- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dual Fluorescence Cytometry Assay to Assess Cellular Protein Levels
(*contributed equally to this work) Published: Vol 10, Iss 8, Apr 20, 2020 DOI: 10.21769/BioProtoc.3597 Views: 6328
Reviewed by: Chiara AmbrogioMaría Antonia Sánchez RomeroValerian DORMOY
Original research article
The authors used this protocol in:

Sep 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Expression levels of cellular proteins can be affected by various perturbations, such as genetic knockout of interactors, drug treatments or cell stress. To specifically measure the effects on protein levels post-synthesis under different experimental conditions, it is important to compensate for transcriptional and other upstream changes. Here, we provide a protocol for a dual-fluorescence flowcytometry-based assay to determine protein levels. The protein of interest is genetically linked to enhanced GFP (eGFP) followed by a viral 2A self-cleaving peptide sequence and mCherry. As a result, translation of the reporter construct leads to two fluorescent protein products from the same mRNA template, which enables unambiguous protein expression analysis with normalization across samples.
Keywords: Protein expressionBackground
Synthesis and maintenance of cellular proteins depend on multiple processes, from transcriptional regulation, processing and degradation of mRNA to translation, folding, localization, post-translational modification and protein degradation (Vogel and Marcotte, 2012). To specifically study the effects of cellular perturbations on protein levels post-synthesis, it is important to compensate for variability in upstream steps of protein expression. Here, we provide a protocol for a dual-fluorescence flow cytometry-based assay to determine protein levels at steady state as previously described (Itakura et al., 2016; Chitwood et al., 2018; Ngo et al., 2019). The protein of interest is genetically fused to enhanced GFP (eGFP) followed by a viral 2A self-cleaving peptide sequence and a second fluorescent protein, mCherry (Figure 1). Translation of the fusion construct generates two protein products at a 1:1 ratio due to peptide bond skipping by the ribosome at the 2A site: the protein of interest fused to eGFP and mCherry. As both proteins are produced from the same transcribed mRNA molecule and under control by the same promoter, this system allows assessment of the abundance of the protein with normalization of transcriptional differences or variability in transfection efficiency. Furthermore, the flow cytometry-based single-cell readout may enable the detection of more subtle differences in protein levels between experimental conditions, which could otherwise be masked in bulk readouts, such as immunoblot.
We demonstrated the use of this method to study the expression levels of two multi-pass transmembrane proteins, beta1-adrenergic receptor (ADRB1) and the dengue virus non-structural proteins (NS) 4A and 4B, in relation to functional ER membrane protein complex (EMC) expression as previously published (Chitwood et al., 2018; Ngo et al., 2019). The obtained data shows that EMC is important in facilitating stable expression of these transmembrane proteins (Figure 2).
This method can thus be used to study the expression levels of specific proteins of interest under different cellular conditions, such as genetic perturbations, drug treatments or other cellular stress.
