- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Lipid Mixing Assay for Murine Myoblast Fusion and Other Slow Cell-cell Fusion Processes
Published: Vol 10, Iss 5, Mar 5, 2020 DOI: 10.21769/BioProtoc.3544 Views: 5115
Reviewed by: Khyati Hitesh Shahsujan kumar mondalLiang LiuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Lipid mixing (redistribution of lipid probes between fusing membranes) has been widely used to study early stages of relatively fast viral and intracellular fusion processes that take seconds to minutes. Lipid mixing assays are especially important for identification of hemifusion intermediates operationally defined as lipid mixing without content mixing. Due to unsynchronized character and the slow rate of the differentiation processes that prime the cells for cell-cell fusion processes in myogenesis, osteoclastogenesis and placentogenesis, these fusions take days. Application of lipid mixing assays to detect early fusion intermediates in these very slow fusion processes must consider the continuous turnover of plasma membrane components and potential fusion-unrelated exchange of the lipid probes between the membranes. Here we describe the application of lipid mixing assay in our work on myoblast fusion stage in development and regeneration of skeletal muscle cells. Our approach utilizes conventional in vitro model of myogenic differentiation and fusion based on murine C2C12 cells. When we observe the appearance of first multinucleated cells, we lift the cells and label them with either fluorescent lipid DiI as a membrane probe or CellTrackerTM Green as a content probe. Redistribution of the probes between the cells is scored by fluorescence microscopy. Hemifused cells are identified as mononucleated cells labeled with both content- and membrane probes. The interpretation must be supported by a system of negative controls with fusion-incompetent cells to account for and minimize contributions of fusion-unrelated exchange of the lipid probes. This approach with minor modifications has been used for investigating fusion of primary murine myoblasts, osteoclast precursors and fusion mediated by a gamete fusogen HAP2, and likely can be adopted for other slow cell-cell fusion processes.
Keywords: Membrane fusionBackground
Fusion of membrane lipid bilayers in cell biological processes as diverse as fusion of intracellular membranes in exocytosis, and fusion of viral and cell membranes in enveloped virus infection, and myoblast fusion in skeletal muscle development apparently involves similar lipid rearrangements (Figure 1) (Brukman et al., 2019). First, a merger of the apposing, contacting monolayers of two fusing bilayers generates a hemifusion connection and allows redistribution of lipid probes between these monolayers. A subsequent merger of the distal monolayers generates a fusion pore and allows redistribution of aqueous probes between fusing membrane compartments.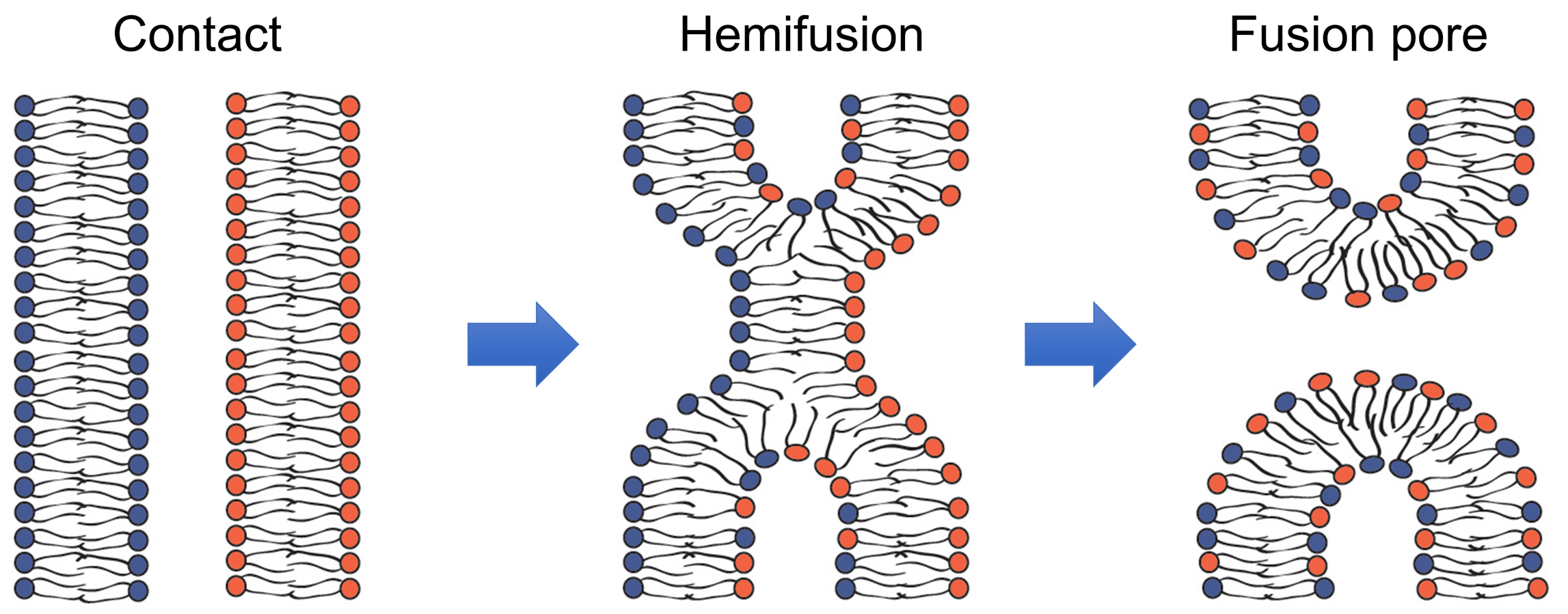
