- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Bimolecular Fluorescence Complementation (BiFC) for Studying Sarcomeric Protein Interactions in Drosophila
Published: Vol 10, Iss 7, Apr 5, 2020 DOI: 10.21769/BioProtoc.3569 Views: 6734
Reviewed by: David PaulPradeep Kumar BhaskarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Protein-protein interactions in Drosophila myofibrils are essential for their function and formation. Bimolecular Fluorescence Complementation (BiFC) is an effective method for studying protein interactions and localization. BiFC relies on the reconstitution of a monomeric fluorescent protein from two half-fragments when in proximity. Two proteins tagged with the different half-fragments emit a fluorescent signal when they are in physical contact, thus revealing a protein interaction and its spatial distribution. Because myofibrils are large networks of interconnected proteins, BIFC is an ideal method to study protein-protein interactions in myofibrils. Here we present a protocol for generating transgenic flies compatible with BiFC and a method for analyzing protein-protein interactions based on the fluorescent BiFC signal in myofibrils. Our protocol is applicable to the majority of Drosophila proteins and with few modifications may be used to study any tissue.
Keywords: MusclesBackground
Sarcomeres are the smallest contractile units in striated muscle and extend in a repeating pattern along the length of a myofibril (Reedy and Beall, 1993). The ability of sarcomeres to produce muscle contractions relies on two myofibril components: thin filaments and thick filaments. Myosin thick filaments are anchored at the M-line, located at the center of the sarcomere, whereas actin thin filaments are anchored at the Z-discs, which border either side of the sarcomere. Z-discs are therefore essential to the maintenance of myofibril structure and contractility and their failure to form can result in drastic defective muscle phenotypes underlying various human myopathies (Lemke and Schnorrer, 2017).
As multi-protein complexes, Z-discs are a hub for various protein-protein interactions, which are key to the assembly and stabilization of Z-disc structure. The indirect flight muscles of Drosophila melanogaster share many features with human striated muscle and are thus an ideal model to study Z-disc protein interactions (Weitkunat and Schnorrer, 2014). Previous studies on protein interactions in Drosophila indirect flight muscles took advantage of biochemical methods, such as co-immunoprecipitation, or used yeast-two hybrid assays (Katzemich et al., 2013; Liao et al., 2016; González-Morales et al., 2017 and 2019b). However, although these methods can indicate the presence of an interaction, they do not show the location of these interactions inside cells. Furthermore, such isolated ex vivo systems do not consider potential disruptions and complexities that can occur within an organism and additional difficulties often arise with co-immunoprecipitation due to the differences in solubility of Z-disc proteins.
On the other hand, BiFC is an in vivo approach used not only to prove the presence of a protein-protein interaction, but also to show its location (Kerppola, 2008). BiFC relies on the reconstitution of a fluorescent molecule to indicate an interaction between two proteins (Figure 1). In this approach, the N-terminal half of a fluorescent molecule is fused to one protein of interest, known as a bait, while the C-terminal half of the fluorescent molecule is expressed with a second potentially interacting protein, known as the prey. An interaction between the bait and prey reconstitutes a detectable fluorescing molecule (Kerppola, 2008). In flies, BiFC has previously been used to detect interactions between cytoskeletal proteins (Gohl et al., 2010), transcription factors (Hudry et al., 2012; Altamirano-Torres et al., 2018; Bischof et al., 2018), and myofibril proteins (González-Morales et al., 2019b).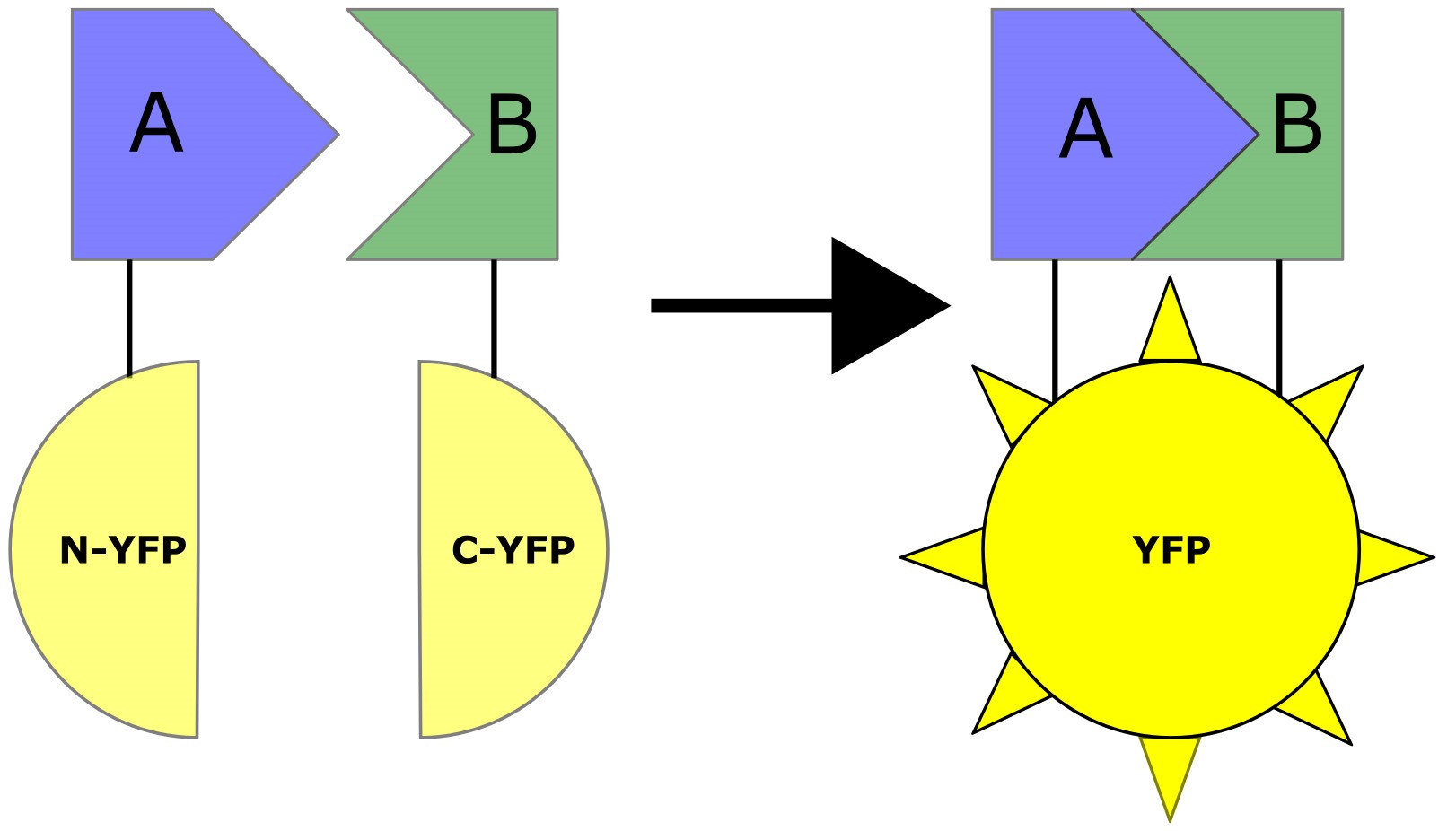
Figure 1. Principle of BiFC. N-terminal and C-terminal halves of a fluorescent molecule (YFP) are fused separately to two proteins of interest. An interaction between proteins results in reconstitution of a functional YFP and fluorescent signal.
