- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High-throughput Flow Cytometry Assay to Investigate TDP43 Splicing Function
Published: Vol 10, Iss 8, Apr 20, 2020 DOI: 10.21769/BioProtoc.3594 Views: 5814
Reviewed by: Steven BoeynaemsBede PortzEmilie Besnard
Original research article
The authors used this protocol in:

Oct 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Mutations in RNA-binding proteins (RBPs) such as TDP43 are associated with transcriptome-wide splicing defects and cause severe neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The impact of RBP mutations on splicing function is routinely studied using PCR-based bulk measurements. However, the qualitative and low-throughput nature of this assay make quantitative and systematic analyses, as well as screening approaches, difficult to implement. To overcome this hurdle, we have developed a quantitative, high-throughput flow cytometry assay to investigate TDP43 splicing function on a single-cell level
Background
RNA-binding proteins (RBPs) such as TDP43 regulate post-transcriptional gene regulation by orchestrating RNA stability, transport and processing, including mRNA splicing (Gerstberger et al., 2014). Impairment of RBP function as a result of mutations and/or aggregation has been implicated in the etiology of many neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Harrison and Shorter, 2017). Notably, ALS-associated mutations in RBPs like TDP43 can cause transcriptome-wide splicing defects, suggesting that misregulation of RBP splicing function may be a key driver of disease (Arnold et al., 2013; Sun et al., 2015). Thus, an important goal in ALS research is to uncover the molecular mechanisms of RBP function, requiring experimental approaches for the systematic interrogation of RBP splicing function.
Widely-used methods for studying splicing are reverse transcription-PCR (RT-PCR) and RNA sequencing (RNA-seq). Both methods can be used to study the splicing of endogenous transcripts; however, typically only as a bulk measurement of an entire population of cells. RT-PCR is simple and affordable, yet efficient only to study splicing of a select few endogenous transcripts. Moreover, RT-PCR read-outs are often only semi-quantitative. In contrast, RNA-seq provides quantitative insights into global splicing, but is time- and cost-intensive and requires sophisticated data analysis pipelines (Wang et al., 2009). Thus, both RT-PCR and RNA-seq are poorly suited for high-throughput applications, for example to systematically compare the splicing function of multiple RBP variants.
A powerful tool to study splicing are minigenes, which typically are plasmid-based, simplified genes containing introns and exons that recapitulate defined splicing events outside of the native gene context (Baralle and Baralle, 2005). In particular, minigenes have been instrumental in discovering cis- and trans-acting splicing elements (Cooper, 2005). Instead of endogenous targets, minigenes are also routinely used to streamline investigation of RBP splicing function (D'Ambrogio et al., 2009; Kino et al., 2011). Conventionally, the splicing of minigenes is evaluated with analytical or quantitative RT-PCR. However, the low-throughput and bulk read-out of this method precludes leveraging the full potential of minigene-based splicing reporters.
To overcome these hurdles, we developed a minigene that encodes for a fluorescent protein (FP minigene), thus allowing us to convert a defined splicing event into a sensitive, inherently quantitative and readily detectable signal on a single-cell level by flow cytometry (Schmidt et al., 2019). To normalize splicing of the FP minigene to the overall abundance of the parent transcript, we fused it to another, intron-free fluorescent protein (constitutive FP). While this approach is broadly applicable (Gurskaya et al., 2012; Sorenson and Stevens, 2014; Gonatopoulos-Pournatzis et al., 2018), we adapted it specifically to study TDP43-dependent splicing by interrupting the FP minigene with exon 9 of the CFTR gene, which is skipped only in the presence of functional TDP43 (Buratti et al., 2001). To rapidly compare the splicing efficiency of multiple TDP43 variants in TARDBP (i.e., the gene encoding TDP43) knock-out cells, we used a bidirectional promoter to drive the coordinated expression of both our splicing reporter and a given BFP-tagged TDP43 variant from the same plasmid. A graphical overview of our splicing reporter design in shown in Figure 1.
