- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Real-time Fluorescence Measurement of Enterovirus Uncoating


Published: Vol 10, Iss 7, Apr 5, 2020 DOI: 10.21769/BioProtoc.3582 Views: 4853
Reviewed by: David PaulMoona HuttunenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Viruses need to open, i.e., uncoat, in order to release their genomes for efficient replication and translation. Especially for non-enveloped viruses, such as enteroviruses, the cues leading to uncoating are less well known. The status of the virus has previously been observed mainly by transmission electron microscopy using negative staining, cryo electron microscopy, X-ray crystallography or gradient separation (reviewed in Tuthill et al., 2010, Myllynen et al., 2016, Ruokolainen et al., 2019). However, monitoring of uncoating has been limited by the lack of methods detecting dynamic changes of the virions. Here, we present a real-time fluorescence based protocol, which detects the viral genome (RNA) during various stages of uncoating in vitro, while RNA is still inside the particle that has been expanded before the actual RNA release, and when the RNA has been totally released from the viral particle. Our method allows to explore how various molecular factors may promote or inhibit virus uncoating.
Background
In our previous study, we found that infectious intermediate echovirus 1 particle allows SYBR Green II, a RNA intercalating dye, to enter the virus particle (Myllynen et al., 2016). This can be observed as an increase of fluorescence and the recorded fluorescence is not susceptible to RNase digestion (Myllynen et al., 2016). Using this information, we developed a real-time method to monitor virus opening using the SYBR Green II dye and RNase in fluorescence spectroscopy. We could follow the fenestration of the particles in real-time at +37 °C, or other temperature of interest, in a 96-well plate format by adding SYBR Green II and factors triggering the uncoating, and observing the increase of SYBR Green II fluorescence. Addition of RNase into parallel wells allowed us to monitor the extent of RNA release from the virions, as RNase readily degrades RNA from the solution, but not from inside of the virion (RNAse cannot enter through the small fenestrations inside to the virus particle, Myllynen et al., 2016). In case of intact virus particles, only very low amount of fluorescence was observed. As an example, in our previous study, a DPBS solution supplemented with 0.01% fatty acid free BSA produced high amounts of intermediate echovirus 1 particles. For more details see the original publication (Ruokolainen et al., 2019).
Materials and Reagents
- Pipette tips
- Sarstedt 96-well plate (Sarstedt, catalog number: 83.3924 ) (or similar)
- 1.5 ml tubes
- Purified virus stock (1 μg of virus per measured well; stored at -80 °C)
Purification of enteroviruses can be done using either 5-20% or 10-40% sucrose gradient and is described in detail in the original publication (Ruokolainen et al., 2019). Also, CsCl purification may be used but it was observed by us to have more variation from batch-to-batch than sucrose purified virus. The protocol has been tested and observed to work with echovirus 1 and coxsackieviruses A9 and B3 suggesting its wide applicability for enteroviruses and probably for picornaviruses in general. - Buffer/solution of your interest
In our paper we used a wide spectrum of concentrations of different ions and albumin to study the virus priming and opening. As an example, DPBS solution supplemented with 0.01% BSA resulted in high amount of intermediate virus particles. - SYBR Green II RNA gel stain (Invitrogen; ThermoFisher Scientific, catalog number S7564 ; stored at -20 °C)
- RNase A, 10 mg/ml (ThermoFisher Scientific catalog number EN0531 ; stored at -20 °C)
- 150 mM NaCl solution
- Ice
Equipment
- Pipette with a volume range including 100 µl
- 8-channel multipipette with a volume range including 50 µl, or similar (optional)
- Perkin Elmer 2030 Multilabel Reader Victor X4 (or similar fluorescence plate reader with suitable filter options)
Software
- Perkin Elmer 2030 Manager
- Microsoft Excel
Procedure
- Sample preparation
- Place the 96-well plate on ice to cool down.