Figure 1. Schematic representation of the lipid rearrangements during hemifusion and fusion pore formation (modified from Figure 1 in Brukman et al., 2019).
Hemifusion can either transition into a fusion pore or represent a dead end of the fusion reaction aborted before fusion pore formation. In the latter case, hemifusion connections can then dissociate yielding two distinct bilayers. In their turn, nascent fusion pores can either close or expand advancing fusion towards its completion with full unification of the membrane compartments. The rates of formation and dissociation of the key fusion intermediates, hemifusion and fusion pores, and the rates of the transition between these intermediates vary between different fusion processes and are determined by the activity of the proteins involved and lipid compositions of the fusing membranes.
In most of the experimental studies, pores large enough to pass content probes in the range from ~1 kDa to ~100 kDa are detected by fluorescence microscopy. Hemifusion, operationally defined as lipid mixing without content mixing, is detected using fluorescence microscopy or spectrofluorometry. Different modifications of lipid mixing assays have been developed in studies on fusion of protein-free lipid bilayers, and fusion mediated by viral fusogens and intracellular fusogens (Brukman et al., 2019). The successful application of the lipid mixing assay in all these systems has been facilitated by a relative rapidness of these fusion processes with characteristic times of lipid mixing varying from seconds to minutes.
Application of lipid mixing assays to developmental cell-cell fusion, the subject of this work, is critically important for clarification of the pathways of the membrane rearrangements but more challenging than for faster viral and intracellular fusion reactions. For instance, formation of multinucleated myotubes, one of the best characterized examples of cell-cell fusion processes (Sampath et al., 2018), is preceded by myogenic differentiation that prepares the cells for fusion and takes days. As a result, by the time fusion is evaluated, fluorescent lipid probes added before fusion to label plasma membranes are already partially internalized. Moreover, labeling of intracellular membrane compartments containing internalized probes may appear brighter than labeling of the plasma membrane because of a higher membrane content and contrast. An additional concern in developing lipid mixing assays for very slow fusion processes is related to the ability of lipid probes to be transferred from membrane to membrane by lipid-exchanging proteins, lipid micelles or extracellular vesicles (reviewed in Merklinger et al., 2016), i.e., by mechanisms that neither involve hemifusion or fusion nor depend on the fusion machinery.