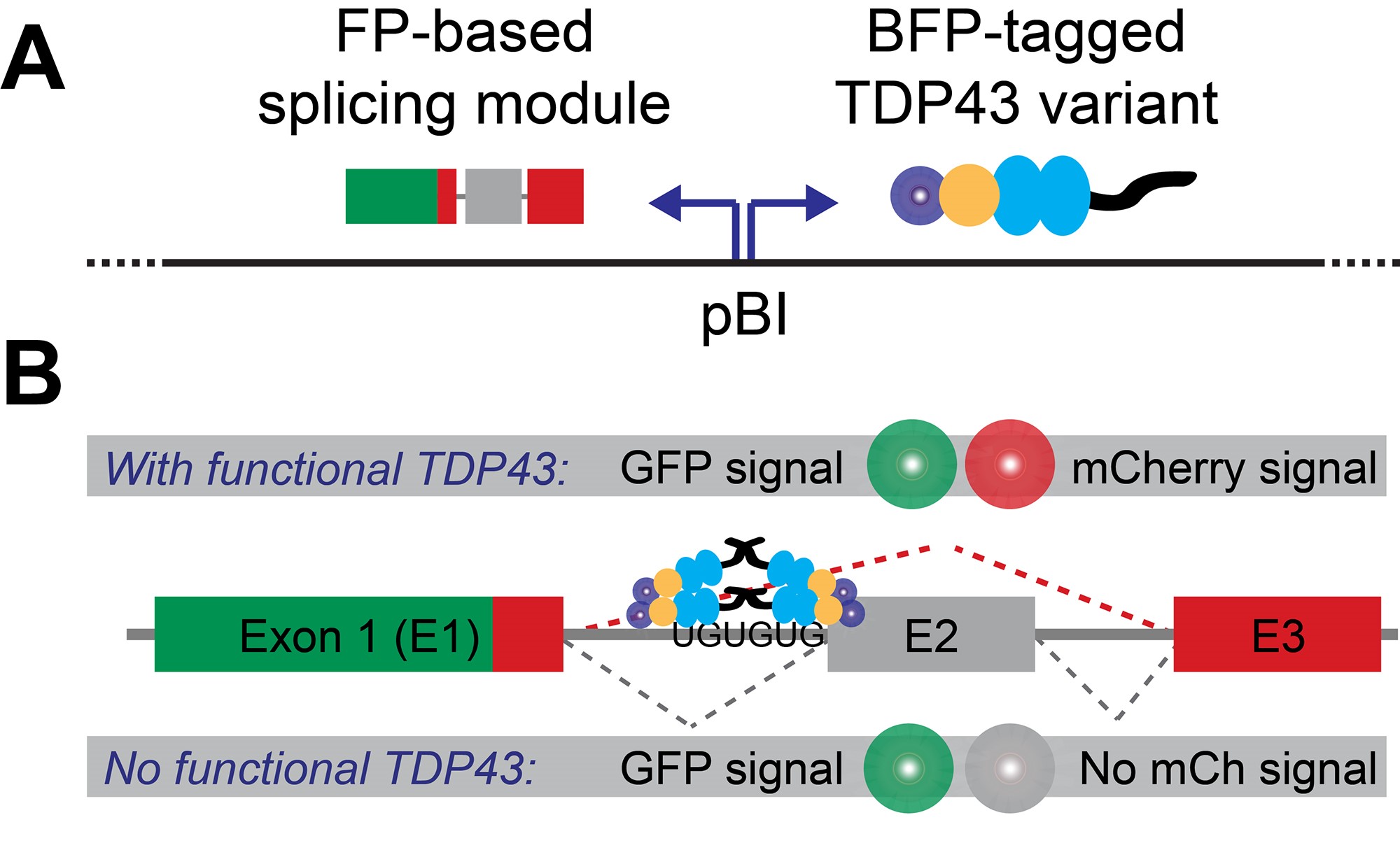
Figure 1. Splicing reporter design. A. The fluorescent protein (FP)-based splicing module and BFP-tagged TDP43 variants are expressed from the same plasmid via a bi-directional promoter (pBI). B. Production of a dually-fluorescent GFP-mCherry fusion protein depends on the presence of functional TDP43, which is required to mediate the skipping of CFTR gene-derived exon (labeled E2).
Here, we describe in detail how to perform and analyze our flow cytometry-based splicing assay. This protocol is optimized for use with HEK-293T wild-type and HEK-293T TARDBP knock-out cells (Schmidt et al., 2019), yet in principle suitable for other recently published constitutive or inducible TARDBP knock-out cell lines as well (Chiang et al., 2010; Gasset-Rosa et al., 2019; Roczniak-Ferguson and Ferguson, 2019).