Note: You can use a metal plate on top of ice to make the surface even and the plate easy to handle. - Cool down the solution(s) of interest on ice.
- Make 1:10 dilution from the 10,000x SYBR Green II stock solution. You can use ddH2O or the solution of interest to make the dilution. If the solution of interest is very exactly defined, then preferably use it for the dilution to keep the conditions unchanged (as ddH2O will change/dilute the solution). Avoid exposing the SYBR Green II to bright light.
- Pipette 450 µl of the solution of interest into two 1.5 ml tubes. You can have many different solutions of interest in one 96-well plate measurement. i.e., many pairs of 1.5 ml tubes. Up to seven different solutions of interest can be applied into one 96-well plate (see Figure 1 for example).
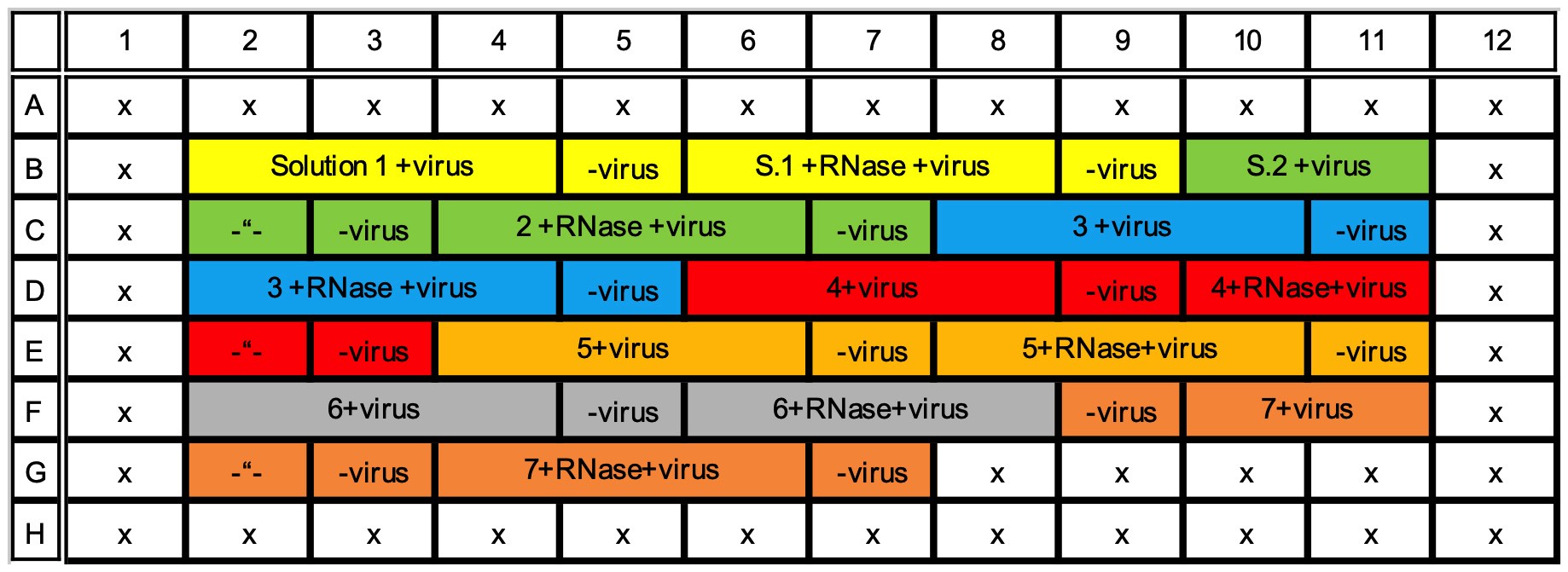
Figure 1. Example of 96-well plate with 7 different solutions of interest (highlighted with different colors) and three replicates of virus with a background control without virus, all measured with and without RNase. Rows A and H, and columns 1 and 12 are left empty. - Add 4.5 µl of the previously diluted SYBR Green II into both 1.5 ml tubes.