Here we present a protocol used to assay lipid mixing in fusion between C2C12 cells, immortalized mouse myoblasts that proliferate in high-serum medium and differentiate and fuse in low-serum medium. We also discuss applications of the modified versions of this protocol to other slow cell-cell fusion processes.
Materials and Reagents
Materials
- Falcon® 15 ml Polystyrene Centrifuge Tube, Conical Bottom, with Dome Seal Screw Cap (Corning, catalog number: 352095 )
- Tissue culture dishes, 35 x 10 mm REF (Corning, catalog number: 353001 )
- Tissue culture dishes, 60 x 15 mm (Corning, catalog number: 353002 )
Cells
- C2C12 cells (ATCC, catalog number: CRL-1772TM)
- Myomaker-deficient C2C12 cells and Myomerger-deficient C2C12 cells were generated in (Millay et al., 2016; Quinn et al., 2017) and grown on collagen coated substrates
Reagents
- VybrantTM DiI Cell-Labeling Solution (Thermo Fisher Scientific, catalog number: V22885 )
- CellTrackerTM Green CMFDA (5-chloromethylfluorescein diacetate) (Thermo Fisher Scientific catalog number: C7025 )
- Orange CMRA CellTrackerTM (Thermo Fisher Scientific, catalog number: C34551 )
- Trypsin-EDTA 0.05% (Thermo Fisher, catalog number: 25300054 )
- Hoechst 33342 Trihydrochloride, Trihydrate- 10 mg/ml Solution in Water (Thermo Fisher Scientific, catalog number: H3570 )
- Formalin 10% Buffered in Phosphate (Electron microscopy Sciences, catalog number: 15740 )
- Collagen from calf skin (Sigma, catalog number: C8919-20 ml )
- Dimethyl sulfoxide (DMSO, Sigma, catalog number: D2650-100 ml )
- The proliferation medium (PM): DMEM, high glucose, GlutaMAXTM Supplement (Thermo Fisher, catalog number: 10566-016 ) + 10% Fetal Bovine Serum, Penicillin/Streptomycin (Thermo Fisher, catalog number: 10378016 )
- The differentiation medium (DM): DMEM, high glucose, GlutaMAXTM Supplement (Thermo Fisher, catalog number: 10566-016 ) + 5% Horse Fetal Serum, Penicillin/Streptomycin
- Fetal Bovine Serum, FBS (GIBCO Life Technologies, catalog number: 10437-028 )
- Horse Serum, heat inactivated, New Zealand origin (Thermo Fisher, catalog number: 26050088 )
- PBS, Corning® Dulbecco’s Phosphate-Buffered Saline (Life Sciences, catalog number: 21-030-CV ), 1x with calcium and magnesium
Equipment
- Zeiss Axioscope microscope
- Camera (Manufacturer pco-tech inc.; pco.edge 3.1 sCMOS)
- F-LD 32/0.4 Zeiss objective lens
- Single fluorophore bandpass filter for green cell tracker: excitation 472 nm/30 nm, emission 520 nm/35 nm, dichroic 495 nm LP from Semrock
- Single fluorophore bandpass filter for DiI: excitation 545 nm/25 nm, emission 605 nm/70 nm, dichroic 570 LP from Zeiss
Software
- Micro-Manager software (Edelstein et al., 2014)
- The open-source platform, ImageJ (National Institute of Health, Rockville Pike, Bethesda, MD