Materials and Reagents
- Cell lines
- HEK-293T wild-type cells (ATCC, catalog number: CRL-3216 )
- HEK-293T TARDBP knock-out cells (available upon request)
- Cell culture
- 1.5 ml microtubes (Eppendorf, catalog number: 0 22363204 )
- 60 µm scepter tips for cell counting (Milipore-Sigma, catalog number: PHCC60050 )
- 10 cm tissue culture plates for propagating cell lines (Thermo Scientific, catalog number: 12-556-002 )
- 6-well tissue culture plates for splicing assays (Thermo Scientific, catalog number: 14-832-11 )
- 70 µm cell strainer (Corning, Falcon, catalog number: 08-771-2 )
- 5 ml round-bottom polystyrene tubes (Corning, Falcon, catalog number: 14-959-1A )
- SH800S flow cytometer setup beads (Sony Biotechnology, catalog number: LE-B3001 )
- HyClone DMEM high glucose (GE Healthcare, catalog number: SH30081FS ) supplemented with:
- 10% FBS (Atlanta Biologicals, catalog number: S11150H )
- 1 mM sodium pyruvate (Gibco, catalog number: 11-360-070 )
- 2 mM L-glutamine (Gemini Biosciences, catalog number: 400106 )
- 1x MEM non-essential amino acids (Gibco, catalog number: 11-140-076 )
- 40 U/ml penicillin and 40 µg/ml streptomycin (Gemini Biosciences, catalog number: 400109 )
- Sterile 1x PBS at pH 7.4 (Sigma-Aldrich, catalog number: P3813-5x10PAK )
- Trypsin-EDTA (Gemini Biosciences, catalog number: 400150 )
- X-tremeGENE9 transfection reagent (Roche, catalog number: 0 6365787001 )
- OptiMEM (Gibco, catalog number: 31985-070 )
- Plasmids (available at https://www.addgene.org/Rajat_Rohatgi/, unless otherwise noted)
- Splicing reporter only: pHBS1389 IBB-GFP-mCherry3E (Addgene, catalog number: 118803 )
- Positive control: pHBS1503 [IBB-GFP-mCherry3E]-[BFP-TDP43 WT] (Addgene, catalog number: 133327 )
- Negative control: pHBS1501 [IBB-GFP-mCherry3E]-[BFP-TDP43 ∆RRM] (Addgene, catalog number: 133325 )
- Extensive collection of TDP43 CTD mutants in the splicing reporter (e.g., Addgene, catalog numbers: 133328-133333 )
- Backbone for subcloning of CTD mutants into splicing reporter: pHBS1224 [IBB-GFP-mCherry3E]-[BFP-TDP43 ∆CTD EcoRV] (available upon request)
- Compensation probes: mammalian expression vectors with BFP, GFP and mCherry inserts (e.g., Addgene, catalog numbers: 54665 , 54759 and 54563 )
Equipment
- Single-channel pipettes (Rainin or equivalent)
- Cell culture incubator (Thermo Scientific or equivalent)
- Scepter cell counter (Milipore-Sigma) or equivalent manual/automatic cell counter
- Sony SH800S cell sorter (Sony Biotechnology) or equivalent cell sorter equipped with 405 nm, 488 nm and 561 nm lasers, as well as appropriate filters
- Computer for data analysis (e.g., 3.1 GHz Intel i7 processor with 16 GB RAM)
Software
- SH800S operating software for data collection (Sony Biotechnology) or operating software for equivalent flow cytometers
- Mathematica Version 11.3 or higher (Wolfram Research) + custom data analysis pipelines:
- Script ‘CSA_Gating.nb’ (available at https://github.com/RohatgiLab/TDP43-analysis)
- Script ‘CSA_Plotting.nb’ (available at https://github.com/RohatgiLab/TDP43-analysis)
Procedure
- Seeding of cells
Note: Work in a tissue culture hood to prevent contaminations. Culture all HEK-293T wild-type and TARDBP knock-out cells in DMEM high-glucose plus supplements (see Materials).- Harvest HEK-293T wild-type and TARDBP knock-out cells by trypsinization and determine cell numbers using the Scepter (or equivalent manual/automatic cell counters).
- For setup and calibration of the flow cytometer, seed four wells of a standard six-well tissue culture plate with 400,000 HEK-293T wild-type cells each (compensation samples).
- For positive and negative controls, seed another four wells of a standard six-well tissue culture plate with 400,000 HEK-293T TARDBP knock-out cells each (control samples).
