- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Sensitive Coupled Enzyme Assay for Measuring Kinase and ATPase Kinetics Using ADP-Specific Hexokinase

Published: Vol 10, Iss 9, May 5, 2020 DOI: 10.21769/BioProtoc.3599 Views: 8404
Reviewed by: Juan Facundo Rodriguez AyalaKumiko OkazakiJose Antonio Reyes-Darias
Original research article
The authors used this protocol in:

Oct 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Kinases and ATPases perform essential biological functions in metabolism and regulation. Activity of these enzymes is commonly measured by coupling ATP consumption to the synthesis of a detectable product. For most assay systems the ATP concentration during the reaction is unknown, compromising the precision of the assay.
Using the ADP-specific hexokinase (ADP-HK) from the thermophilic archaeon Thermococcus litoralis the protocol outlined here allows real time coupling of ATP consumption to downstream signal change enabling accurate kinetic measurements. ADP-HK phosphorylates glucose that is then used by glucose-6-phosphate dehydrogenase to reduce NAD+ to NADH which can be measured at 340 nm. We have shown this assay to be sensitive to the detection of micromole quantities of ADP with no detectable background from ATP.
Background
Kinases and ATPases can be measured by coupling the production of ADP to a spectrophotometrically detectable signal. Commercial vendors provide kits that detect the amount of ADP produced or ATP depleted by coupling them to the production of a fluorescent or bioluminescent signal (examples include ATP-Glo; Promega, ADP-sensor; Biovision). These kits are sensitive but are hard to apply to kinetic questions because they are end-point assays, measuring ADP amounts at a single time point. Furthermore, the black box nature of such kits make it difficult to determine if reaction mixtures are in the steady-state conditions required for Michaelis-Menten kinetics.
Continuous assays measure enzyme activity over time allowing the determination of enzyme rate in a single run. The classic continuous coupled-enzyme assay is the pyruvate kinase and lactate dehydrogenase system (Kornberg and Pricer, 1951). During the reaction, pyruvate kinase uses the ADP produced by the target enzyme to convert phosphoenolpyruvate to pyruvate and in the process regenerate ATP. Pyruvate is then used by lactate dehydrogenase to oxidize NADH to NAD+ which is measured by the decrease of absorbance at 340 nm (Figure 1A). A drawback of this method is that in regenerating the ATP continuously, the ATP concentration at any given point is unknown which means that Michaelis-Menten kinetics for ATP cannot be calculated.
An optimal coupled reaction proceeds in a linear pathway so that the ADP produced is converted stoichiometrically into the component producing the signal. ADP-specific hexokinase (ADP-HK) from Thermococcus litoralis uses ADP to phosphorylate glucose into glucose-6-phosphate, which can be used by glucose-6-phosphate dehydrogenase (G6PDH) to reduce NAD+ to NADH (Ito et al., 2001; Sakuraba and Ohshima, 2002) (Figure 1B). ADP-HK is an ADP-specific enzyme from thermophilic archaea that is adapted to consume ADP instead of ATP, probably because of the higher thermostability of ADP over ATP. ADP-HK has previously been used in conjunction with G6PDH and diaphorase I to measure the abundance of dNDPs from cellular extracts with a colorimetric signal but has yet to be demonstrated in a continuous assay to measure kinetics (Kumagai et al., 2014). Here, we have used the specificity of ADP-specific hexokinase to develop a method for assaying ADP production by kinases and ATPases.

Figure 1. Schematic of two coupled enzyme assays to measure ATPase/kinase activity. A. The traditional method for measuring ATPase/kinase activity uses pyruvate kinase to couple ADP production to the conversation of phosphoenolpyruvate (PEP) to pyruvate and then lactate dehydrogenase (LDH) to convert pyruvate to lactate–which simultaneously oxidizes NADH to NAD+ resulting in a decrease of absorbance at 340 nm. B. Our alternative method uses ADP-HK to couple ADP production to the phosphorylation of glucose which is then used by glucose-6-phosphate dehydrogenase (G6PDH) to convert glucose-6-phosphate to gluconate-6-phosphate–which simultaneously reduces NAD+ to NADH resulting in an increase of absorbance at 340 nm.