Materials and Reagents
- 12-well cell culture plates (Fisher Scientific, catalog number: 12556005 )
- Falcon 5 ml Round-Bottom Tubes with 35 μm strainer lids (Corning, catalog number: 352235 )
- 293FT cells (Thermo Fisher, catalog number: R70007 )
- EMC4-KO 293FT cells (generated as described in PMID: 31516121)
- Stbl3 competent E. coli (MacroLab, catalog number: Stbl3 )
- eGFP coding sequence, amplified from pX458 (gift from Feng Zhang; Addgene plasmid, catalog number: 48138 )
- mCherry coding sequence, amplified from pCAGGS-mCherry (gift from Phil Sharp; Addgene plasmid, catalog number: 41583 )
- ADRB1 coding sequence, amplified from pcDNA3 Flag beta-1-adrenergic-receptor (gift from Robert Lefkowitz; Addgene plasmid, catalog number: 14698 )
- Dengue virus serotype 2 16681 strain NS4A-NS4B coding sequence, amplified from infectious clone (gift from Karla Kirkegaard, NCBI Reference Sequence: NC_001474)
- Kapa HiFi HotStart ReadyMix (Kapa Biosystems, catalog number: KK2602 )
- DNA oligos (Integrated DNA Technologies, see Table 1 for sequences)
Table 1. DNA oligo sequences
- QIAquick Gel Extraction Kit (Qiagen, catalog number: 28706 )
- Molecular Biology Agarose (Bio-Rad, catalog number: 1613102 )
- NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs, catalog number: E2621L )
- pLenti CMV Puro DEST (w118-1) (gift from Eric Campeau and Paul Kaufman; Addgene plasmid, catalog number: 17452 )
- EcoRV-HF (New England Biolabs, catalog number: R3195S )
- Quick CIP (New England Biolabs, catalog number: M0525S )
- S.O.C. Medium (Teknova, catalog number: S1657 )
- LB agar plates with carbenicillin (Teknova, catalog number: L1010 )
- LB Broth (Teknova, catalog number: L8050 )
- Carbenicillin 100 mg/ml (Teknova, catalog number: C2135 )
- QIAfilter Plasmid Midi Kit (Qiagen, catalog number: 12243 )
- Dulbecco's modified Eagle medium (DMEM) (Thermo Fisher Scientific, catalog number: 11965118 )
- Fetal bovine serum (FBS) (Omega Scientific, catalog number: FB-11 )
- Opti-MEM reduced serum medium (Life Technologies, catalog number: 31985062 )
- Penicillin-Streptomycin 100x (Life Technologies, catalog number: 15140122 )
- L-glutamine 100x (Thermo Fisher Scientific, catalog number: 25030081 )
- Dulbecco's phosphate-buffered saline (DPBS) (Thermo Fisher Scientific, catalog number: 14190-250 )
- Trypsin-EDTA 0.05% (Life Technologies, catalog number: 25300120 )
- TransIT®-LT1 Transfection Reagent (Mirus Bio, catalog number: MIR2300 )
- Lipofectamine 3000 Transfection Reagent (Fisher Scientific, catalog number: L3000008 )
- Culture medium for 293FT (see Recipes)
Equipment
- Nanodrop One Microvolume UV-Vis Spectrophotometers (Thermo Fisher, catalog number: ND-ONE-W )
- CytoFLEX S V4-B2-Y4-R3 Flow Cytometer with a 488 and 561nm lasers (Beckman Coulter, catalog number: B96621 )
- Shaker
- Waterbath
Software
- CytExpert Version 2.3 (Beckman Coulter Inc., https://www.beckmancoulter.com/)
- FlowJo v10.6.1 (FlowJo, LLC, https://www.flowjo.com/)
- Microsoft Excel (Microsoft Corp., https://www.microsoft.com/en-us/)
- Prism 8 (GraphPad Software, LLC, https://www.graphpad.com/)
Procedure
- Cloning of dual fluorescence reporter constructs
- Design primers for the amplification of the gene of interest (here: ADRB1 or dengue NS4A-4B), eGFP and mCherry with the addition of 20-30 bp overlaps between the neighboring fragments for assembly of multiple DNA fragments. The forward primer for the gene of interest contains an overlap with left arm of EcoRV-linearized pLenti CMV Puro DEST (w118-1), a Kozak sequence and an ATG start codon. The reverse primer overlaps with the 5′ end of the eGFP coding sequence. The reverse primer for eGFP has no stop codon and contains an overhang encoding the first ~40 bp of T2A thus creating an overlap with the T2A-mCherry fragment. mCherry is amplified with a forward primer containing parts of the T2A sequence and a reverse primer with stop codon and overlap to the right arm of EcoRV-linearized pLenti CMV Puro DEST (w118-1). A control construct, which only encodes eGFP-T2A-mCherry, can be generated, where the forward primer for eGFP contains an overlap with left arm of EcoRV-linearized pLenti CMV Puro DEST (w118-1), a Kozak sequence and an ATG start codon.