The Drosophila Genomics Resource Center (DGRC) offers a big collection of cDNA clones from D. melanogaster. The FlyBi ORFeome collection at the DGRC contains open reading frames cloned into the pDONR223 Gateway vector, facilitating the cloning of BiFC-tagged proteins. This collection represents two thirds of the fly proteome.
Here, we present a detailed BiFC protocol to study protein-protein interactions in Drosophila myofibrils. We describe in detail the cloning process using the FlyBi ORFeome collection together with destination vectors that incorporate the BiFC-required tags (Gohl et al., 2010). We will describe in detail generation of BiFC flies, as well as necessary controls. We also suggest various strategies for imaging and analysis of BiFC-marked Z-discs and propose ways in which our methodology can be extended to other myofibril structures.
Materials and Reagents
- Medium-sized pointed brush
- 1.5 ml microcentrifuge tube
- 15 ml glass test tube
- Pipette tips
- FisherbrandTM Petri dishes with clear lid (Fisher Scientific, catalog number: 0875713 )
- Subcloning efficiencyTM DH5αTM competent cells (Thermo Fisher Scientific, catalog number: 18265017 )
- M13 primers, M13 primers can be acquired from Alpha DNA (http://alphadna.com/)
- Potassium chloride (KCl) (BioShop®, catalog number: POC308 )
- Calcium chloride (CaCl2) (BioShop®, catalog number: CCL302 )
- Magnesium chloride, hexahydrate (MgCl2) (BioShop®, catalog number: MAG510 )
- BactoTM tryptone (BD Biosciences, catalog number: 211699 )
- BactoTM yeast extract (BD Biosciences, catalog number: 212720 )
- Sodium chloride (NaCl) (BioShop®, catalog number: SOD001 )
- Agar (BioShop®, catalog number: AGR003 )
- Spectinomycin dihydrochloride pentahydrate (Sigma-Aldrich, catalog number: S9007 )
- Ampicillin (Sigma-Aldrich, catalog number: A9393 )
- Ethyl alcohol anhydrous (Commercial Alcohols, catalog number: P000EAAN )
- D-Glucose, anhydrous (VWR, catalog number: 200003-818 )
- UltraPureTM 1 M Tris-HCI, pH 8.0 (Thermo Fisher Scientific, catalog number: 15568025 )
- Ethylenediaminetetraacetic acid (EDTA), pH 8.0 (Thermo Fisher Scientific, catalog number: AM9260G )
- Ribonuclease A from bovine pancreas (RNase) (Sigma-Aldrich, catalog number: R4642-10MG )
- Sodium hydroxide (NaOH) (VWR International, catalog number: BDH9292 )
- Sodium dodecyl sulfate (SDS) electrophoresis grade (BioShop®, catalog number: SDS001 )
- Potassium acetate (Fisher Scientific, catalog number: BP364-500 )
- Glacial acetic acid (Sigma-Aldrich, catalog number: ARK2183 )
- TE, pH 8 (Thermo Fisher, catalog number: AM9849 )
- GatewayTM LR ClonaseTM (Thermo Fisher, catalog number: 11791100 )
- Proteinase K solution (Thermo Fisher, catalog number: AM2548 )
- InvitrogenTM PureLinkTM HiPure plasmid midiprep kit (Fisher Scientific, catalog number: K210004 )
- Phalloidin-tetramethylrhodamine B isothiocyanate (Sigma-Aldrich, catalog number: P1951 )
- 5x KCM buffer (see Recipes)
- LB broth (see Recipes)
- Spectinomycin- or ampicillin-selective LB agar plates (see Recipes)
- Solution 1 for small-scale preparation of plasmid DNA (100 ml) (see Recipes)
- Solution 2 for small-scale preparation of plasmid DNA (4 ml) (see Recipes)
- Solution 3 for small-scale preparation of plasmid DNA (100 ml) (see Recipes)
Equipment
- Water bath
- 250 ml Erlenmeyer flask
- IEC Micromax RF refrigerated microcentrifuge
- VWR® analog vortex mixer (VWR International, catalog number: 10153-838 )
- VWR® digital dry block heaters
- Shaker
- CO2 flypad, standard size (8.1 x 11.6 cm) (Genesee Scientific, Flystuff, catalog number: 59-114 )
- P2, P20, P100, and P1000 micro pipettes (e.g., Gilson, catalog numbers: F144801 , F123600 , F123615 and F123602 )
- Stereomicroscope (Leica Microsystems, model: Leica MS5 )
- Point-scanning confocal system (Leica Microsystems, model: Leica SP8 )
Software
- Fiji (https://fiji.sc/)
- R Statistics package (https://www.r-project.org/)
Procedure
- General cloning
- Obtain open reading frame (ORF) clones of the proteins whose interactions will be tested in pDONR223 entry gateway vectors from the Drosophila Genomics Research Center (https://dgrc.bio.indiana.edu).