Materials and Reagents
- Proteosec 6-600 column (Generon)
- Thermococcocus litoralis gDNA (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Strain number: DSM-5473)
- pRSET-A expression plasmid (Thermo Fisher)
- Competent E. coli KRX (Promega)
- ADP-HK expressed and purified from E. coli, stored at -80 °C
- G6PDH from baker's yeast (Sigma-Aldrich, catalog number: G6378-100UN ), stored at 4 °C
- D-Glucose, powder stored at room (Sigma-Aldrich, catalog number: G8270-100G ), stored as powder at room temperature
- NAD+ (Sigma-Aldrich, catalog number: N1636-25MG ), stored in aliquots of 50 mM reaction buffer (see Recipes) at -80 °C
- ADP (Sigma-Aldrich, catalog number: A2754-100MG ), stored in aliquots of 100 mM reaction buffer (see Recipes) at -80 °C
- ATP (Sigma-Aldrich, catalog number: A2383-1G ), stored in aliquots of 100 mM in reaction buffer (see Recipes) at -80 °C
- Tris Base (Melford, catalog number: T60040-5000 )
- NaCl (Sigma-Aldrich, catalog number: S9888-1KG )
- MgCl2 (Sigma-Aldrich, catalog number: 208337-1KG )
- Terrific Broth (Melford, catalogue number: 7758-11-4 )
- Rhamnose (Melford, catalogue number: 10030-85-0 )
- Ni-NTA resin (Generon NB-45-00042-100)
- Imidazole (Sigma-Aldrich, catalogue number: I5513-100G )
- BCA (bicinchoninic acid) (Thermo Fisher, catalogue number: 23225 )
- Reaction buffer (see Recipes)
- ATPase/kinase (see Recipes)
- 1,000 Units/ml G6PDH (see Recipes)
- 50 mM NAD+ (see Recipes)
- 100 mM ADP (see Recipes)
- 100 mM ATP (see Recipes)
- 1 M Glucose (see Recipes)
- 2x Assay reagent (see Recipes)
Equipment
- UV spectrophotometer (Shimadzu, model: UV-1601 )
- 100 µl quartz microcuvette (Hellma, model: 105-201-15-40 )
- Ti 45 Fixed-Angle Titanium Rotor (Beckman Couler, model: Ti 45 )
- Centrifuge (Beckman Couler, model: Allegra® J2-21)
- Äkta Purifier (GE Healthcare)
- -80 °C freezer
Software
- Graphing software (e.g., Origin Pro, OriginLab)
Procedure
- Assay Reagent Formulation (Figure 2)
- The coupled enzyme mix contains 25 µg/ml ADP-HK, 10 mM Glucose, 400 µM NAD+ and 4 units/ml G6PDH.
Notes:- Glucose, G6PDH and NAD+ are available commercially. Individual components are stored as stock solutions at -80 °C and mixed on the day.
- For each measurement, an assay reagent containing ADP-HK, G6PDH, glucose and NAD+ is prepared at 2x concentration (50 µg/ml ADP-HK, 20 mM Glucose, 800 µM NAD+ and 8 units/ml G6PDH) and mixed in the cuvette with an equal volume of 2x concentration substrate. By mixing equal volumes i.e., 100 µl + 100 µl, fast mixing conditions are obtained and concentrations of the assay reagent and substrate are halved.
- The detectable absorbance signal is produced by NADH which has an absorbance peak at 340 nm (A340) that is not present for NAD+. The extinction coefficient for NADH at 340 nm is 6,220 M-1cm-1.
- Clone ADP-HK from T. litoralis gDNA into an E. coli expression plasmid and purified by Ni-affinity purification via an N-terminal hexa-Histidine tag followed by size-exclusion chromatography.
- Produce the ADP-hexokinase in-house by purifying a recombinantly expressed with an N-terminal His-tagged construct.
- Produce the plasmid by cloning the ADP-hexokinase from Thermococcocus litoralis gDNA (DSM-5473, Deutsche Sammlung von Mikroorganismen und Zellkulturen) gene into a pRSET-A expression plasmid (Thermo Fisher).
- For expression, transform the plasmid into chemically competent E. coli KRX (Promega) and grow in 1 L Terrific Broth until OD600 ~0.6-0.8 and then induced with 0.1% (w/v) rhamnose for 15 h at 18 °C.
- Harvest cells by centrifugation at 2,700 x g for 15 min, resuspend in 50 mM Tris, 150 mM NaCl, pH 7.9.
- Cells are lysed by sonication (Bandelin Sonopuls Sonication) with 2 s on/off pulses for 5 min, followed by centrifugation for 1 h at 80,000 x g (Beckman Ti 45 rotor in an Allegra J2-21).
- Mix the supernatant with 3 ml of Ni-NTA resin (Qiagen) and left on a turning rotor overnight at 4 °C.
