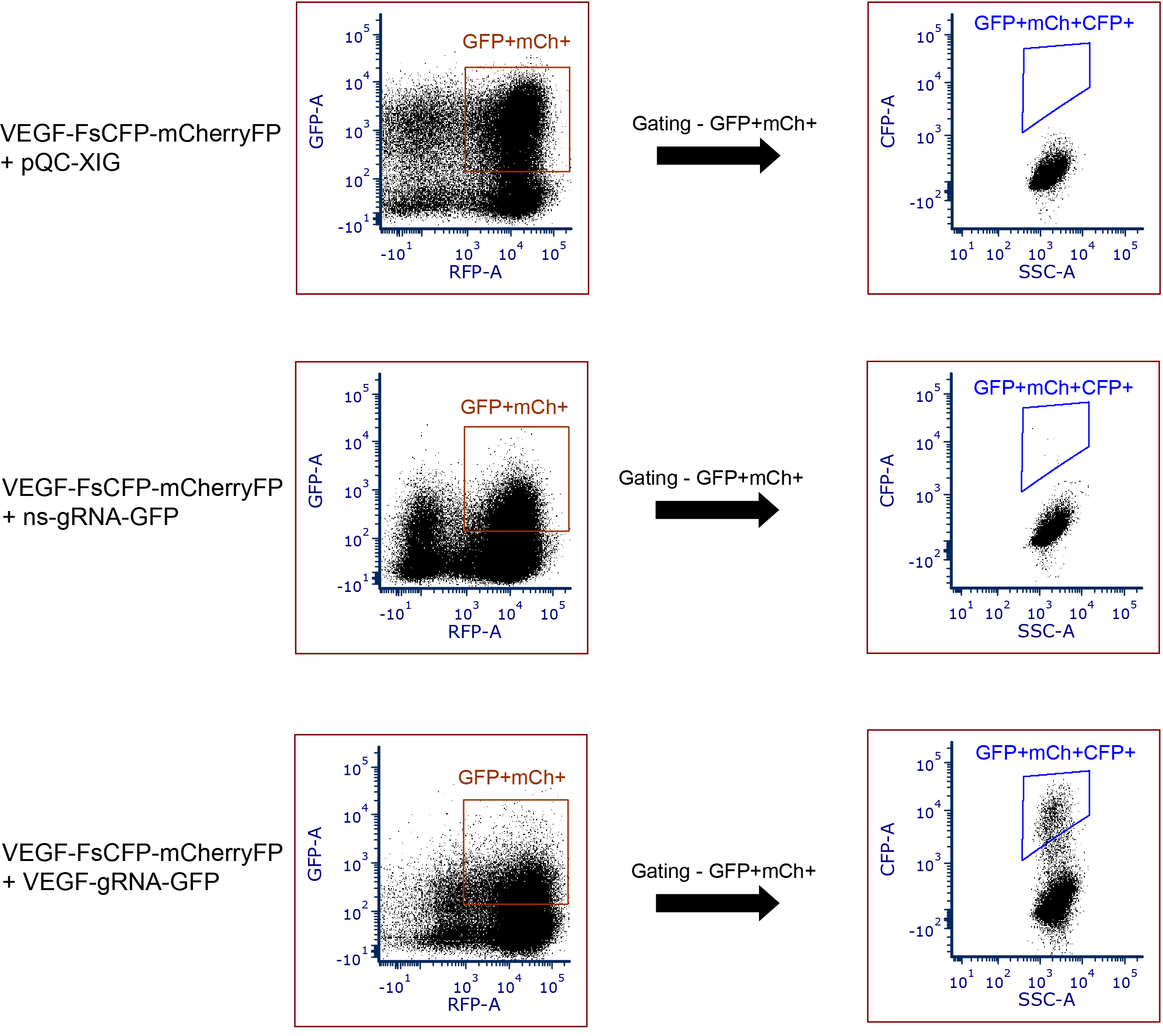
Figure 5. Examples of data to depict the CFP-positive events using this reporter tool in HEK293T cells. Data showing the absence of CFP fluorescence (FsCFP-reporter with target sequence derived from human VEGF gene + pQC-XIG); background CFP fluorescence due to spontaneous mutageneis (reporter + non-specific (ns) gRNA); and presence of CFP-positive events occurred by in/del events (reporter + VEGF gRNA). GFP+mCh+ indicates the gating of double-positive cells (GFP and mCherry). GFP+mCh+CFP+ indicates the presence of CFP+ positive events in the double-positive cells (GFP and mCherry). - The number or the percentage of CFP events among the double positives in each cell population, which is the readout of the genome editing efficiency, can be calculated using MS-Excel or the build-in function of the software (FCS Express 6 in this case) .