- Add 4.5 µl of the RNase A into one of the 1.5 ml tube in a final concentration of 10 µg/ml. Preferably use high concentrated stock to negate the buffer effect to the solution of interest. We use pre-diluted 1 mg/ml RNase stock in 150 mM NaCl, i.e., 1 µl of RNase in 100 µl of solution of interest. Add 4.5 µl of 150 mM NaCl solution without RNase A into the other tube to keep the ion concentrations identical.
- Mix the 1.5 ml tubes well and pipette 100 µl into four adjacent wells of the 96-well plate. Keep the plate on ice. Do not use rows A and H and columns 1 and 12 since the proximity of the plate edge might affect the measurement. See Figure 1 for an example. Avoid bubbles. Easiest way to avoid them is to use reverse pipetting for mixing and placing the liquid into the wells. In reverse pipetting, press the pipette cylinder all the way down before taking the adjusted volume inside the tip, resulting in extra volume inside the tip when mixing the solution. Then the volume only up to the first step is pipetted out in the well before sucking in the next volume etc. (For further details, see for example Good Laboratory Pipetting Guide https://assets.thermofisher.com/TFS-Assets/LSG/brochures/D16542.pdf.)
- Add 1 µg of virus into each of the three first wells of a solution and leave the fourth well without the virus to measure the background fluorescence of the solution. Somewhat lower virus amount, at least down to 0.5 µg, may also be applicable. However, if the solution of interest contains molecules that cause background fluorescence (such as BSA in our case) the signal-to-noise ratio might be low and the results less reliable. Also, preferably use a concentrated virus stock so that adding of the virus in a buffer will not change the chemical concentrations too much. On the other hand, too concentrated stock may add to the pipetting error. Optimal virus stock concentration would be around 1 µg/µl, i.e., 1 µl of the virus stock would be added per well.
- Mix the wells. Avoid bubbles. Easiest way is to use 8-channel multipipette and reverse pipetting.
- Keep the plate on ice, cover with aluminium folio and transfer to the fluorescence plate reader.
- Sample preparation in short:
- Take 450 µl of solution of interest in two 1.5 ml tubes, tube A and tube B. Keep the tubes on ice.
- Add 4.5 µl of pre-diluted 1,000x SYBR Green II in both tubes A and B.
- Add 4.5 µl of RNase A from 1 mg/ml stock only into tube B to a final concentration of 10 µg/ml.
- Mix both tubes carefully and transfer 100 µl of the solution into four adjacent wells from each tube, i.e., 8 wells in total.
- Pipette 1 µg of virus into 3 first wells transferred from tube A and leave the fourth well without virus. Do the same for the wells transferred from tube B that has the RNase A.
- Mix well and avoid air bubbles. Protect from light, keep on ice and transfer to the measurement device.
- Place the 96-well plate on ice to cool down.
- Fluorescence measurement
- Prepare a measuring protocol using appropriate filters to measure SYBR Green II. With the Perkin Elmer 2030 Multilabel Reader Victor X4 use CW-Lamp Filter F485 and emission filter F535. Use CW-Lamp energy of 14,592 (or similar value around 15,000) and counting time of 1 second. Use measurement height with “user defined” -option of 13 mm and Plate type of “Generic 8x12 size plate”. Save the protocol for later use.
- Mark the wells to measure, measure each plate 90 times (Using 90 repeats and two-minute gap between the repeats results in total of 3-hour measurement. To use some other total measuring time adjust the field of “measure each plate” accordingly) and delay between repeats 0 s. Pre-run the protocol and time how long one round of measurements takes. To measure each well every two min, subtract the time taken by one round of measurements from 120 s and place the result into “delay between repeats” -field. For example, if the time to measure the wells in your experiment takes 30 s, subtract this from 120 s and place the resulting 90 s into “delay between repeats”. In this way the plate reader waits 90 s after the first round of measurements before starting to take the next ones and, as a result, every well is measured exactly every 2 min. Note, that when changing the amount of wells in your measurement, the “delay between repeats” needs to be adjusted accordingly.