- Amplify DNA fragments from plasmid templates using the KAPA HiFi HotStart ReadyMix (or any other high-fidelity polymerase). Use PCR cycling conditions as recommended by the manufacturer. After PCR, run amplified fragments on a 1% agarose gel followed by extraction using a gel purification kit.
- Assemble purified PCR fragments together with EcoRV-linearized and CIP-treated plenti-CMV-Puro plasmid using NEBuilder® HiFi DNA Assembly Master Mix according to the manufacturer’s instructions. Note that other mammalian expression vectors can be used as recipient plasmid but you need to modify the overhang sequences for assembly of multiple DNA fragments in Step A1 accordingly.
- Transform Stbl3 E. coli with assembled plasmids by heat shocking at 42 °C waterbath for 45 s. Recover heat shocked bacteria in S.O.C. medium and 30 °C shaker for 1 h at 225 RPM. Spread transformed bacteria on 10 mm agar plates that contain 100 μg/ml Carbenicillin and incubate at 30 °C or 37 °C overnight.
- The next day, pick a single colony from the plate and inoculate 50 ml of LB broth that contains 100 μg/ml Carbenicillin. Shake inoculated LB broth in 30 °C (if plasmid sequence is prone to recombination, such as lentiviral plasmids) or 37 °C shaker at 225 RPM overnight.
- Extract plasmid DNA from E. coli using a plasmid DNA Midiprep kit according to the manufacturer’s instruction. Determine concentration and purity (260/280 ratio) of nucleic acid using Nanodrop. Sequence-verify plasmid insert by Sanger sequencing using a forward primer upstream of eGFP and a reverse primer downstream of mCherry.
- Transfection of cell lines
- Plate ~3 x 105 293FT cells (WT and EMC4 KO genotypes) in 12-well plates the day before transfection using 293FT culture medium (see Recipes). Cells should be 70%-80% confluent 24 h after plating.
- The next day, transfect dual fluorescence constructs into 293FT cells using Mirus TransIT®-LT1 Transfection Reagent or Lipofectamine 3000. To do so, dilute required amount of plasmid DNA in serum-free media or OptiMEM and add the transfection reagent according to the manufacturer’s instructions. Then, add DNA-lipid complexes dropwise to cells.
- Flow cytometry analysis
- After 24-48 h, detach cells by adding 100 μl 0.05% Trypsin-EDTA per well followed by incubation at 37 °C for 3 min. Inactivate trypsin by adding an equal volume of culture medium containing FBS. Pass cells through a 35 μm cell strainer to obtain a single-cell suspension.
- Analyze cells on a flow cytometer. First, use untransfected cells to draw negative gates. Gate for forward (FSC) and side scatter (SSC), then height and area of FSC for a singlet gate, then mCherry fluorescence (mCherry positive cells are the successfully transfected cells) and finally eGFP fluorescence. Then run your samples (eGFP-2A-mCherry control and protein of interest constructs). Record 5 x 103-5 x 104 mCherry-positive events per transfected sample.