Procedure
Figure 2 gives a schematic presentation of the timeline for preparing the cells for lipid mixing experiments.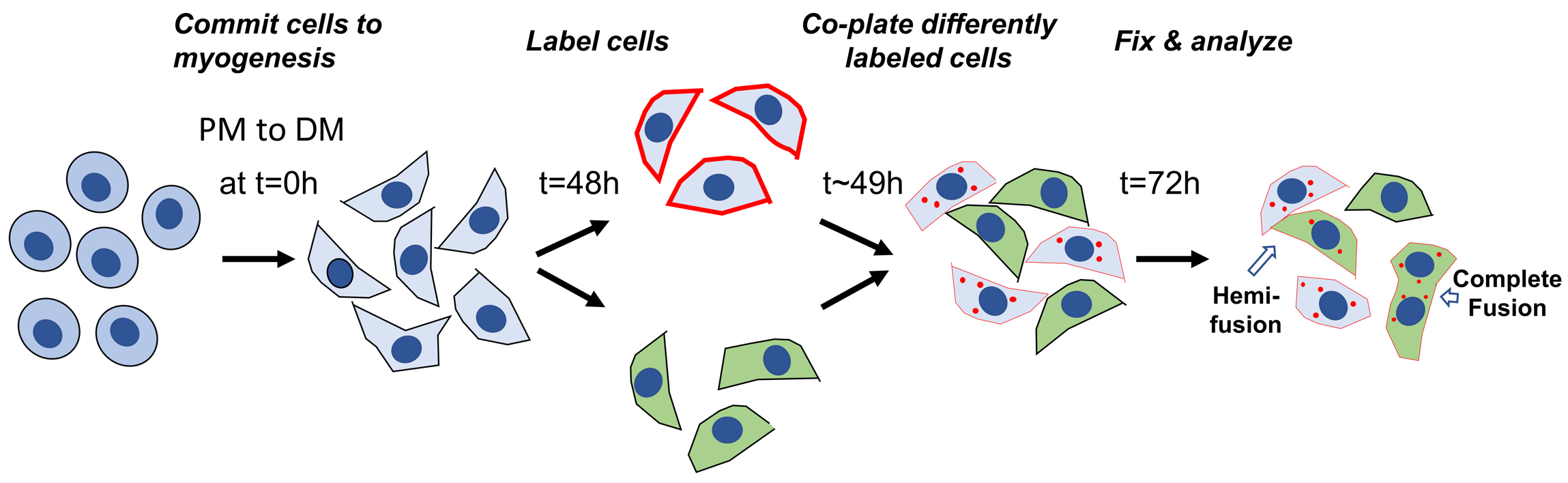
Figure 2. Schematic presentation of the timeline for preparing and labeling the cells for lipid mixing experiments. The labeling of the cells is started 48 h post differentiation (i.e., 48 h after placing the cells into DM). Differently labeled cells are mixed 1 h later at 49 h post differentiation.
- Collagen-coating dishes
Coat the dishes (60 x 15 mm) (two dishes for each condition) and 35 x 10 mm dishes (5 dishes for each condition) with collagen by covering the dishes with 3 ml or 1.5 ml of 1:10 solution of collagen in sterile water for 60 x 15 mm dish and 35 x 10 mm dish, respectively, and, after overnight incubation at room temperature and two washes (3 ml and 2 ml each for large and small dishes, respectively) with water, dry the dishes in the biosafety cabinet. - Cells
Grow C2C12 cells in the proliferation medium (PM) in the collagen-coated dishes to 75% confluency. Transfer the cells into the differentiation medium (DM) (t = 0). By 48 h post differentiation (t = 48 h), the cells spread to ~90% confluency and the first myotubes are observed. At this time, wash the cells in each of the two dishes with 2 ml PBS and place the cells in 2 ml fresh PBS. - Cell labeling
Label the cells in dish 1 with CellTrackerTM Green and cells in dish 2 either with Orange CMRA CellTracker or with DiI. To label with CellTrackerTM Green; add 4 μl of the CellTrackerTM Green stock solution (50 μg of the probe in 20 μl of DMSO) to 2 ml of PBS and incubate the cells in this medium for 15 min at 37 °C. Then place the cells into complete DM for 30 min at 37 °C. Use the same procedure for labeling with Orange CMRA CellTracker. To label cells with DiI, inject 4 μl of 1 mM DiI stock solution into 2 ml of PBS at 37 °C and incubate the cells for 45 min. Wash the cells in the dishes 1 and 2 three times with 2 ml of DM and two times with 2 ml of PBS. - Co-plating differently labeled cells
- Gently lift the cells in dishes 1 and 2 with 1 ml of trypsin-EDTA 0.05% applied for 1 min at 37 °C. During the subsequent 2-3 min incubation already at the room temperature, check whether the cells started to round up and lift. When they do, remove EDTA-trypsin and lift the cells into 4 ml of complete DM at 37 °C. Collect the cells from dishes 1 and 2 into the same 15 ml tube and mix the cells by vortexing. Adjust the volume to 10 ml. Then plate the cells (2 ml of cell suspension) onto each of five collagen-treated small dishes (35 x 10 mm). Two hours later, remove debris and non-attached cells by changing the medium for the fresh DM.