- Pre-heat the plate reader to +37 °C or to any other temperature you wish to measure in. The plate reader takes some time to heat up, so do this before starting to prepare the samples and the plate.
- Place the previously prepared plate inside the plate reader with the lid on, but without the aluminium folio.
- Start the protocol.
- Data handling
- Export the data into Excel.
- Subtract the fluorescence background of each solution at each time point from the fluorescence of the same solution with the virus. Do this for all three replicates.
- Calculate average of the three replicates. Calculate the standard deviation and the standard error of the mean.
- Plot the data into a graph.
Data analysis
After plotting the graphs, first of all the results with the same solution with and without RNase should be compared. Fluorescence without the RNase presents the fluorescence originating from the empty and expanded particles. Fluorescence with the RNase presents the fluorescence originating from the expanded particles and the difference between the conditions with and without RNase originates from the empty capsids. Also, a measurement in virus storage buffer, or in another stabile environment for the virus, should be performed to verify the stability status of the unmodified virus, which should result in low amount of fluorescence. After comparing the measurements with and without RNase in one solution, one can also compare the results between different solutions. At least the proportion of different virus states can be compared. See Figure 2 as an example of measurements monitoring stable virus (only small increase in fluorescence), virus priming (increased fluorescence and only small decrease of signal with RNase treatment) and virus opening (increased fluorescence but loss of fluorescence after RNase addition).
When comparing the absolute fluorescence values, one must remember that the fluorescence potency of SYBR Green II might be somewhat affected by the surrounding conditions. In order to verify the proportional share of different states of the virions, other methods are recommended such as negative staining with TEM or cryo-EM.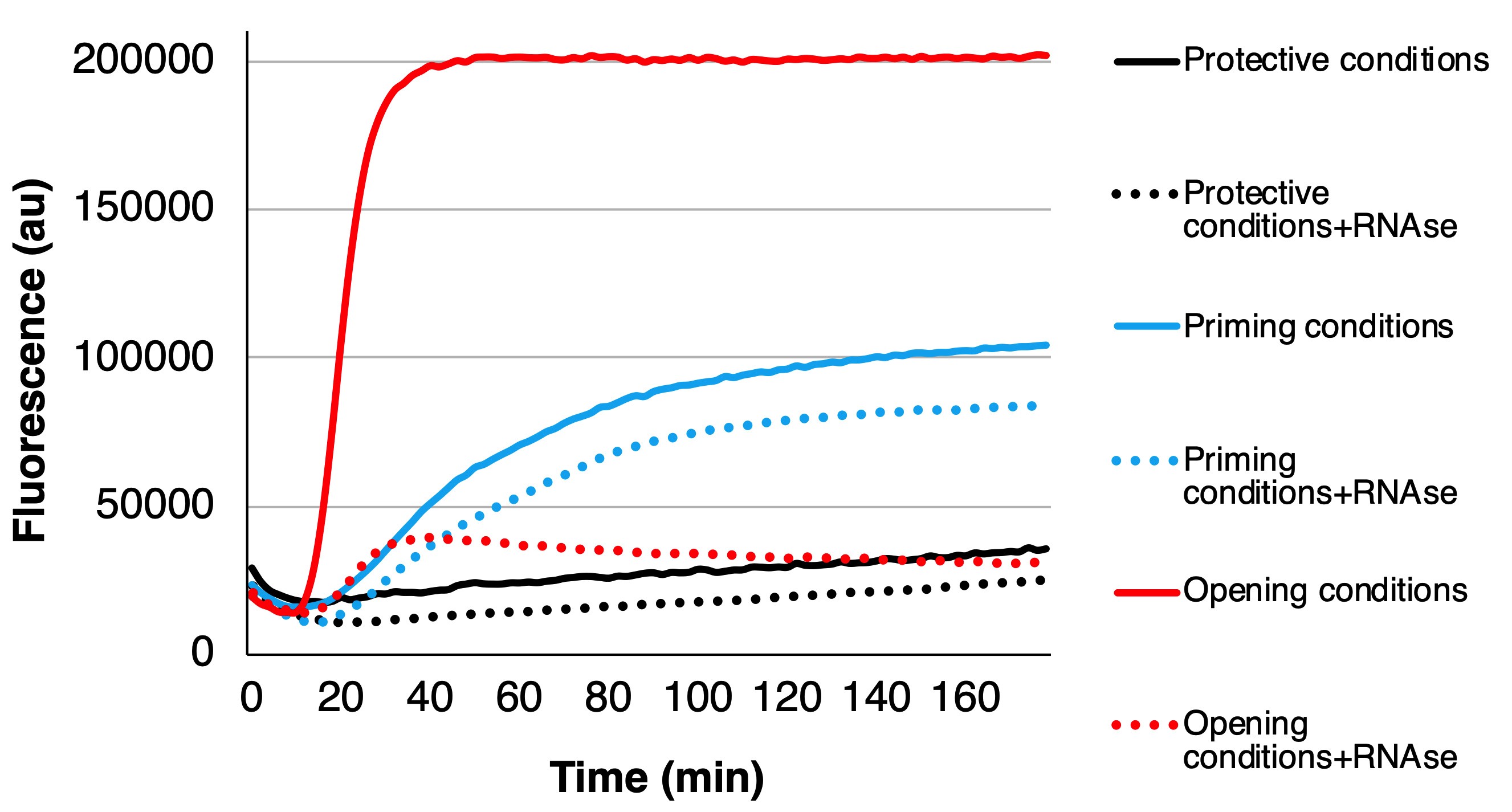