- Wash the resin three times by resuspending in 15 ml of 50 mM Tris, 150 mM NaCl, 30 mM imidazole, pH 7.9 followed by centrifugation at 1,000 x g for 5 min.
- Elute the protein by washing three times with 15 ml of 50 mM Tris, 150 mM NaCl, 300 mM imidazole, pH 7.9.
- Concentrate the eluted protein to 1 ml and further purify using size-exclusion chromatography using a Proteosec 6-600 column (Generon) and FPLC on an Äkta Purifier (GE Healthcare).
- Concentrate the protein to 5 mg/ml with 20% (v/v) glycerol, dispense into 50 µl aliquots and flash freeze in liquid nitrogen.
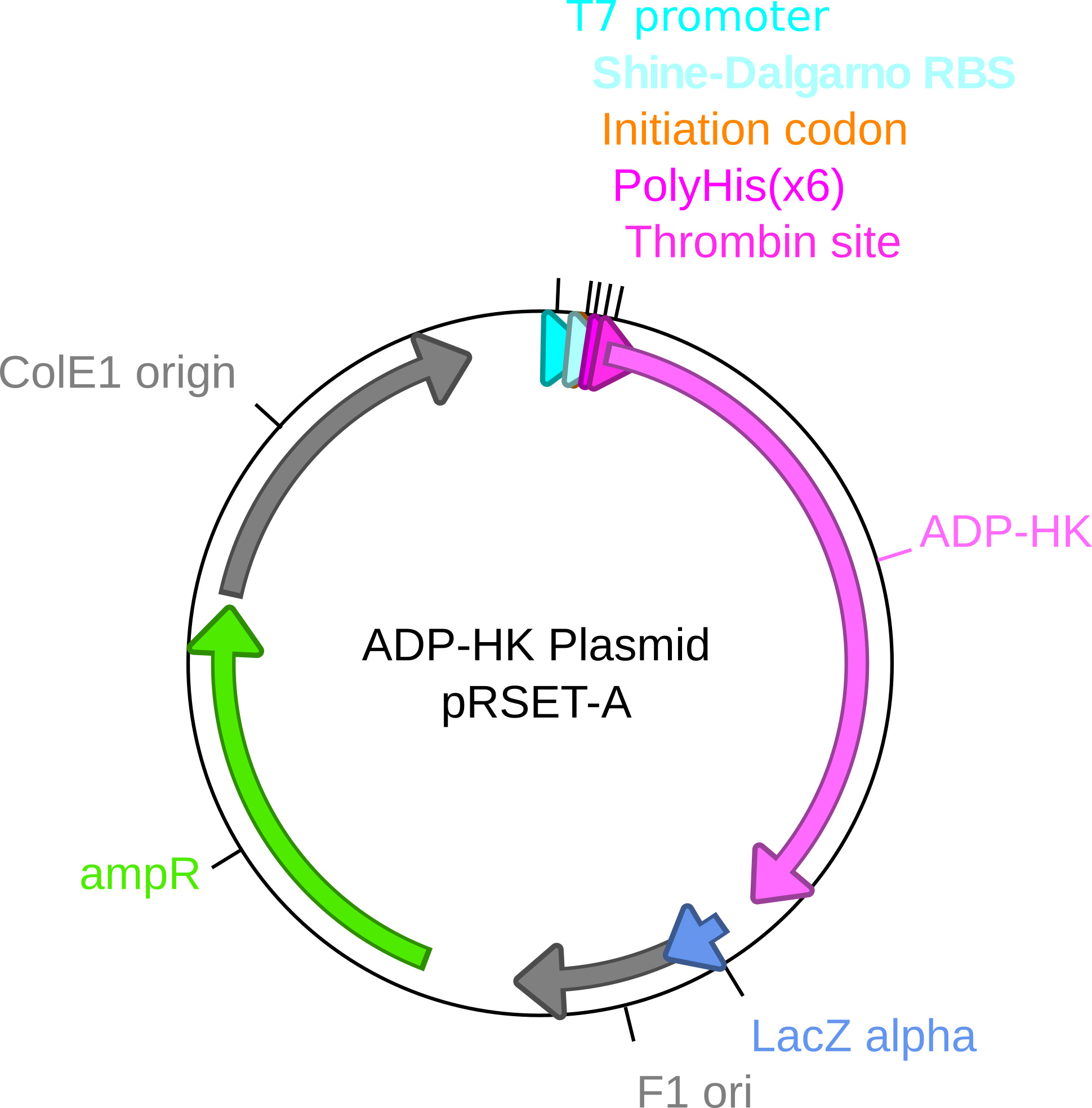
Figure 2. ADP-HK expression plasmid map. The expression plasmid for ADP-HK is a modified prSET-A plasmid. The ADP-HK gene from Thermococcocus litoralis (DSM5473) was cloned into the plasmid with a thrombin cleavable hexa-Histidine tag. The insert is under the control of an inducible T7 promoter and a strong ribosome binding site (Shine-Dalgarno RBS). - The coupled enzyme mix contains 25 µg/ml ADP-HK, 10 mM Glucose, 400 µM NAD+ and 4 units/ml G6PDH.