- Use coplating of the cells labeled with different CellTrackers to analyze content mixing between the cells. In the absence of content mixing, use coplating of the cells labeled with CellTrackerTM Green and cells labeled with DiI to analyze lipid mixing.
- At t = 72 h, fix the cells with 10% Formalin in phosphate buffer for 10 min at room temperature. Stain the nuclei by replacing Formalin with PBS supplemented with Hoechst (10 mg/ml stock diluted 1,000-fold) applied for 30 min at room temperature. Replace Hoechst-supplemented PBS with PBS. Take the images on a fluorescence microscope using appropriate excitation and emission filters.
Data analysis
- We analyze the images in ImageJ. First, using plates with only green and only red cells we verified that bleed-through between green and red channels is negligible at the used settings. In the images from the experiments with co-plated CellTrackerTM Green -labeled cells and Orange CMRA CellTracker -labeled cells, the cells that aborted fusion at the fusion pore opening stage would be detected as mononucleated cells co-labeled with both probes. So far, we have observed this phenotype only for fusions mediated by viral fusogens.
- In the images from the experiments with co-plated DiI-labeled cells and CellTrackerTM Green -labeled cells, the cells that aborted fusion at the hemifusion stage are seen and scored as mononucleated cells co-labeled with both probes. Figures 3, 4 and 5 show representative fluorescence microscopy images that we used to analyze the redistribution of lipid probe (DiI) and content probe (CellTrackerTM Green) in Myomerger−/− (Figure 3); Myomaker−/− (Figure 4) and WT (Figure 5) C2C12 cells.
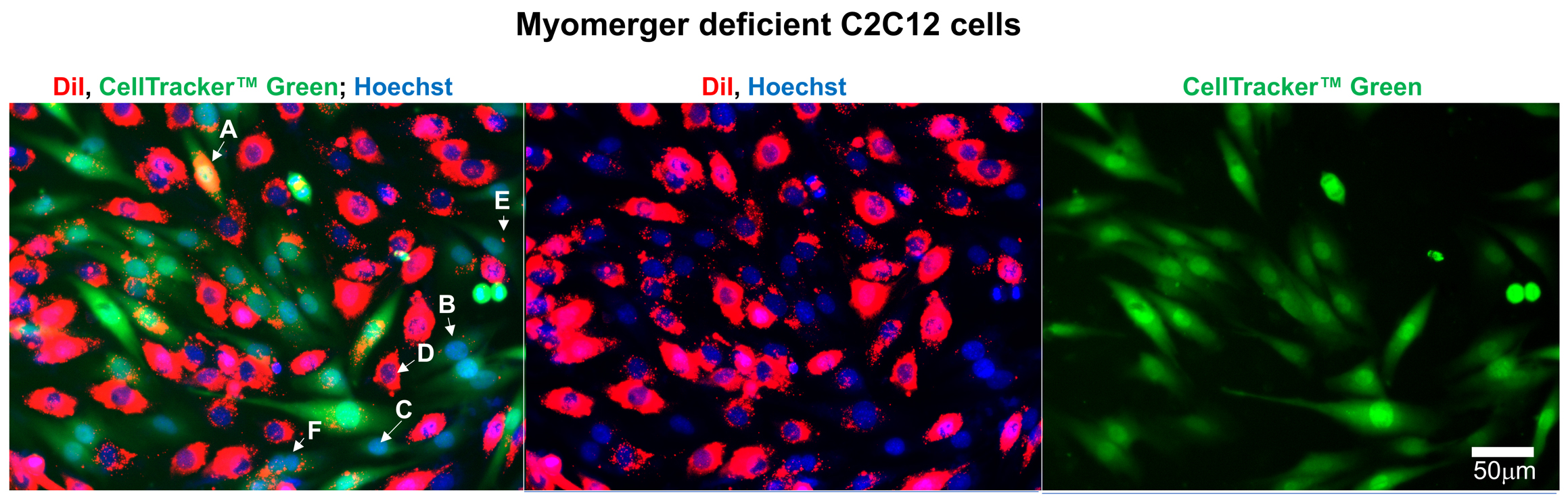