- Transform entry vector into DH5α competent bacterial cells
- Centrifuge entry vector at 2,100 x g for 1 min at 4 °C.
- Add 20 µl of autoclaved water.
- Vortex for 1 min.
- Heat at 50 °C on a heat block for 10 min.
- Keep prepared DNA on ice.
- Add 80 µl of autoclaved water, 20 µl of 5x KCM buffer, and 1 µl of prepared DNA (above) with 100 µl of competent DH5α bacterial cells in a 1.5 ml microtube.
- Place tube on ice for 30 min.
- Heat shock cells at 42 °C by placing the tube in a water bath for 90 s. If a water bath is not available, any standard heating block will do as well.
- Add 500 µl of LB broth into the tube and let them recover for 1 h at 37 °C while shaking.
- Plate on spectinomycin selective LB agar plates.
- Incubate bacteria overnight at 37 °C.
- Make alkaline lysis minipreparations of the pDONR223 entry gateway vectors
- Make a stock of spectinomycin and LB medium in a 1:1,000 ratio.
- Select one successfully transformed colony from the spectinomycin selective LB agar plates using a 200 µl pipet tip.
- Place colony in 3 ml of the spectinomycin/LB stock in a sterile 15 ml tube and pipet up and down five times to mix.
- Loosely cap the tube and incubate the culture overnight in a 37 °C shaker.
- The next day, pour 1.5 ml of the culture into a 1.5 ml microtube.
- Centrifuge at ~13,000 x g for 30 s at 4 °C.
- Discard supernatant.
- Add the remaining 1.5 ml of the culture into the same tube.
- Centrifuge at ~13,000 x g for 30 s at 4 °C.
- Discard supernatant. Remove all liquid from the bacterial pellet, using a pipet to aspirate any remaining liquid if necessary.
- Add 100 µl of ice-cold Solution 1 to the bacterial pellet in the 1.5 µl microtube.
- Vortex for 30 s.
- Add 200 µl of Solution 2.
- Mix the contents by inverting the tube rapidly five times. Do not vortex. Store tube on ice.
- Add 150 µl of ice-cold Solution 3 and invert the tube rapidly five times.
- Store the tube on ice for 3-5 min.
- Centrifuge at ~13,000 x g for 5 min at 4 °C.
- Transfer supernatant to a fresh 1.5 µl microtube. Discard the pellet.
- Add 2 ml of 100% room-temperature ethanol and vortex for 30 s.
- Allow mixture to stand for 2 min at room temperature to precipitate the double-stranded DNA.
- Centrifuge at ~13,000 x g for 5 min at 4 °C.
- Discard the supernatant and allow the pellet to air-dry for 10 min.
- Rinse the pellet with 1 ml of 70% ice-cold ethanol.
- Remove the supernatant and allow pellet to air-dry for 10 min.
- Dissolve the DNA pellet in 50 µl of ultra-pure water.
- Confirm ORF identities by Sanger sequencing using m13 primers.
- Perform Gateway cloning from pDONR223 entry vector to pUAS-BiFC destination vectors (Figure 2; Table 1).
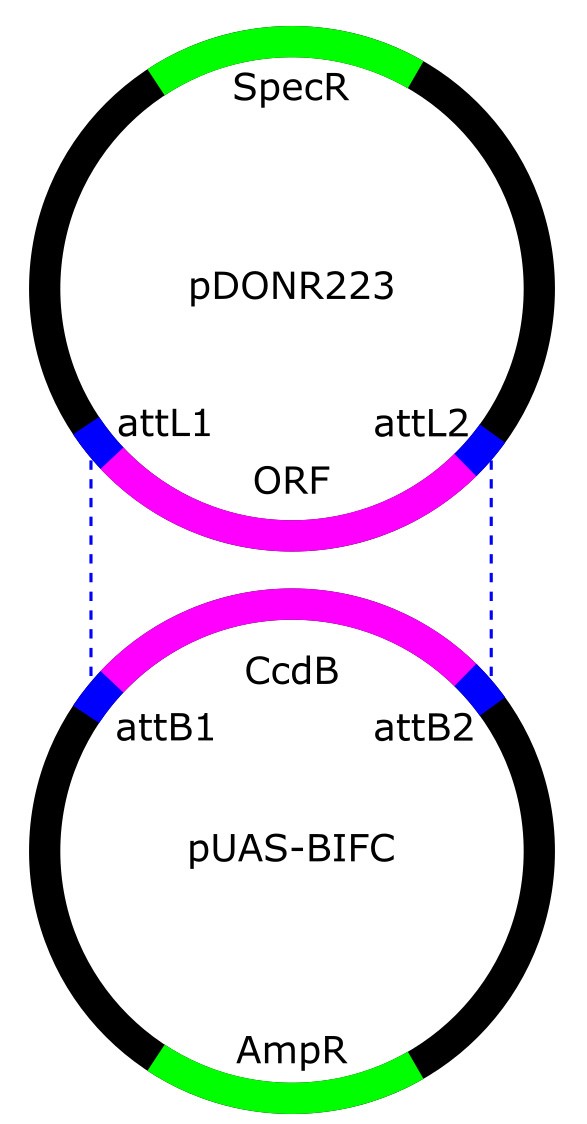
Figure 2. Gateway cloning of pDONR223 vector sequences into pUAS-BiFC vectors. pDONR223 vector confers spectinomycin resistance and contains the gene of interest between attL recombination sites. pUAS-BiFC vectors confer ampicillin resistance and contain the CcdB toxin between attB recombination sites. Recombination between the attB and attL sites mediates the transfer of the gene of interest into pUAS-BiFC.