- Assay Procedure
Note: The volumes stated are for twenty-five 200 µl reactions, enough to perform controls and produce a 5-point Michaelis-Menten curve in triplicate.- Prepare 2.5 ml of assay reagent containing 50 µg/ml ADP-HK, 20 mM Glucose, 800 µM NAD+ and 8 Units G6PDH in reaction buffer.
- Divide the assay reagent into two unequal parts:
- 2 ml in which the kinase/ATPase being measured is added. It is recommended that the final concentration of enzyme is 1 µM in the first instance. If steady-state conditions are not met, i.e., the reaction is too fast or if the reaction is too slow to be measured, then the concentration of enzyme can be adjusted.
- 0.5 ml in which assay reaction buffer 5 µl of assay buffer is added.
- Prepare ATP over the range of concentrations to be measured in reaction buffer. For each concentration prepare ATP at 2x concentration, e.g., for a 10 µM ATP measurement prepare the solution at 20 µM. A recommended concentration range is from 0 mM ATP to 10 times above the expected enzyme KM. For kinases with a second substrate, ensure this is present in large excess to achieve pseudo-Michaelis-Menten conditions (100-fold greater than KM).
- For each measurement, quickly mix 100 µl of assay reagent with 100 µl of substrate within the quartz cuvette, aspirate 3 times to mix and start measuring the absorbance at A340 for 200 s.
Note: Adjust measurement time in accordance with the speed of the reaction so that curve fitting is accurate. Slower kinases may require longer measurement times. - Perform positive and negative controls for each assay. Positive control; mixing 100 µM ADP and assay reaction reagent and ensuring the increase at A340 is as expected, corresponding to a stoichiometric amount of NADH. Negative control; mixing 100 µM ATP and assay reagent without the kinase/ATPtase and ensuring there is no increase in A340.
- Measure activity starting at 0 µM ATP then at increasing ATP concentrations. It is recommended to measure activity at at least 5 different ATP concentrations with the highest concentration at 10-fold above the expected KM.
Data analysis
- A340 timecourses were recorded in triplicate and plotted using graphing software (here we have used the graphing software Origin Pro).
- For Michaelis-Menten analysis, the steady-state rate (A340/s) were determined by plotting a straight line over the first 30 s of each plot and calculating the slope. The average rate with standard deviation (S.D.) was plotted for each concentration of ATP and fitted to a Michaelis-Menten equation [1] to calculate Vmax and KM.
- The concentration of NADH, which is stoichiometric to the conversion of ATP to ADP, was calculated using the Beer-Lambert law [2].
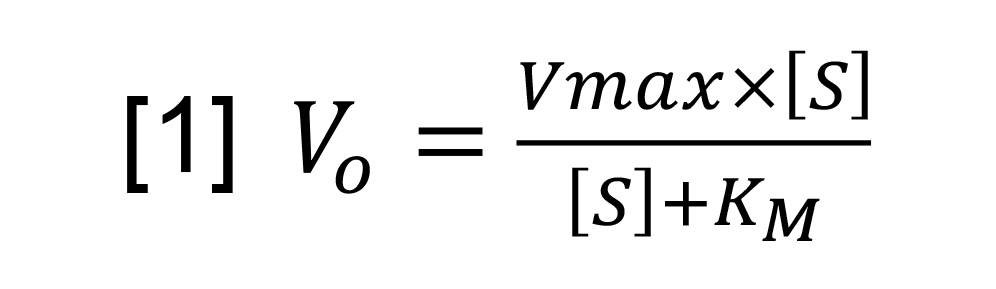
Vo is the initial rate, [S] the substrate concentration, Vmax is the maximal velocity and KM is the substrate concentration which produces that half-maximal velocity.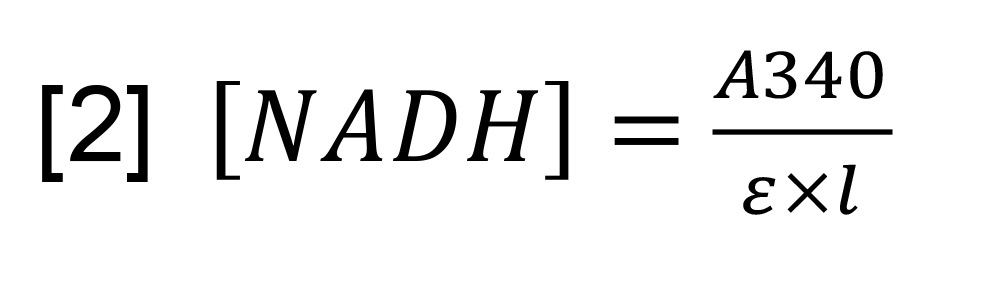
[NADH] is the concentration of NADH, A340 is the UV absorbance at 340 nm, ε is the extinction coefficient of NADH (6,220 M-1cm-1) and l is the cuvette pathlength (in this case 1 cm).