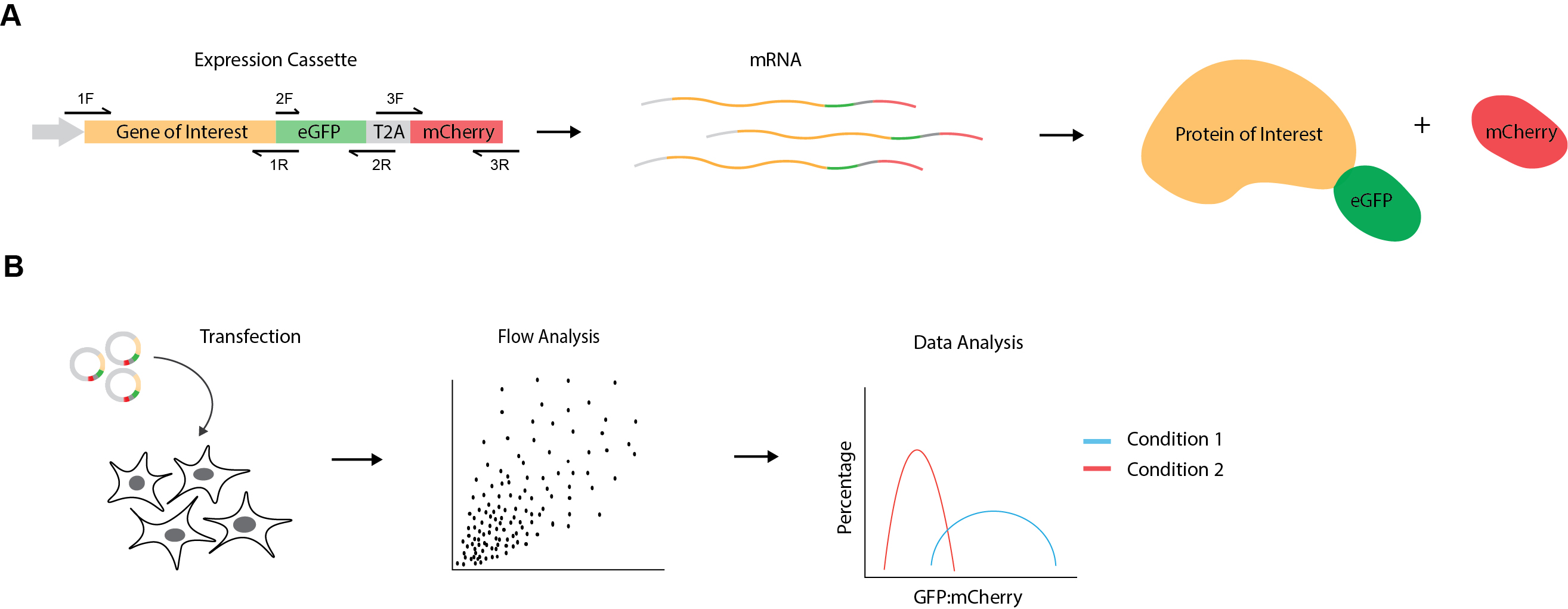
Figure 1. Experimental design for dual fluorescence cytometry assay. A. Schematic representation of mammalian expression cassette showing gene of interest followed by eGFP, a viral T2A sequence and mCherry. The primers to generate DNA fragments containing overhangs for plasmid construction are indicated by black arrows (1F = PLENTI-CMV-ADRB1-F or PLENTI-CMV-DENV_NS4A-F; 1R = ADRB1(NOSTOP)_GFP-R or DENV_NS4B_GFP-R; 2F = EGFP-F; 2R=EGFP-T2A_GIBSON-R; 3F = T2A-MCHERRY-F; 3R = MCHERRY-PLENTI-R). Transcribed mRNA molecules encode both fluorescent proteins, which are then translated into two protein products by peptide bond skipping at the T2A site. This results in the production of the GFP fusion protein and mCherry at an initial 1:1 stoichiometry. Thus, the eGFP:mCherry ratio allows measuring effects on protein levels post-synthesis. B. Experimental workflow. eGFP-T2A-mCherry reporter constructs are transfected into cells of different experimental conditions (e.g., wild-type vs knockout genotypes, drug treatments, cell stress etc.). Cells are then analyzed for GFP and mCherry fluorescence levels using flow cytometry. Lastly, eGFP:mCherry ratios are calculated for the protein of interest under the different experimental conditions and graphed as frequency distributions.
Data analysis
- FCS files are exported from CytExpert cytometer software and imported in FlowJo 10 for FACS analysis. The gating order is as follows: FSC-H/SSC-H (cells vs debris), FSC-H/FSC-A (singlets), ECD-H (mCherry channel)/FSC-H and the FITC-H (eGFP channel)/FSC-H. First, use untransfected cells to set the negative gates for mCherry and eGFP fluorescence. Then apply all gates onto the remaining samples. The mCherry-positive cells represent the successfully transfected cells. As GFP and mCherry are expressed from the same construct/promoter, all mCherry positive cells also have GFP fluorescence. Export their relative intensity values of the mCherry positive gated populations as comma-separated values (CSV) files by right-click on the “mCherry positive” group and then “Export/Concatenate Group”.