Table 1. Cloning vectors required to make BiFC transgenes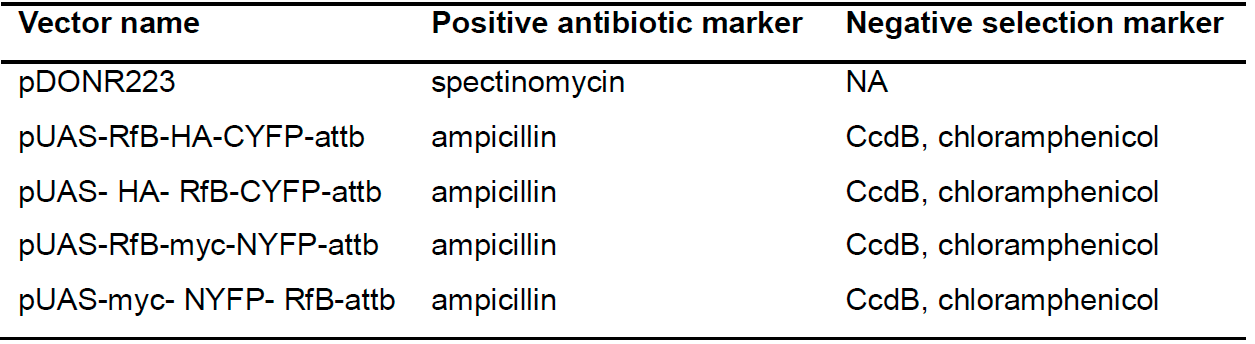
- Add 75 ng of pDONR223 entry vector from minipreparations (see Step A3) and 75 ng of pUAS-BiFC destination vector to a 1.5 ml microtube.
- To the same tube, add TE buffer, pH 8 to 4 µl.
- Thaw the LR ClonaseTM II enzyme on ice for 2 min. Vortex the enzyme mix for 2 s.
- Add 1 µl of LR ClonaseTM II enzyme to the microtube containing the DNA mix and TE buffer. Vortex for 2 s.
- Incubate at 25 °C for 1 h.
- Add 0.5 µl of Proteinase K to the mix and vortex for 2 s.
- Incubate at 37 °C for 10 min.
- Transform pUAS-BiFC destination vectors into DH5α competent bacterial cells.
Note: Procedure is identical to what has been described above with one exception: bacteria must be plated on ampicillin selective LB agar plates. - Perform a midiprep of the pUAS-BiFC destination vectors from successfully transformed bacterial cells using the PureLinkTM HiPure plasmid midiprep kit.
- In a 250 ml Erlenmeyer flask, add 100 ml of LB medium and 100 µl of ampicillin.
- Select one successfully transformed colony containing pUAS-BiFC destination vector from the ampicillin-selective LB agar plates using a 200 µl pipet tip.
- Add colony to the LB/ampicillin mix in the 250 ml Erlenmeyer flask and pipette up and down five times to mix.
- Incubate overnight in a 37 °C shaker.
- Follow instructions as specified in the PureLinkTM HiPure plasmid midiprep kit.
- Resuspend the purified plasmid DNA in 100 µl of water and place in a 1.5 ml microtube. Store at -20 °C.
- Confirm ORF identities by Sanger sequencing using the primers in Table 2 to confirm the insert and orientation.
Table 2. Primers for pUAS-BiFC destination vector sequencing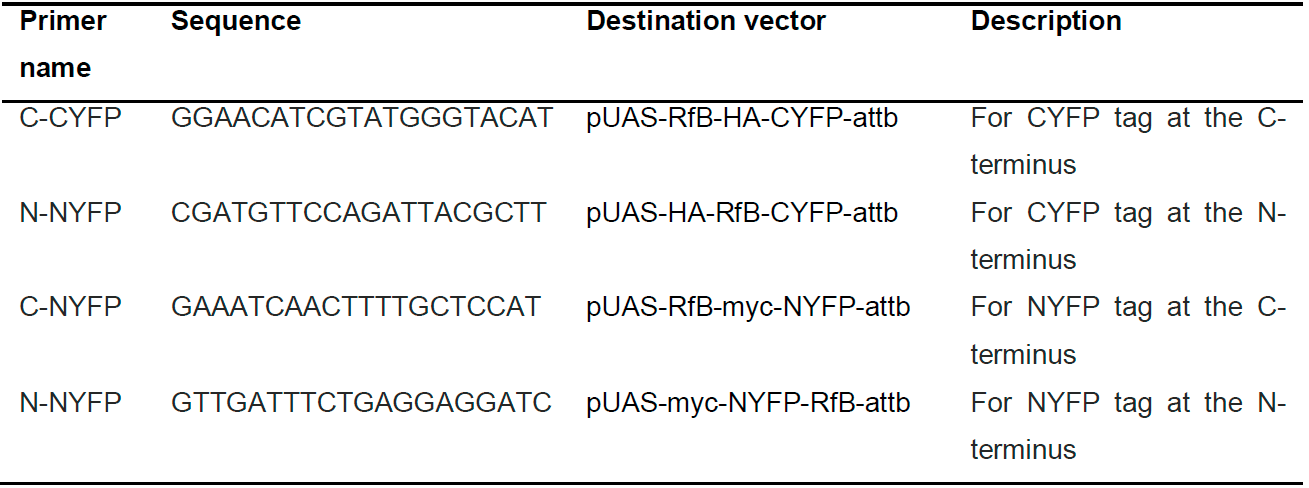
- Making transgenic lines and genetic crosses
- Transgenic flies are generated by microinjections using the ΦC31 Integrase method. We suggest outsourcing this part to a company that handles microinjections like BestGene or Genome ProLab.
- We suggest using the following landing sites to create transgenic fly lines:
- ZH-58A BDSC#244844: M[3xP3-RFP.attP]ZH-58A on the second chromosome.
- ZH-86Fb BDSC#24749: M[3xP3-RFP.attP]ZH-86Fb on the third chromosome.
- To activate expression of pUAS BiFC-tagged proteins, it is necessary to incorporate a Gal4 driver.
Prepare a stock of each pUAS BiFC construct crossed with a Gal4-containing line (Table 3).