Statistics
- Mean and Standard Deviation
Each plotted rate was the mean rate (x), calculated by dividing the sum of rates (∑xi) with the number of data points (n), which is this case is 3. The standard deviation of the rate was estimated using the standard formula [3].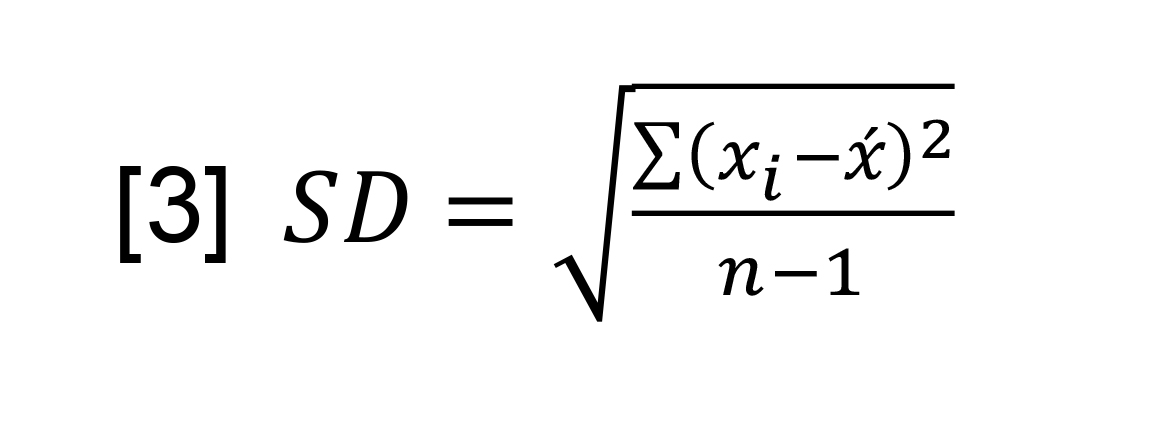
- Error KM and Vmax
The error for KM and Vmax are calculated by Origin Pro (OriginLab) software by determining the KM and Vmax of each individual data set and calculating the standard deviation.
Notes
- Validation of sensitivity and specificity
The assay was tested for its range of sensitivity and specificity for ADP (Figure 3A). For each substrate concentration the A340 change was recorded in triplicate and the corresponding difference between the curve baseline and plateau was measured. The initial rate, an approximation of the steady-state rate, was determined by calculating the curve slope for the first 30 s of the reaction.
The assay is sensitive from low micromolar concentration of ADP to millimolar concentration. Recordings using 10 µM to 3,000 µM ADP showed that at increasing ADP concentrations the reaction rate increased such that at up to 3,000 µM ADP, glucose and NAD+ were not rate limiting.