- Analysis of genome sequence cell colonies
If desirable, single-cell colonies can be produced by dilution and spreading of the sorted cells in 96-well plates for DNA analysis. See Figure 6 for a summary of all steps in the test.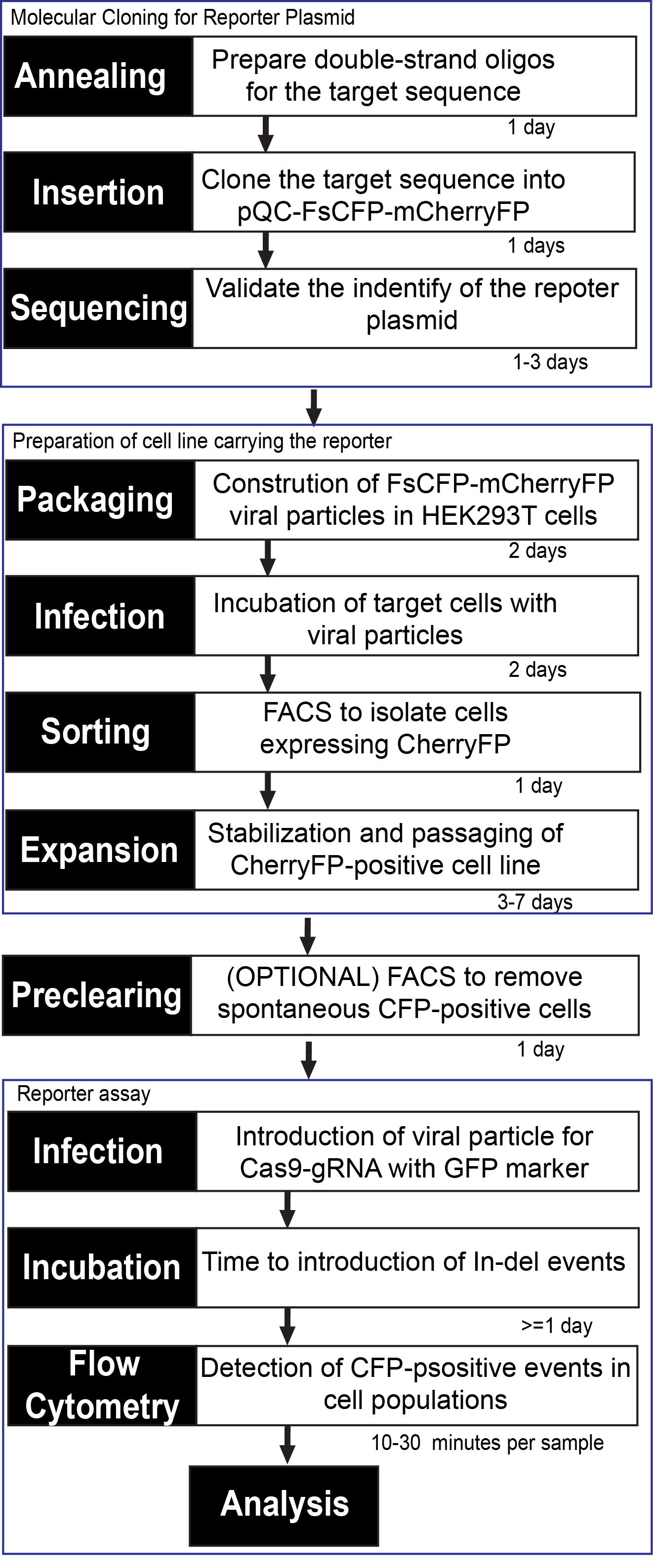
Figure 6. Diagram representing workflow and estimated times to establish a cell line carrying the FsCFP-mCherryFP reporter and perform the genomic editing assay (with Cas9 as an example)
Notes:- If the target sequence contains an internal START codon, care should be taken to ensure that it would not lead to in-frame translation of the CFP. If this is the case, one additional nucleotide can be inserted after the PAM site (preferably after the premature STOP codon) to further shift the reading frame.