Table 3. Example of some suggested experimental and control experiments. We also suggest similar experiments using N-terminally-tagged proteins of interest (i.e., YFP-Protein1).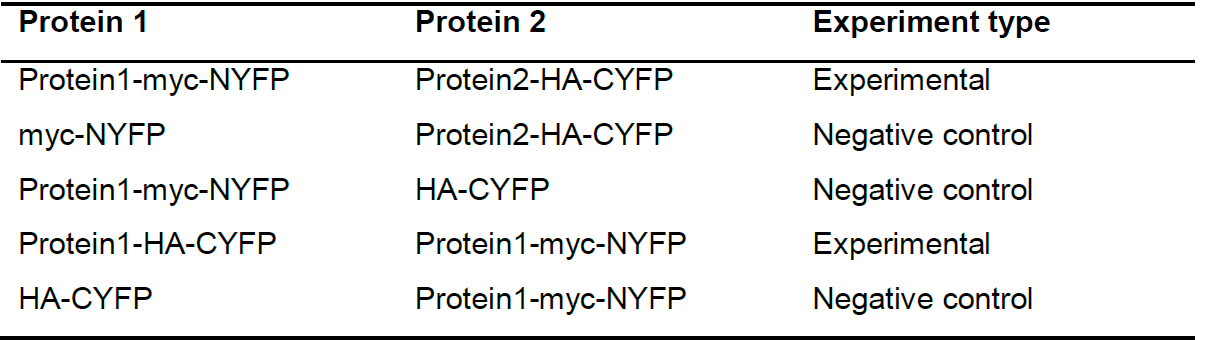
- Set the appropriate crosses to obtain both YFP halves and the Gal4 driver in a single fly. Set crosses for the necessary controls at this time.
- Example crossing scheme and some possibilities of combinations with controls are provided in Figure 3. We also suggest testing two proteins whose interactions are known as a positive control and for a baseline fluorescence measurement.
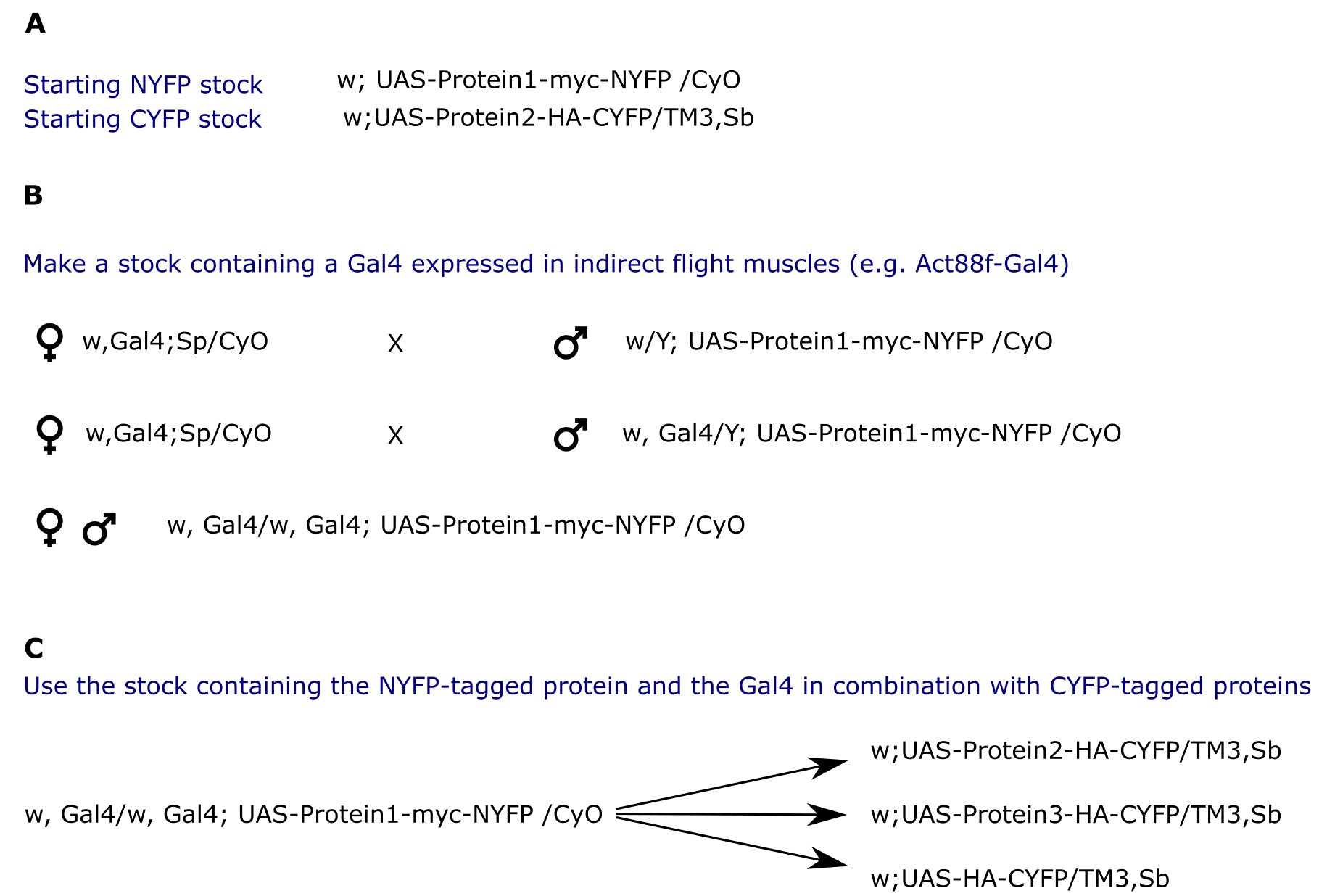
Figure 3. Example crossing scheme. A. Establish stocks carrying versions of the proteins of interest with NYFP and CYFP tags. B. Incorporation of an indirect flight muscle specific Gal4 driver on the X chromosome. C. Use the established stock in B to test for interactions and negative controls.
- Dissection and imaging of indirect flight muscles
- Collect flies of the correct genotype.
- Dissect the indirect flight muscle for imaging as described previously (Xiao et al., 2017).
We suggest using tetramethylrhodamine (TRITC)-conjugated phalloidin for actin staining. - Image indirect flight muscles by confocal microscopy.
- Use the Leica SP8 point-scanning confocal system. Other confocal microscopes should work as well.
- Image muscle fibers using the 63x/1.4 oil objective and a pinhole of 1 airy unit (AU).
- Initially locate the muscle fibers using brightfield to not bleach the sample.
- Set appropriate filters for YFP and TRITC detection.
- Take images at 9x magnification 1,024 x 1,024 pixels (~20 µm x ~20 µm).
- Sample images are provided in Figure 4.