To determine ADP specificity over ATP, 100 µM of each substrate was measured for the reaction rate and the total A340 change (Figure 3B). ATP produced no measurable activity with a total A340 change of 0.006 ± 0.000 at an initial rate of 0.002 ± 0.000 A340/s. Conversely, ADP produced a measurable reaction with a total A340 change of 0.7 ± 0.1 at an initial rate of 1.42 ± 0.00 A340/s. Therefore ADP produced 116 times higher final signal than ATP at 705 times faster rate. The A340 implied 113 µM NADH, suggesting that the 100 µM ADP produced NADH stoichiometrically in the assay, and that the reaction had gone to completion.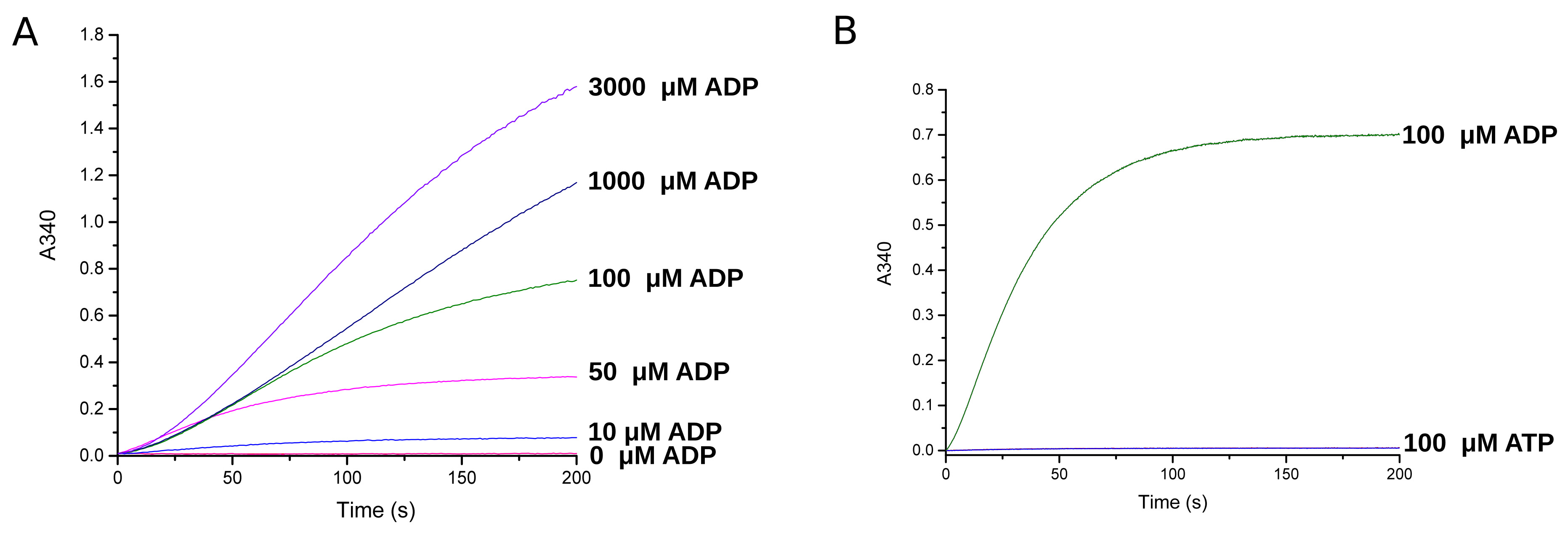
Figure 3. Validation of ADP concentration dependence and specificity. A. A340 recordings for increasing ADP concentrations. B. The A340 difference for ADP versus ATP. The absorbance was measured spectrophotometrically (Shimadzu, UV-1601 ) at 340 nm at 1 Hz and with a slit-length of 1 nm. Traces were plotted and analysed using Origin Pro software (OriginLab). - Michaelis-Menten kinetics of GAPDH-CP12-PRK
Michaelis-Menten curves of phosphoribulokinase (PRK) activity were measured for oxidized and reduced GAPDH-CP12-PRK to determine the redox dependent inactivation of the kinase (Figure 4) (McFarlane et al., 2019). PRK is a sugar kinase that phosphorylates ribulose-5-phosphate to ribulose-1,5-bisphosphate using ATP. The GAPDH-CP12-PRK complex was purified to homogeneity and the activity of PRK was measured at increasing concentrations of ATP to determine the Michaelis-Menten constants of PRK within the GAPDH-CP12-PRK complex under oxidizing and reducing conditions.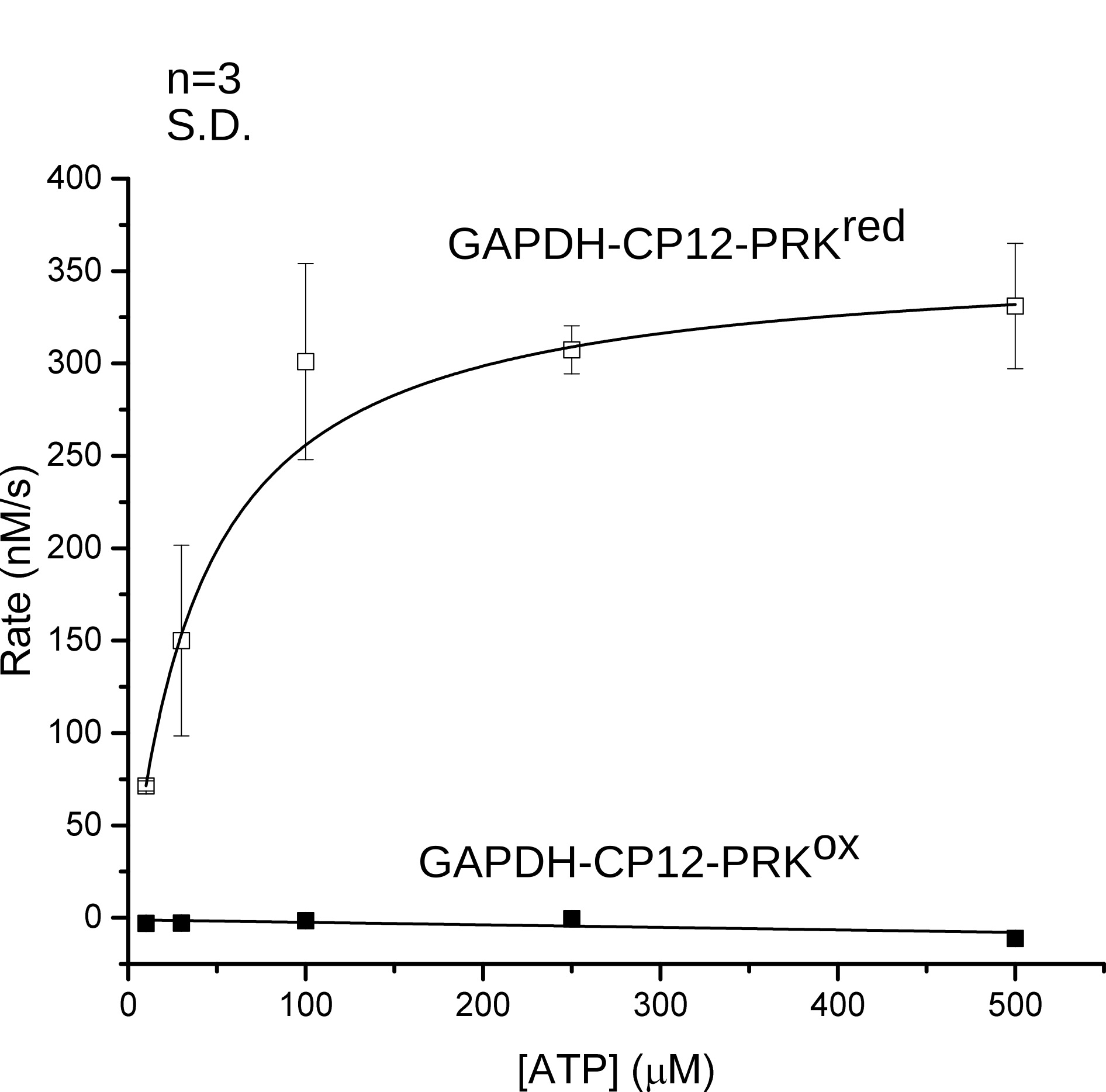