Figure 3. Representative fluorescence microscopy images of Myomerger-deficient C2C12 cells used to evaluate the efficiency of lipid mixing by analysis of redistribution of lipid probe (Dil) and content probe (CellTrackerTM Green). Images of the cells after co-incubation of Dil-labeled cells with CellTrackerTM green-labeled cells in the differentiation medium. Nuclei are stained by Hoechst. Arrows indicate examples of several distinct phenotypes counted or not counted as mononucleated cells co-labeled with Dil and CellTrackerTM Green. A and B mark colabeled cells with stronger (A) and weaker (B) levels of Dil fluorescence. Cells like the ones marked as A and B were counted as co-labeled mononucleated cells. Cells labeled with only CellTrackerTM Green (C), or only Dil (D), or green cells with just one red point (E) or cells with more than one nucleus (F) were not counted as co-labeled mononucleated cells. Scale bar = 50 μm.
Figure 4. Representative fluorescence microscopy images of Myomaker-deficient C2C12 cells used to evaluate the efficiency of lipid mixing by analysis of redistribution of lipid probe (Dil) and content probe (CellTrackerTM Green). Images of the cells after co-incubation of Dil-labeled cells with CellTrackerTM Green-labeled cells in the differentiation medium. Nuclei are stained by Hoechst. Arrow indicates an example of mononucleated cell co-labeled with Dil and CellTrackerTM Green. Scale bar = 50 μm.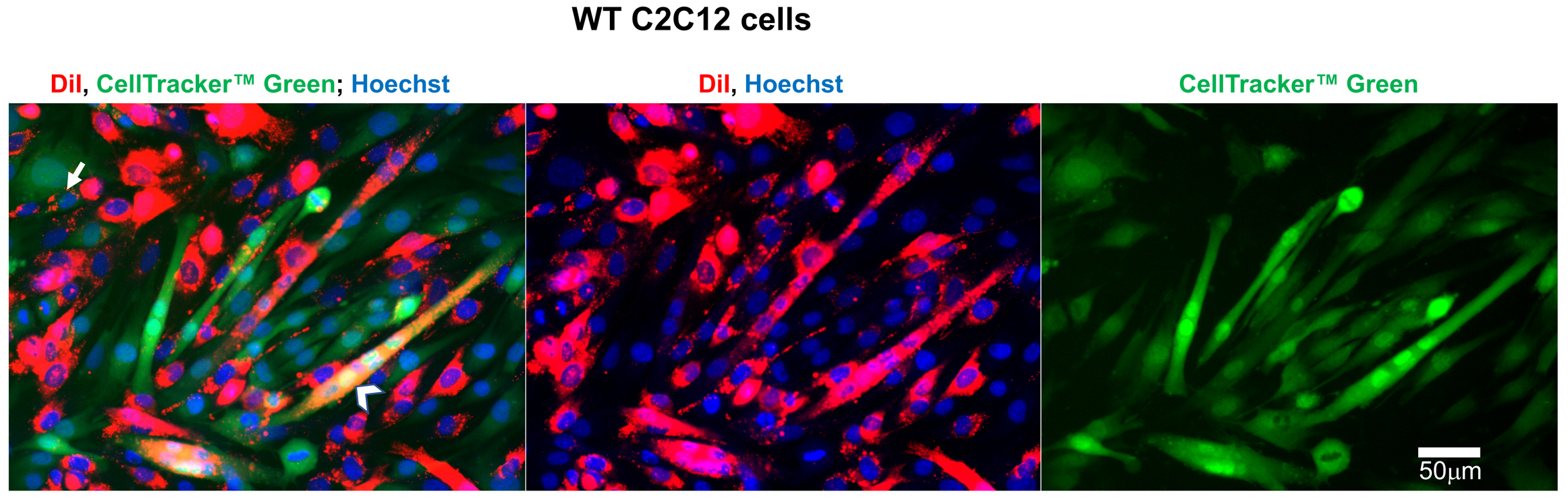