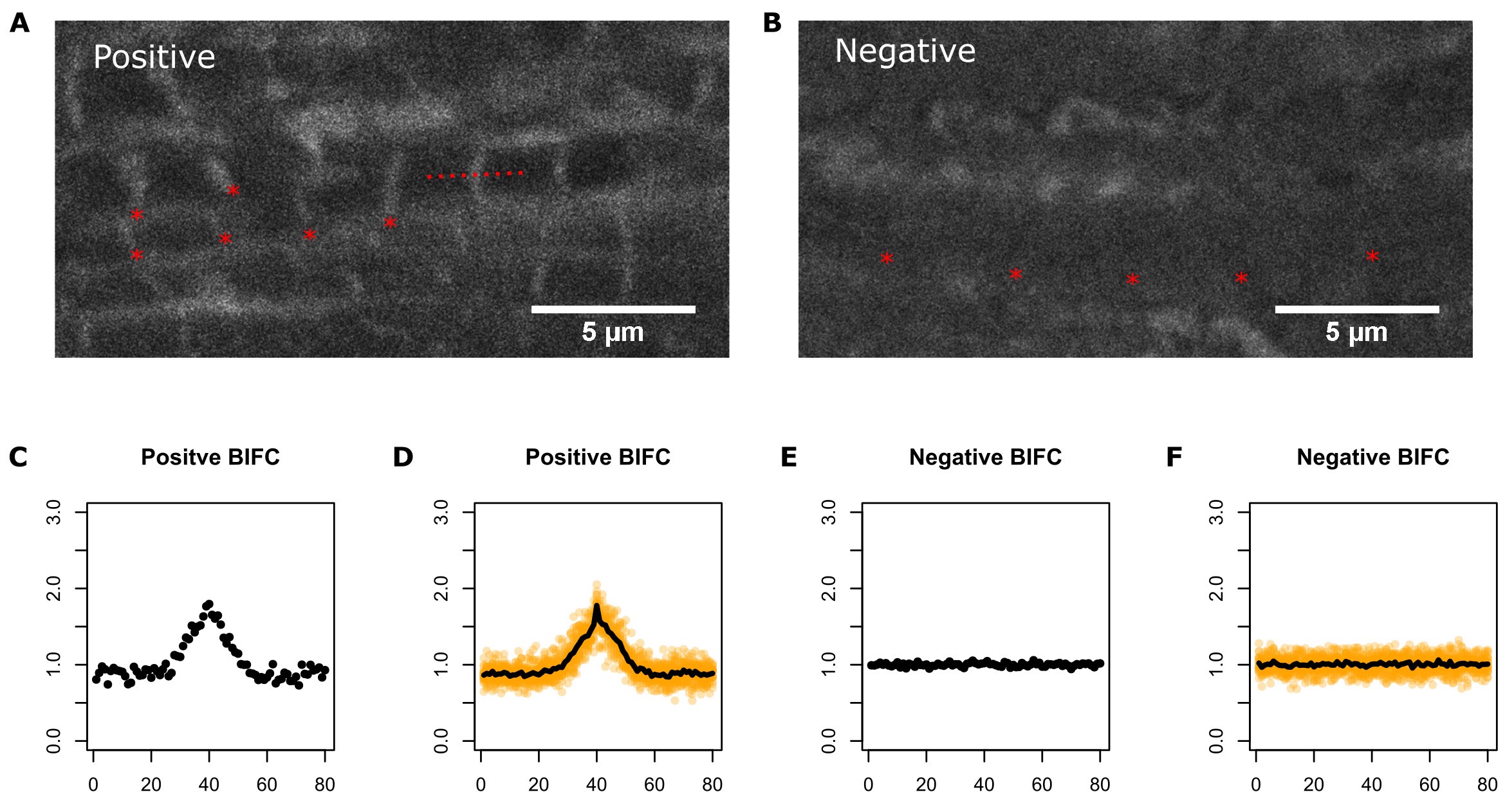
Figure 4. Example BIFC images and plots. A. Myofibrils with positive fluorescence signal at the Z-disc (red asterisks below selected Z-discs), indicating YFP reconstitution and interaction between proteins of interest. Dotted red line shows the Plot Profile tool in ImageJ software. B. Myofibrils from a negative control sample without detectable YFP at the Z-discs (red asterisks). C. Plot profile of a single sarcomere with positive BiFC signal. D. Plot profile of several sarcomeres with positive BiFC signal. In both C and D, the intensity peak corresponds to the Z-disc. E. Plot profile of a single sarcomere with negative BiFC signal. D. Plot profile of several sarcomeres with negative BiFC signal. In C-F, the fluorescence intensity values correspond to the y-axis and Z-disc position on the x-axis. A peak of fluorescence at the Z-disc is an indication of a positive BiFC signal.
Data analysis
- Measure YFP intensities in Fiji/ImageJ
- Open myofibril images in Fiji/ImageJ program.
- Select the straight-line tool and set line width to 30 pixels.
- Draw a straight line on each Z-disc from M-line to M-line, making sure to encompass the Z-disc in the center.
- Create a plot of fluorescence against distance using the Plot Profile tool, which can be accessed in the Analyze menu.
- Repeat for at least thirty different Z-discs from different myofibril images per genotype.
- Collapse plots from several Z-discs into one using R Statistics package (R version 3.5.1).
- Save all the Plot profiles from the previous step into a single folder.
- Open R Statistics package
- Set the folder containing all the Plot profiles as the working directory
- Run the R script (Code 1).
- Example of plots for positive and negative BiFC signals are provided in Figure 4.