- For each TDP43 variant to be tested, seed two wells of a standard six-well tissue culture plate with 400,000 HEK-293T TARDBP knock-out cells each (experimental samples).
- Let the cells adhere overnight in a cell culture incubator at 37 °C and 5% CO2.
- Transfection of cells
General workflow: Work in a tissue culture hood to prevent contaminations. Per transfection, carefully add 3 µl of X-tremeGENE9 transfection to 250 µl OptiMEM without touching the walls of the microtube and incubate for 20 minutes at room temperature. Per transfection, mix 1 µg of reporter plasmid with 250 µl OptiMEM in a separate microtube. After incubation, combine the reactions and carefully spread the mix dropwise across the cells in a given well.- To prepare the compensation samples, transfect one well of HEK-293T cells with 1 µg of either BFP, GFP or mCherry mammalian expression constructs. Note that one well will purposely remain untransfected.
- To prepare control samples, transfect two wells of HEK-293T TARDBP knock-out cells with 1 µg each of either pHBS1501 or pHBS1503.
- To prepare experimental samples, transfect two wells of HEK-293T TARDBP knock-out cells with 1 µg each of the desired construct. A reference collection of TDP43 CTD mutants in the splicing reporter background is available at https://www.addgene.org/Rajat_Rohatgi/.
- Incubate all samples for 24 h in a cell culture incubator at 37 °C and 5% CO2.
- Flow cytometer setup and calibration
Note: Only a basic outline of the SH800S setup and calibration procedure is given below. Please consult the manual or contact the technical service for further questions and troubleshooting.- Before starting the flow cytometer, check the sheath fluid and ddH2O levels (refill if necessary) and empty the waste.
- Make sure that the sheath fluid container and lines are sealed, then turn on the compressor.
- Turn on the SH800S flow cytometer and start the operating software.
- Follow the prompts during the chip loading and fluidics check steps. We use 100 µm chips for analyzing HEK-293T cells. Select the 405 nm, 488 nm and 561 nm lasers when prompted.
- Follow the prompts during the chip alignment step. Sort calibration is not required, as analyzer mode is sufficient.
- During setup, harvest the four compensation samples by trypsinization. Make sure to keep the sample volumes low to prevent unnecessary dilution of the cell suspensions (we typically use 250 µl trypsin per well and quench by adding 250 µl complete DMEM). Filter each sample through a cell strainer into a 5 ml round-bottom polystyrene tube. Keep tubes on ice until further use.
- Once setup is finished, create a new experiment in the operating software. In the measurement settings pane, select only the FSC (forward scatter), BSC (side scatter), FL1 (BFP), FL2 (GFP) and FL3 (mCherry) channels. When prompted, start the compensation wizard and follow the steps. The compensation samples will be needed at this point.
- Upon completion of the compensation step, setup is finished and the remaining samples can be prepared.
- Prepare and analyze samples by flow cytometry
- Harvest the control and experimental samples by trypsinization as described in Step C6. Filter each sample through a cell strainer into a 5 ml round-bottom polystyrene tube. Keep samples on ice until further use.
- Start by analyzing the control samples to set the gates and get an idea of the expected effect sizes.
- In the BSC-A vs. FSC-A density plot, ensure that the gate for the cell population set during compensation still applies (Gate 1; Figure 2A).
- In Gate 1, create a FSC-H vs. FSC-A density plot to further gate for single cells (Gate 2; Figure 2B). This filter makes use of disproportions between the area and height of cell clumps compared to single cells (Hazen et al., 2018).
- In Gate 2, create FL1-A (BFP), FL2-A (GFP) and FL3-A (mCherry) histogram plots. The BFP signal is proportional to the total TDP43 amount. The GFP signal scales with total splicing reporter transcript levels, whereas the mCherry signal reflects TDP43 splicing activity.
- To get a preview of TDP43 splicing efficiency, first gate for BFP-positive cells in the FL1-A histogram (Gate 3; Figure 2C). This step is to select for successfully transfected cells only. The transfection efficiency (i.e., fraction of BFP-positive cells) should be 50% or greater. Then in Gate 3, create a FL2-A (GFP) vs. FL3-A (mCherry) density plot. There should be a significant difference in the position of the positive and negative control populations in this plot (Figure 2D).