- Calculate the eGFP-to-mCherry ratios for each row in all datasheets (each row represents an individual recorded cell) using Microsoft Excel. As eGFP and mCherry have high structural similarity and are expressed under the same promoter, it can be assumed that their protein levels are identical. Thus, use the sample with the eGFP-2A-mCherry only construct and calculate the mean of all eGFP-to-mCherry ratios. The mean of the ratios will be applied as scaling factor to the eGFP-to-mCherry ratios of all other samples to normalize for differences of laser intensities and detector settings between the eGFP and mCherry channels.
- Import the normalized eGFP-to-mCherry values for each sample to a column table in Prism 8. Select “Analyze” and “Frequency distribution” in order to generate a histogram of all calculated fluorescence ratios. Choose “Frequency distribution” under Create and select “XY graph” as output. Alternatively, histograms can be generated using other software packages, such as R or Python.
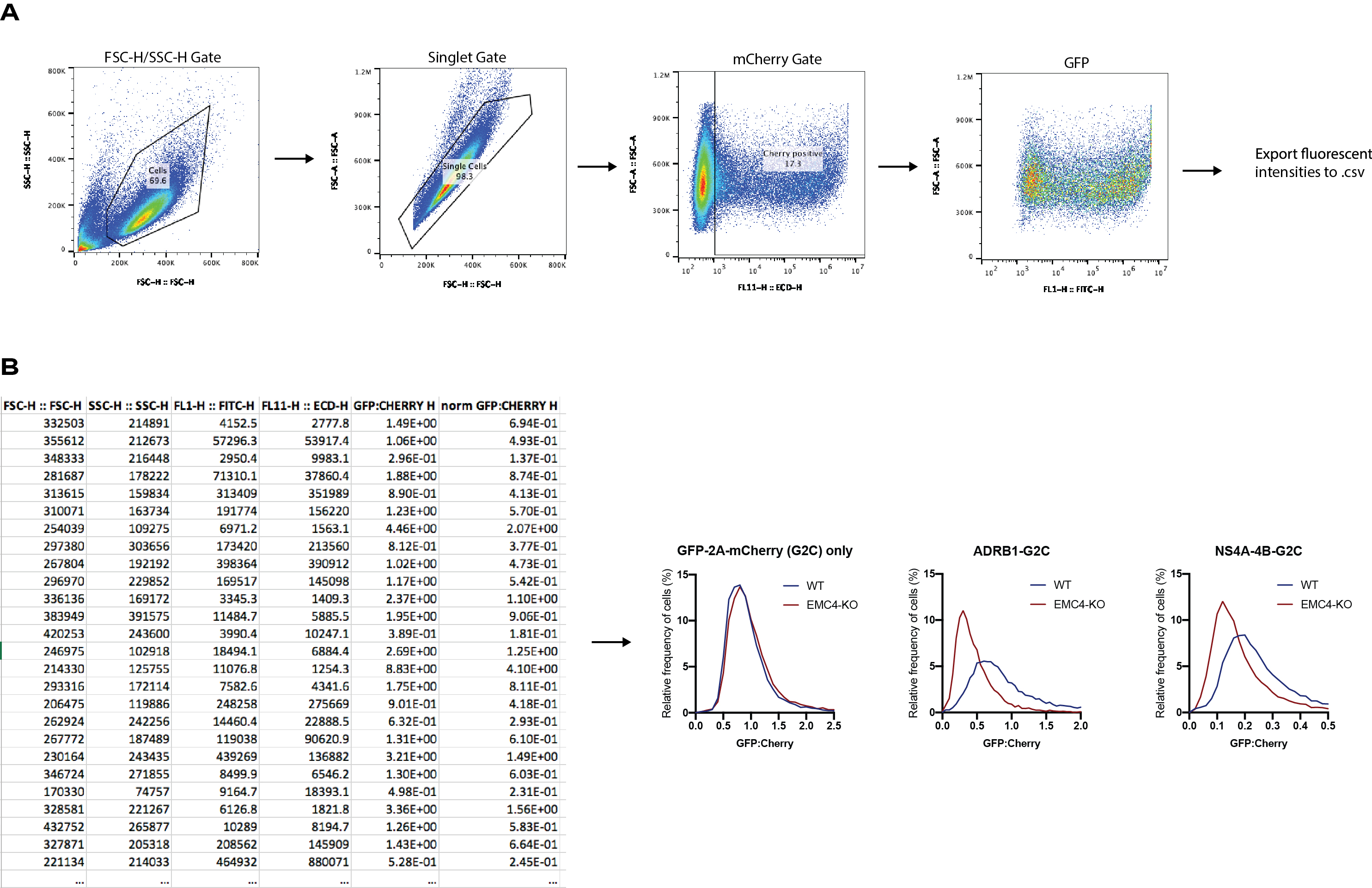
Figure 2. Example data for dual fluorescence cytometry assay. A. Gating strategy for transfected cells in FlowJo: forward and side scatter, singlets, mCherry fluorescence and eGFP fluorescence. The fluorescence intensity values for the individual mCherry-positive cells are exported for downstream analysis. B. Example of CSV table containing individual fluorescence intensity values for ADRB1-eGFP-T2A-mCherry construct transfected into wild-type 293FT cells. eGFP:mCherry ratios are calculated from intensity values and normalized by the mean of the eGFP:mCherry ratio of the eGFP-T2A-mCherry only construct. Similar tables are generated for other samples. eGFP:mCherry ratios for all samples are normalized to the mean of the ratios in the eGFP-T2A-mCherry sample. Finally, normalized ratios are imported to Prism 8 and histograms are generated. While eGFP-only levels are not affected by knockout of EMC4, ADRB1 and dengue NS4A-4B display reduced expression in cells lacking EMC4.