Figure 4. GAPDH-CP12-PRK Michaelis-Menten curves. Michaelis-Menten fits of oxidized (ox) and reduced (red) GAPDH-CP12-PRK (McFarlane et al., 2019).
Measuring Michaelis-Menten kinetics of the complex allowed detection of any changes of PRK activity in a substrate concentration dependent manner and the low background of the ADP-HK assay allowed accurate determination of the extent of inactivation. In the presence of excess Ru5P (2.5 mM), the rate of ADP production was measured at increasing ATP concentrations. Initial rates for each trace was measured by fitting a straight line over the initial 30 s. Rates were then plotted and fitted to a Michaelis-Menten equation.
Pre-formed oxidized complex showed zero measurable activity for all ATP concentrations and the reduced complex showed activity with a KM of 40 ± 2 µM and and Vmax of 13.6 ± 0.3 s−1. These values correspond to the fact that the oxidized complex is completely inactive for PRK activity and that the inactivation is independent of ATP concentration. When the complex is chemically reduced, phosphoribulokinase is activated and conforms to Michaelis-Menten kinetics by showing increased activity at increased ATP concentration until the maximal velocity in reached.
Conclusion
The protocol described here demonstrates how the specificity of ADP-hexokinase from T. litoralis can be used in a coupled enzyme reaction to measure ATPase or kinase activities. The materials and equipment required for this assay are commonly accessible allowing for sensitive measurements to be obtained without purchasing expensive commercial kits. The assay produces a rapid and stoichiometric conversion of ADP to an absorbance signal that can be measured continuously and the high specificity and sensitivity ensures low background and therefore accurate kinetic measurements.