Note: These gates are only for data preview, but not final data analysis. - Collect at least 50,000 events in Gate 3 for each control and experimental sample. Make sure to hit the ‘Record’ button to ensure that data is saved during data collection. As a reference, we recommend to take a screen shot of the above specified plots for each sample after data collection. Unfortunately, this has to be done manually for each sample. Right-click on the worksheet area to bring up the quick access menu and select ‘Copy Worksheet Picture’. Paste the picture into a simple image processor such as Microsoft Paint and save.
- After all samples have been analyzed, export the data as an fcs-file by right-clicking on the experiment in the ‘Active Experiments’ panel.
- Shut-down the flow cytometer and software following the instructions of the shutdown wizard, which can be found in the ‘Cytometer’ tab.
- After the instrument shut-down, turn-off the compressor and release the pressure from the sheath tank.
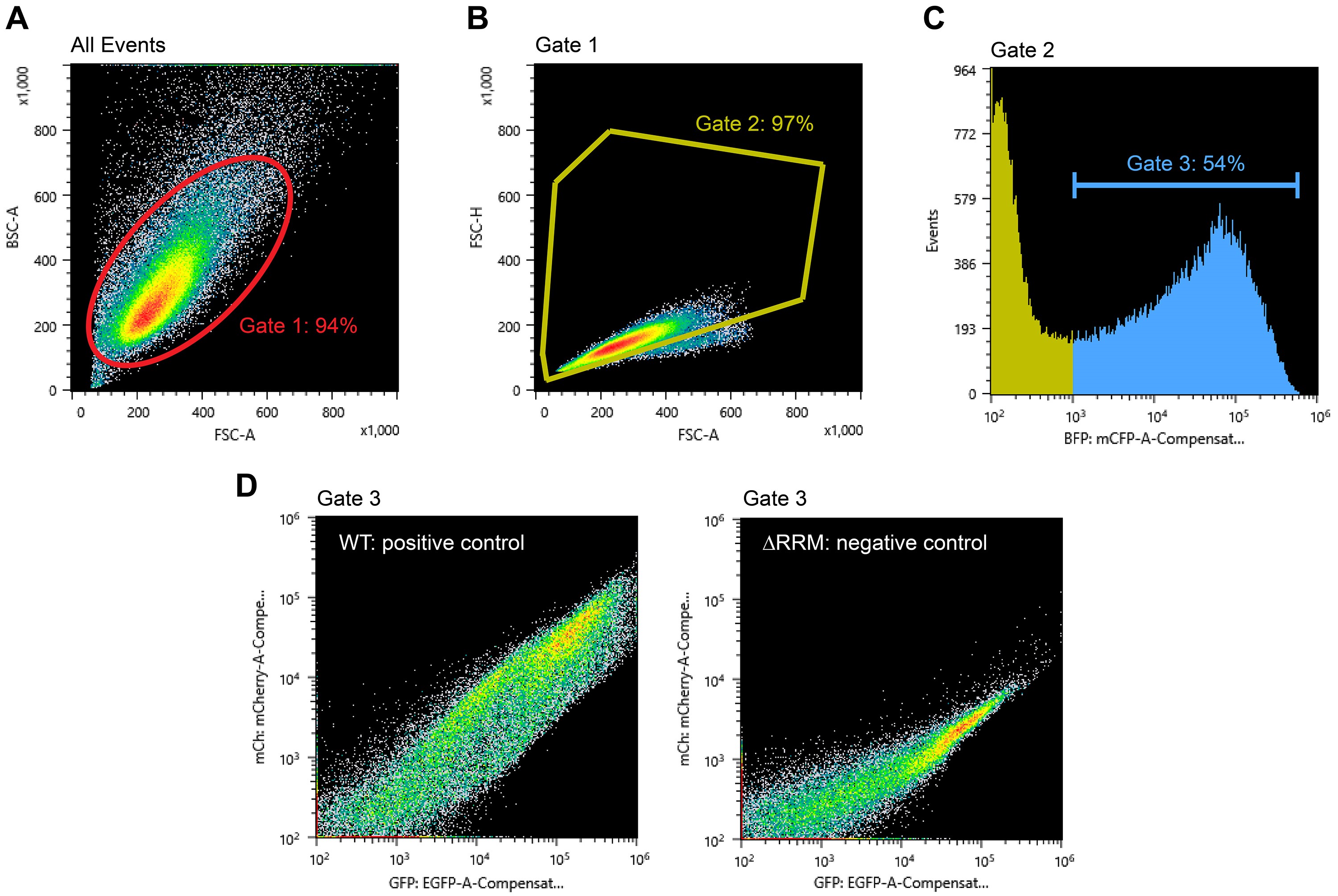
Figure 2. Example data and gating strategy during data acquisition. A. BSC-A vs. FSC-A density plot showing all detected events and Gate 1 that selects for cells. B. FSC-H vs. FSC-A density plot and Gate 2 to select for single cells. C. FL1-A (BFP) histogram and Gate 3 to select for transfected cells. D. FL3-A (mCherry) vs. FL2-A (GFP) density plots for full-length, wild-type (WT) TDP43 and TDP43 lacking its RNA-binding domains (∆RRM).