- This method is sensitive for low-frequency in/del events too. We have tested this by mutating two central nucleotides in the targeting gRNA, which is expected to reduce the targeting efficiency significantly. When this mutated gRNA is co-expressed along with the reporter, a much reduced yet still statistically significant CFP signal was detected in the cells expressing both GFP and mCherryFP.
- Spontaneous mutagenesis, such as those resulted from base excision, may also lead to the reactivation of the CFP signal. However, in our experience, the magnitude of such spontaneous events (as demonstrated cells with the reporter and pQC-XIG vector) are at least one order lower than the non-specific action of Cas9 supplemented with a non-specific gRNA. Therefore, in our experience, it is safe to use the cells carrying the reporter (with mCherryFP marker) and the pQC-XIG vector (with GFP marker) to set up the detection gate for CFP. However, if a lower background is desired for setting up the CFP detection gate, a cell that only expressing GFP and mCherryFP and not carrying any coding sequence of CFP can be used as the control.
- The gate for CFP detection can be moved up or down for a few (1-5) folds, as long as the threshold is above the border of the main population in CFP-negative control cells (with GFP and mCherryFP double-positive) and as long as the gate is applied universally to all sampling groups, the differences between each group will still exist.
- Production of lentiviral particles in HEK 293T cell line
Acknowledgments
We thank the flow cytometry core facility at the Sylvester Comprehensive Cancer Center for providing the service. We also thank the funding resources–NIGMS/NIH, R01#GM107333; DoD (CDMRP), Idea Award, PC140622. The salary of the authors and the cost of experiments are supported in part by these funding resources. These funding bodies were not directly involved in any part of the design of study, the collection, analysis, or interpretation of the data, or writing of the manuscript. This protocol was adapted from this published work (Kumar et al., 2019).
Competing interests
The authors declare that they have no competing financial interests.
References
- Christian, M., Cermak, T., Doyle, E. L., Schmidt, C., Zhang, F., Hummel, A., Bogdanove, A. J. and Voytas, D. F. (2010). Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186(2): 757-761.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Epinat, J. C., Arnould, S., Chames, P., Rochaix, P., Desfontaines, D., Puzin, C., Patin, A., Zanghellini, A., Paques, F. and Lacroix, E. (2003). A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res 31(11): 2952-2962.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Kim, Y. G., Cha, J. and Chandrasegaran, S. (1996). Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93(3): 1156-1160.
- Kumar, A., Birnbaum, M. D., Moorthy, B. T., Singh, J., Palovcak, A., Patel, D. M. and Zhang, F. (2019). Insertion/deletion-activated frame-shift fluorescence protein is a sensitive reporter for genomic DNA editing. BMC Genomics 20(1): 609.
- Maeder, M. L. and Gersbach, C. A. (2016). Genome-editing technologies for gene and cell therapy. Mol Ther 24(3): 430-446.
- Sander, J. D. and Joung, J. K. (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4): 347-355.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Moorthy, B. T., Kumar, A., Lotenfoe, L. X. and Zhang, F. (2020). Evaluation of the Efficiency of Genome Editing Tools by a Frameshift Fluorescence Protein Reporter. Bio-protocol 10(10): e3622. DOI: 10.21769/BioProtoc.3622.
Category
Biochemistry > Protein > Fluorescence
Cancer Biology > Genome instability & mutation > Cell biology assays > DNA structure and alterations
Molecular Biology > DNA > DNA damage and repair
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.