Recipes
- Reaction buffer
50 mM Tris
150 mM NaCl
10 mM MgCl2
pH 7.9
Dissolve 6.02 g of Tris, 8.76 g of NaCl and 0.95 g of MgCl2 in 1 L of ultrapure water - ATPase/kinase
Determine the ATPase/Kinase protein concentration for example using a Pierce BCA (bicinchoninic acid) assay (Thermo Fisher) and prepare the ATPase/Kinase in a stock concentration of 100 µM in the reaction buffer. - 1,000 Units/ml G6PDH
- Dissolve lyophilized G6PDH powder in 100 µl of reaction buffer
- Store at 4 °C and use within 1 month
- 50 mM NAD+
- Dissolve contents of a 25 mg vial in 753 µl of ultrapure water
- Dispense into 80 µM aliquots and stored at -80 °C
- 100 mM ADP
- Dissolve 100 mg of ADP into 2.34 ml of ultrapure water
- Dispense into 100 µl aliquots and store at -80 °C
- 100 mM ATP
- Dissolve 100 mg of ADP into 1.81 ml of ultrapure water
- Dispense into 100 µl aliquots and store at -80 °C
- 1 M Glucose
Dissolve 180 mg of D-glucose into 1 ml of reaction buffer on the day of the experiment - 2x Assay reagent (2.5 ml)
50 µg/ml ADP-HK
20 mM Glucose
800 µM NAD+
8 Units G6PDH
Add 25 µl of 5 mg/ml ADP-HK, 50 µl of 1 M Glucose, 40 µl of 50 mM NAD+, 20 µl of 1,000 Units/ml G6PDH and 2.36 ml of buffered saline solution
Note: The assay was developed under the conditions given, however it is likely that some variation can be tolerated. G6PDH is stable at a few hours at pH 4.0 (Souza et al., 2002), and ADP-HK is stable up to 100 °C (Kengen et al., 1995).
Acknowledgments
This work was supported by a BBSRC Doctoral Training Programme grant (BB/J014575/1) and was the subject of previous work by McFarlane et al. (2019).
Competing interests
The authors declare no competing interests.
References
- Ito, S., Fushinobu, S., Yoshioka, I., Koga, S., Matsuzawa, H. and Wakagi, T. (2001). Structural basis for the ADP-specificity of a novel glucokinase from a hyperthermophilic archaeon. Structure 9(3): 205-214.
- Kengen, S. W., Tuininga, J. E., de Bok, F. A., Stams, A. J. and de Vos, W. M. (1995). Purification and characterization of a novel ADP-dependent glucokinase from the hyperthermophilic archaeon Pyrococcus furiosus. J Biol Chem 270(51): 30453-30457.
- Kornberg, A. and Pricer, W. E., Jr. (1951). Enzymatic phosphorylation of adenosine and 2,6-diaminopurine riboside. J Biol Chem 193(2): 481-495.
- Kumagai, K., Kojima, H., Okabe, T. and Nagano, T. (2014). Development of a highly sensitive, high-throughput assay for glycosyltransferases using enzyme-coupled fluorescence detection. Anal Biochem 447: 146-155.
- McFarlane, C. R., Shah, N. R., Kabasakal, B. V., Echeverria, B., Cotton, C. A. R., Bubeck, D. and Murray, J. W. (2019). Structural basis of light-induced redox regulation in the Calvin-Benson cycle in cyanobacteria. Proc Natl Acad Sci U S A 116(42): 20984-20990.
- Sakuraba, H. and Ohshima, T. (2002). Novel energy metabolism in anaerobic hyperthermophilic archaea: a modified Embden-Meyerhof pathway. J Biosci Bioeng 93(5): 441-448.
- Souza, M. A., Ribeiro, M. Z., Silva, D. P., Pessoa, A., Jr. and Vitolo, M. (2002). Effect of pH on the stability of hexokinase and glucose 6-phosphate dehydrogenase. Appl Biochem Biotechnol 98-100: 265-272.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
McFarlane, C. R. and Murray, J. W. (2020). A Sensitive Coupled Enzyme Assay for Measuring Kinase and ATPase Kinetics Using ADP-Specific Hexokinase. Bio-protocol 10(9): e3599. DOI: 10.21769/BioProtoc.3599.
Category
Microbiology > Microbial biochemistry > Protein > Activity
Cell Biology > Cell-based analysis > Enzymatic assay
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.