Code 1. General R script to read Plot Profiles and collapse them into a single plot
library(data.table)
#Get the data
setwd("Plot Profile containing folder")
temp = list.files(pattern="*.csv")
myfiles = lapply(temp, read.csv)
a = lapply(myfiles,"[",2,drop=FALSE)
a = lapply(a, scale, center=F)
b=matrix(nrow = 80, ncol = length(a))
#Align the profiles
for (i in 1:length(a)) {
x=unlist(a[i])[1:80]
n=length(x)/2 - which( x == max(x))
n=sqrt(n*n)
s=shift(x,n=n, fill = NA,type = "lead")
s[is.na(s)]=x[1:n]
b[,i]=s
}
#Make a x/y plot
plot(-1,-1,xlim = c(1,80), ylim = c(0,3), ann=FALSE)
for (i in 1:length(b[1,])) {points(b[,i],pch=16,col= adjustcolor("orange", .3))}
lines(rowMeans(b), type = "l", lwd=3)
title(main="BiFC")
Notes
- The importance of negative controls
False positive signal can occur when tagged proteins bind either the CYFP or the NYFP tag. To rule this out, it is crucial that the selected proteins are tagged with CYFP and NYFP and then tested against the complementary tag alone. It is possible that the tagged protein binds only one of the YFP halves. In this scenario, the interacting protein and the interacting YFP half should be fused together. As this information is only available after the experiment is complete, to avoid wasting time, all possible combinations between YFP halves and proteins should be done in the cloning step. - BiFC during indirect flight muscle development
We have only analyzed adult indirect flight muscles, but our protocol is compatible with pupa dissections. Protocols that employ GFP to mark the indirect flight muscle during development are useful to dissect the IFM during pupa stages (Kao et al., 2019). Using GFP as a counter stain is not compatible with BiFC protocol, but other fluorescent proteins like Zasp52-mCherry can be used in combination with BiFC (Gonzalez-Morales et al., 2019b). Importantly, protein abundance and isoform usage are highly dynamic during indirect flight muscle development (Spletter et al., 2018). This should be considered when doing pupal BiFC experiments. - BiFC in other muscle types
Our protocol is optimized for the analysis of Z-disc proteins in the indirect flight muscle but can be applied to other muscles as well. Protein and isoform distribution vary among muscle types. This is particularly true for the Zasp proteins in flies (Gonzalez-Morales et al., 2019a). A certain interaction is therefore likely to happen in only a subset of muscles. To adjust our protocol to other muscles a different Gal4 line must be used, Mef2-Gal4 for example drives strong expression in all muscle types, Act79B-Gal4 only in jump muscle (Bryantsev et al., 2012; Fernandes and Schöck, 2014; Spletter and Schnorrer, 2014). - Use other techniques or databases to validate the results
It is important to note that our BiFC protocol is based on the overexpression of tagged proteins. Muscles are particularly sensitive to relative protein concentrations (Beall et al., 1989) and thus the overexpression of proteins can have a potential impact on the muscle ultrastructure and protein binding partners. Another independent method for inferring protein-protein interactions should always be used to validate the BiFC results. For the indirect flight muscle, a super-resolved atlas of protein localization using dSTORM is available and serves to estimate if the proteins of interest are in proximity to support direct binding (Szikora et al., 2020). In addition, yeast two hybrid or GST pulldown experiments are good validation tools. - Compatibility of other DNA collections at the DGRC
There are other large collections of plasmids publicly available that allow cloning of the BIFC tags into specific genes and isoforms (MiMIC, Gold collection, Fosmid collection, etc.), but the advantage here is that the ORFeome is in the pDONR223 vector that facilitate Gateway cloning. It would be possible to use other cloning approaches and plasmid collections to create BiFC-tagged proteins for isoform-specific protein tags or ORFs not found in the ORFeome collection. - Discriminate real BiFC signal from sample noise
The BiFC signal comes from the reconstituted YFP that forms once both YFP halves are in proximity. While this is a powerful system that allows visualization of protein interactions in single Z-discs, the YFP signal is sometimes low enough that distinguishing real binding events from the sample noise can be challenging. However, because sarcomeres are well organized structures with well defined regions (Z-disc, M-line, A-band, etc.) we can compare the localization of the endogenous proteins of interest to the localization of the BiFC signal. If the endogenous proteins and the BiFC signal localize to the same structure, it likely represents a real binding event. - Mapping the domains responsible for protein interactions
Identifying the residues or the protein domains that mediate the physical interaction between proteins is an important piece of information. Our BiFC setup can be used for this purpose by introducing mutations directly into the entry vector. It is important to verify the correct localization of mutated protein versions, as a decrease in BiFC signal may come from a mislocalization problem. This can easily be tested using Myc or HA tags in the destination vectors. - The problems or negative results when interpreting BiFC results
Even in the complete absence of detectable BiFC signal it is nearly impossible to completely rule out binding between a pair of proteins based only on BiFC. The addition of the BiFC tags may disrupt protein-protein interaction by steric hindrance or by causing the misfolding of proteins. In addition, even if the proteins are bound together, the two YFP halves must be in proximity for reconstituting a functional YFP. If one of the proteins is rather big, the distance between tags might be large enough to prevent YFP reconstitution. Testing all combinations between N-terminal and C-terminal tags might be required.
Recipes
- 5x KCM buffer
5.1 ml 1 M KCl
1.5 ml 1 M CaCl2
2.5 ml 1 M MgCl2
0.9 ml of ddH2O - LB broth
- Dissolve in 1 L of ddH2O:
50 g of BactoTM tryptone
25 g of BactoTM yeast extract
50 g of NaCl - Autoclave
- Dissolve in 1 L of ddH2O:
- Spectinomycin- or ampicillin-selective LB agar plates
- Prepare spectinomycin 50 mg/ml and ampicillin 100 mg/ml stock solutions
- Dissolve in 1 L of ddH2O:
50 g of BactoTM tryptone
25 g of BactoTM yeast extract
50 g of NaCl
75 g of agar - Autoclave
- Add 1 ml of spectinomycin or ampicillin stock solution (1:1,000 concentration)
- Pour medium into Petri dishes and allow to solidify
- Store, covered at 4 °C
- Solution 1 for small-scale preparation of plasmid DNA (makes 100 ml)
5 ml of 1 M glucose (final concentration is 50 mM of glucose)
2.5 ml of 1 M Tris-HCl pH 8.0 (final concentration is 25 mM of Tris-HCl)
2 ml of 500 mM EDTA pH 8.0 (final concentration is 10 mM of EDTA)
90.5 ml of ddH2O
2 g of RNase A (20 mg/ml) - Solution 2 for small-scale preparation of plasmid DNA (makes 4 ml)
160 µl of 5 M NaOH (final concentration is 0.2 M of NaOH)
400 µl of 10% SDS (final concentration is 1% SDS)
3.44 ml of ddH2O - Solution 3 for small-scale preparation of plasmid DNA (makes 100 ml)
60 ml of 5 M potassium acetate
11.5 ml of glacial acetic acid
28.5 ml of H2O
Acknowledgments
We thank Beili Hu for help with Drosophila injections, the CIAN imaging facility for help with confocal microscopy, the Drosophila Genomics Resource Center, and Sven Bogdan. This work was supported by operating grant MOP-142475 from the Canadian Institutes of Health Research. We have no conflicts of interest to declare.