Figure 5. Representative fluorescence microscopy images of wild type (WT) C2C12 cells used to evaluate the efficiency of lipid mixing by analysis of redistribution of lipid probe (Dil) and content probe (CellTrackerTM Green). Images of the cells after co-incubation of Dil-labeled cells with Cell TrackerTM Green-labeled cells in the differentiation medium. Nuclei are stained by Hoechst. Arrow indicates an example of mononucleated cell co-labeled with Dil and CellTrackerTM Green. Arrowhead marks an example of a co-labeled syncytium. Scale bar = 50 μm. - To identify DiI labeled mononucleated CellTrackerTM Green labeled cells, we use the following procedure. First, we find the median of maximal DiI fluorescence levels Fmax in ~100 DiI-labeled cells that have no green fluorescence. We use this value to set the lower limit of fluorescence for detection of DiI redistribution using the following formula:
Fmin=background+k(Fmax-background)
We found that k = 0.1 works well to exclude cells that acquired DiI fluorescence by fusion-unrelated mechanisms. To count the co-labeled cells, we adjust image contrast by setting in Brightness/Contrast tool minimum to Fmin and select the maximum to reveal the faintest distinct puncta. After that, we count as co-labeled all green cells that have at least 3 distinct perinuclearly located DiI puncta. (1-2 bright red puncta that we occasionally find associated with cell-surface likely represent debris or extracellular vesicles and the corresponding cells are not scored as co-labeled.) - To quantify the efficiency of hemifusion for different conditions we normalize the number of mononucleated double-labeled cells in the field of view by the total number of mononucleated cells in the field. For each condition, we score at least 10 randomly selected fields of view. The characteristic results for Myomerger-deficient cells, Myomaker-deficient cells and wild type (WT) cells are shown in Figure 6.