Recipes
- Culture medium for 293FT
500 ml Dulbecco's modified Eagle medium (DMEM)
50 ml heat-inactivated FBS
5 ml 100x Penicillin and streptomycin (final concentration 100 U/ml)
5 ml 100x L-glutamine (final concentration 2 mM)
Acknowledgments
This protocol is based on previous work and example data published in eLife (Ngo et al., 2019).
Competing interests
The authors do not have any competing interests.
References
- Chitwood, P. J., Juszkiewicz, S., Guna, A., Shao, S. and Hegde, R. S. (2018). EMC is required to initiate accurate membrane protein topogenesis. Cell 175(6): 1507-1519 e1516.
- Itakura, E., Zavodszky, E., Shao, S., Wohlever, M. L., Keenan, R. J. and Hegde, R. S. (2016). ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol Cell 63(1): 21-33.
- Ngo, A. M., Shurtleff, M. J., Popova, K. D., Kulsuptrakul, J., Weissman, J. S. and Puschnik, A. S. (2019). The ER membrane protein complex is required to ensure correct topology and stable expression of flavivirus polyproteins. Elife 8: e48469.
- Vogel, C. and Marcotte, E. M. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13(4): 227-232.
Article Information
Copyright
![]() Ngo et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Ngo et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ngo, A. M., Wang, R. and Puschnik, A. S. (2020). Dual Fluorescence Cytometry Assay to Assess Cellular Protein Levels. Bio-protocol 10(8): e3597. DOI: 10.21769/BioProtoc.3597.
- Ngo, A. M., Shurtleff, M. J., Popova, K. D., Kulsuptrakul, J., Weissman, J. S. and Puschnik, A. S. (2019). The ER membrane protein complex is required to ensure correct topology and stable expression of flavivirus polyproteins. Elife 8: e48469.
Category
Cell Biology > Cell-based analysis > Flow cytometry
Molecular Biology > Protein > Stability
Molecular Biology > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.