Data analysis
Note: The data analysis pipeline outlined below uses Mathematica scripts that have been written specifically to analyze fcs-files generated by the Sony SH800 software and may not be immediately compatible with fcs-files from other machines without tweaking the code.
For analysis of the splicing data, the mCherry signal of each cell is normalized to the GFP signal of the same cell to quantify splicing efficiency (Figure 3A). To further normalize to the TDP43 level of that cell, the splicing efficiency is plotted against the BFP signal. We have developed Mathematica scripts (available at https://github.com/RohatgiLab/TDP43-analysis) that extract the raw measurements for each observation from standard fcs-files, gate for cells and calculate the splicing ratio (script ‘CSA_Gating.nb), as well as plot the data (‘CSA_Plotting.nb’).
First open the ‘CSA_Gating.nb’ script in Mathematica. Under ‘Step 0’, the gates for the analysis are specified (Figure 3B). Try the pre-defined values or refer to the reference screen shots to set appropriate boundary conditions. Confirm by evaluating the cell. In ‘Step 1’, the location of the data files on the hard drive is specified. To the parameter ‘RandomFile’, attribute the file path to a random file in the folder containing the fcs-files from the experiment (Figure 3C). Evaluate the cell to confirm; a list of all fcs-files found will be returned. Finally, evaluate the cell titled ‘Step 2’. In this analysis loop, the data will be loaded, gated and analyzed (Figure 3D). For each sample, a summary of the analysis as well as csv-files with the raw numerical values will be exported.
For further analysis and plotting of the data, open the ‘CSA_Plotting.nb’ script in Mathematica. Start by attributing the file path to the csv-files from the above analysis to the parameter ‘RandomFile’ (Figure 3E) and evaluate the cell to confirm. Then evaluate the next cell to re-format the data for further analysis. To plot TDP43 splicing efficiency, expressed as the ratio of the mCherry-to-GFP signals, versus the total cellular TDP43 concentration (BFP signal), evaluate the cell labeled ‘Plot the data!’