Figure 2. Example of three cases: protective conditions where the virus stays more or less unchanged (black curves), priming conditions where the virus mainly converts to primed and expanded intermediate form (blue curves), and opening conditions where the virus releases its RNA (red curves). For each condition, the fluorescence below the dotted line originates from the expanded particles since the RNase cannot enter inside the primed or intact particles, and the fluorescence from between the solid and dotted lines originates from the externalized genomes as the RNase abolishes the fluorescence.
Notes
Depending on the reproducibility of your virus purification, different batches of purified viruses might show different degree of virus priming and opening. Especially, we observed CsCl purified virus batches to have more variability than sucrose purified. Unpurified culture supernatants contain several contaminants that probably add into background fluorescence and lower the quality of the outcome and they might have also too low a virus concentration. For the reasons mentioned above, always verify the usability of a new batch of virus with some well known controls.
Competing interests
There are no competing interests by the authors of this article.
References
- Myllynen, M., Kazmertsuk, A. and Marjomäki, V. (2016). A novel open and infectious form of echovirus 1. J Virol 90(15): 6759-6770.
- Ruokolainen, V., Domanska, A., Laajala, M., Pelliccia, M., Butcher, S. J. and Marjomaki, V. (2019). Extracellular albumin and endosomal ions prime enterovirus particles for uncoating that can be prevented by fatty acid saturation. J Virol 93(17).
- Tuthill, T. J., Groppelli, E., Hogle, J.M. and Rowlands, D. J. (2010). Picornaviruses. In: Johnson, J. (Ed.). In: Cell Entry by Non-Enveloped Viruses. Current Topics in Microbiology and Immunology, vol 343. Springer, Berlin, Heidelberg.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ruokolainen, V., Laajala, M. and Marjomäki, V. (2020). Real-time Fluorescence Measurement of Enterovirus Uncoating. Bio-protocol 10(7): e3582. DOI: 10.21769/BioProtoc.3582.
Category
Molecular Biology > RNA > RNA detection
Biochemistry > RNA > RNA-protein interaction
Biochemistry > RNA > RNA structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.