Competing interests
We declare no competing interests.
References
- Altamirano-Torres, C., Salinas-Hernandez, J. E., Cardenas-Chavez, D. L., Rodriguez-Padilla, C. and Resendez-Perez, D. (2018). Transcription factor TFIIEbeta interacts with two exposed positions in helix 2 of the Antennapedia homeodomain to control homeotic function in Drosophila. PLoS One 13(10): e0205905.
- Beall, C. J., Sepanski, M. A. and Fyrberg, E. A. (1989). Genetic dissection of Drosophila myofibril formation: effects of actin and myosin heavy chain null alleles. Genes Dev 3(2): 131-140.
- Bischof, J., Duffraisse, M., Furger, E., Ajuria, L., Giraud, G., Vanderperre, S., Paul, R., Bjorklund, M., Ahr, D., Ahmed, A. W., Spinelli, L., Brun, C., Basler, K. and Merabet, S. (2018). Generation of a versatile BiFC ORFeome library for analyzing protein-protein interactions in live Drosophila. Elife 7: 38853.
- Bryantsev, A. L., Baker, P. W., Lovato, T. L., Jaramillo, M. S. and Cripps, R. M. (2012). Differential requirements for myocyte enhancer factor-2 during adult myogenesis in Drosophila. Dev Biol 361(2): 191-207.
- Fernandes, I. and Schöck, F. (2014). The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils. J Cell Biol 206(4): 559-572.
- Gohl, C., Banovic, D., Grevelhorster, A. and Bogdan, S. (2010). WAVE forms hetero- and homo-oligomeric complexes at integrin junctions in Drosophila visualized by bimolecular fluorescence complementation. J Biol Chem 285(51): 40171-40179.
- González-Morales, N., Holenka, T. K. and Schöck, F. (2017). Filamin actin-binding and titin-binding fulfill distinct functions in Z-disc cohesion. PLoS Genet 13(7): e1006880.
- González-Morales, N., Marsh, T. W., Katzemich, A., Marescal, O., Xiao, Y. S. and Schöck, F. (2019a) Different evolutionary trajectories of two insect-specific paralogous proteins involved in stabilizing muscle myofibrils. Genetics 212(3): 743-755.
- González-Morales, N., Xiao, Y. S., Schilling, M. A., Marescal, O., Liao, K. A. and Schöck, F. (2019b). Myofibril diameter is set by a finely tuned mechanism of protein oligomerization in Drosophila. Elife 8: 50496.
- Hudry, B., Remacle, S., Delfini, M. C., Rezsohazy, R., Graba, Y. and Merabet, S. (2012). Hox proteins display a common and ancestral ability to diversify their interaction mode with the PBC class cofactors. PLoS Biol 10(6): e1001351.
- Kao, S. Y., Nikonova, E., Ravichandran, K. and Spletter, M. L. (2019). Dissection of Drosophila melanogaster flight muscles for omics approaches. J Vis Exp (152).
- Katzemich, A., Liao, K. A., Czerniecki, S. and Schöck, F. (2013). Alp/Enigma family proteins cooperate in Z-disc formation and myofibril assembly. PLoS Genet. 9(3): e1003342.
- Kerppola, T. K. (2008). Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37: 465-487.
- Lemke, S. B. and Schnorrer, F. (2017). Mechanical forces during muscle development. Mech Dev 144(Pt A): 92-101.
- Liao, K. A., González-Morales, N. and Schöck, F. (2016). Zasp52, a Core Z-disc protein in Drosophila indirect flight muscles, interacts with α-actinin via an extended PDZ domain. PLoS Genet 12(10): e1006400.
- Reedy, M. C. and Beall, C. (1993). Ultrastructure of developing flight muscle in Drosophila. I. assembly of myofibrils. Dev Biol 160(2): 443-465.
- Spletter, M. L. and Schnorrer, F. (2014). Transcriptional regulation and alternative splicing cooperate in muscle fiber-type specification in flies and mammals. Exp Cell Res 321(1): 90-98.
- Spletter, M. L., Barz, C., Yeroslaviz, A., Zhang, X., Lemke, S. B., Bonnard, A., Brunner, E., Cardone, G., Basler, K., Habermann, B. H. and Schnorrer, F. (2018). A transcriptomics resource reveals a transcriptional transition during ordered sarcomere morphogenesis in flight muscle. Elife 7: 34058.
- Szikora, S., Gajdos, T., Novák, T., Farkas, D., Földi, I., Lenart, P., Erdélyi, M. and Mihály, J. (2020). Nanoscopy reveals the layered organization of the sarcomeric H-zone and I-band complexes. J Cell Biol 219(1): e201907026.
- Weitkunat, M. and Schnorrer, F. (2014). A guide to study Drosophila muscle biology. Methods 68(1): 2-14.
- Xiao, Y. S., Schöck, F. and González-Morales, N. (2017). Rapid IFM dissection for visualizing fluorescently tagged sarcomeric proteins. Bio-protocol 7(22): e2606.
Article Information
Copyright
![]() Marescal et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Marescal et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Marescal, O., Schöck, F. and González-Morales, N. (2020). Bimolecular Fluorescence Complementation (BiFC) for Studying Sarcomeric Protein Interactions in Drosophila. Bio-protocol 10(7): e3569. DOI: 10.21769/BioProtoc.3569.
- González-Morales, N., Xiao, Y. S., Schilling, M. A., Marescal, O., Liao, K. A. and Schöck, F. (2019b). Myofibril diameter is set by a finely tuned mechanism of protein oligomerization in Drosophila. Elife 8: 50496.
Category
Developmental Biology > Morphogenesis > Cell structure
Cell Biology > Cell imaging > Confocal microscopy
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.