. To change the axis of the plots, update the ‘yScale’, ‘xStart’ and ‘xEnd’ parameters (Figure 3F), then re-evaluate the cell. This step generates both individual plots for each sample that is automatically rendered and saved (see example in Figure 3G). The last two cells in the script allow the comparison of TDP43 splicing efficiency in a range of TDP43 levels defined by the parameters ‘start’ and ‘end’, generating a violin plot (Figure 3G). To manually re-arrange the order of the samples in the plot, specify the desired sample order in the list termed ‘OrderedData’ and evaluate the cell (Figure 3H).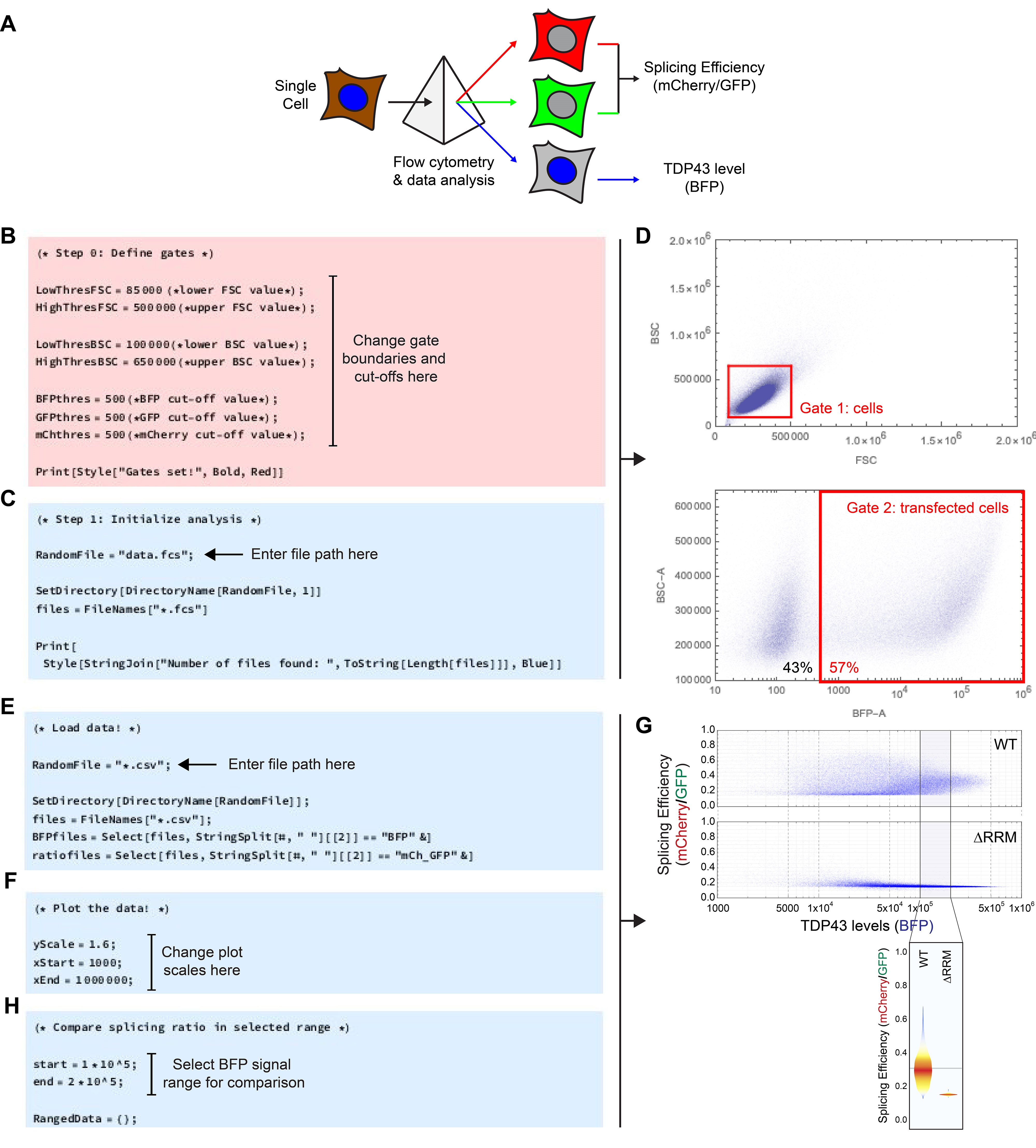
Figure 3. Splicing assay data analysis and plotting. A. Outline of the analysis pipeline. A flow cytometer is used to measure and deconvolute the mCherry, GFP and BFP fluorescence signals of individual cells in a population. Using custom scripts, the splicing efficiency is calculated by taking the ratio of the mCherry to GFP signal of a given cell (Figure 1) and compared to the TDP43 level of the same cell (BFP signal). B. Screenshot highlighting where to change the gate boundaries and cut-offs in the ‘CSA_Gating.nb’ script. C. Screenshot indicating where to provide the file path to the fcs raw data in the ‘CSA_Gating.nb’ script. D. Example density plots generated during evaluation of the ‘CSA_Gating.nb’ script to visualize the gates defined for analysis. E. Screenshot illustrating where in the ‘CSA_Plotting.nb’ script to provide the file path to the csv files generated during evaluation of the ‘CSA_Gating.nb’ script. F. Screenshot highlighting where to adjust the plot scales in the ‘CSA_Plotting.nb’ script. G. Example density plot generated by the ‘CSA_Plotting.nb’ script that depicts splicing efficiency vs. TDP43 levels for wild-type (WT) and ∆RRM TDP43 variants (top) and example violin plot highlighting the splicing efficiencies in the defined BFP signal range (bottom). H. Screenshot of the ‘CSA_Plotting.nb’ script indicating where to define the BFP signal range for the violin plots comparing splicing efficiency.
Acknowledgments
This work has been supported by the Deutsche Forschungsgemeinschaft (SCHM 3082/2-1 to H.B.S.) and the National Institutes of Health (DP2 GM105448 and R35 GM118082 to R. R.). The original protocol was published in Schmidt et al. (2019).
Competing interests
The authors declare no competing interests.
References
- Arnold, E. S., Ling, S. C., Huelga, S. C., Lagier-Tourenne, C., Polymenidou, M., Ditsworth, D., Kordasiewicz, H. B., McAlonis-Downes, M., Platoshyn, O., Parone, P. A., Da Cruz, S., Clutario, K. M., Swing, D., Tessarollo, L., Marsala, M., Shaw, C. E., Yeo, G. W. and Cleveland, D. W. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A 110(8): E736-745.
- Baralle, D. and Baralle, M. (2005). Splicing in action: assessing disease causing sequence changes. J Med Genet 42(10): 737-748.
- Buratti, E., Dörk, T., Zuccato, E., Pagani, F., Romano, M. and Baralle, F. E. (2001). Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20(7): 1774-1784.
- Chiang, P. M., Ling, J., Jeong, Y. H., Price, D. L., Aja, S. M. and Wong, P. C. (2010). Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A 107(37): 16320-16324.
- Cooper, T. A. (2005). Use of minigene systems to dissect alternative splicing elements. Methods 37(4): 331-340.
- D'Ambrogio, A., Buratti, E., Stuani, C., Guarnaccia, C., Romano, M., Ayala, Y. M. and Baralle, F. E. (2009). Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res 37(12): 4116-4126.
- Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., Shorter, J., Da Cruz, S. and Cleveland, D. W. (2019). Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102(2): 339-357 e337.
- Gerstberger, S., Hafner, M. and Tuschl, T. (2014). A census of human RNA-binding proteins. Nat Rev Genet 15(12): 829-845.
- Gonatopoulos-Pournatzis, T., Wu, M., Braunschweig, U., Roth, J., Han, H., Best, A. J., Raj, B., Aregger, M., O'Hanlon, D., Ellis, J. D., Calarco, J. A., Moffat, J., Gingras, A. C. and Blencowe, B. J. (2018). Genome-wide CRISPR-Cas9 interrogation of splicing networks reveals a mechanism for recognition of autism-misregulated neuronal microexons. Mol Cell 72(3): 510-524 e512.
- Gurskaya, N. G., Staroverov, D. B., Zhang, L., Fradkov, A. F., Markina, N. M., Pereverzev, A. P. and Lukyanov, K. A. (2012). Analysis of alternative splicing of cassette exons at single-cell level using two fluorescent proteins. Nucleic Acids Res 40(8): e57.
- Harrison, A. F. and Shorter, J. (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem J 474(8): 1417-1438.
- Hazen, A. L., Bushnell, T. and Haviland, D. L. (2018). The importance of area scaling with FACS DIVA software. Methods 134-135: 130-135.
- Kino, Y., Washizu, C., Aquilanti, E., Okuno, M., Kurosawa, M., Yamada, M., Doi, H. and Nukina, N. (2011). Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res 39(7): 2781-2798.
- Roczniak-Ferguson, A. and Ferguson, S. M. (2019). Pleiotropic requirements for human TDP-43 in the regulation of cell and organelle homeostasis. Life Sci Alliance 2(5).
- Schmidt, H. B., Barreau, A. and Rohatgi, R. (2019). Phase separation-deficient TDP43 remains functional in splicing. Nat Commun 10(1): 4890.
- Sorenson, M. R. and Stevens, S. W. (2014). Rapid identification of mRNA processing defects with a novel single-cell yeast reporter. RNA 20(5): 732-745.
- Sun, S., Ling, S. C., Qiu, J., Albuquerque, C. P., Zhou, Y., Tokunaga, S., Li, H., Qiu, H., Bui, A., Yeo, G. W., Huang, E. J., Eggan, K., Zhou, H., Fu, X. D., Lagier-Tourenne, C. and Cleveland, D. W. (2015). ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 6: 6171.
- Wang, Z., Gerstein, M. and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1): 57-63.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Schmidt, H. B. and Rohatgi, R. (2020). High-throughput Flow Cytometry Assay to Investigate TDP43 Splicing Function. Bio-protocol 10(8): e3594. DOI: 10.21769/BioProtoc.3594.
Category
Molecular Biology > RNA > RNA splicing
Neuroscience > Nervous system disorders > Neurodegeneration
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.