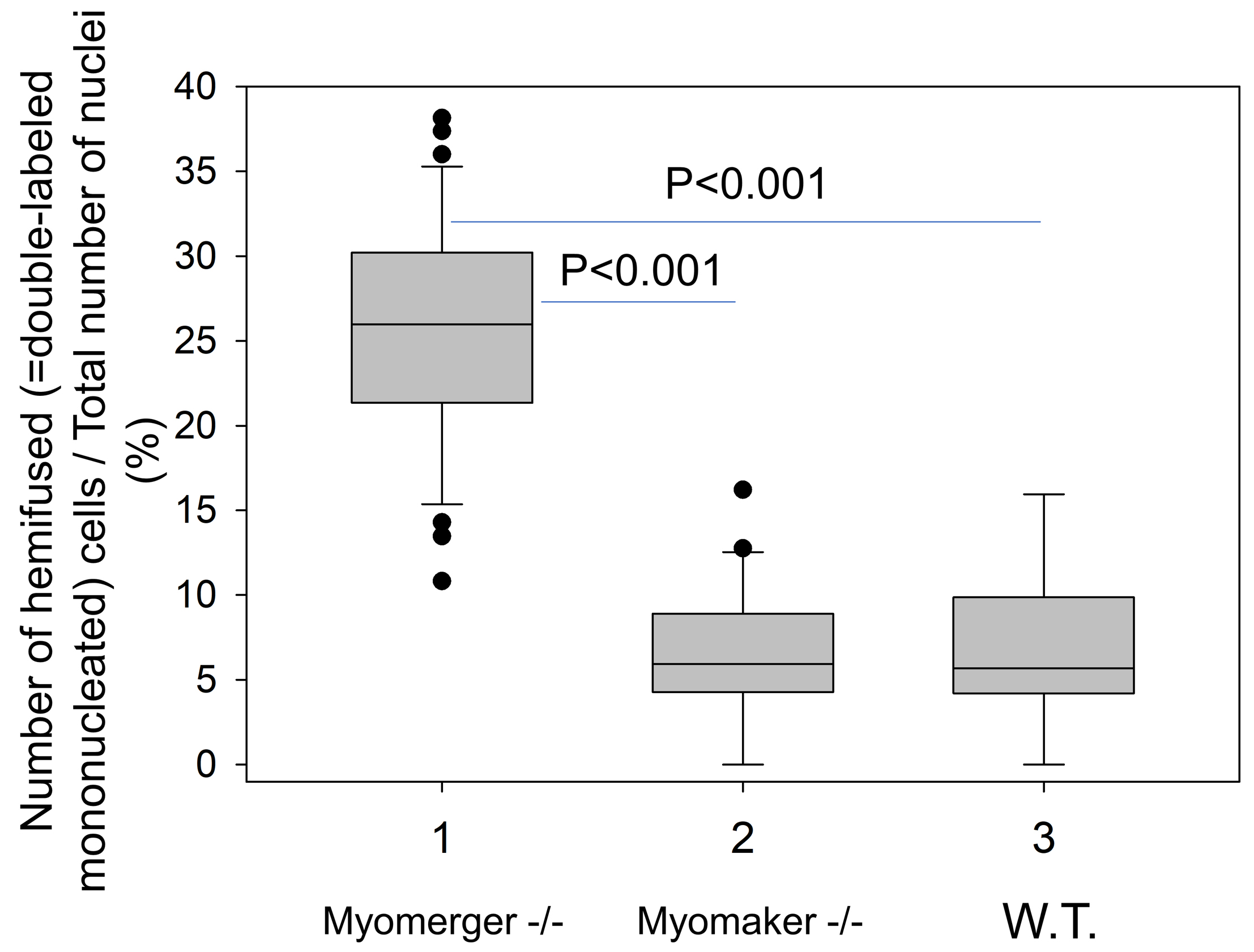
Figure 6. Quantification of hemifusion by lipid mixing assay. Hemifusion was operationally defined as Dil redistribution in the absence of Cell TrackerTM Green and detected as formation of mononucleated cells labeled with both Dil and CellTrackerTM Green. Data presentation and statistical analyses: box-and-whisker plots show median (center line), 25th-75th percentiles (box) and minimum and maximum values (whiskers); statistical significance was evaluated with Mann-Whitney test. - Syncytia are seen as bright double-labeled or single-labeled multinucleated (n ≥ 2) cells generated by fusion involving differently-labeled and similarly-labeled cells, respectively. Hemifusion efficiency, i.e., the probability that under given conditions the cells will hemifuse but will not proceed to fusion completion is quantified by normalizing the numbers of hemifusion events in the field of view (mononucleated double-labeled cells to the total number of nuclei in this field). Assuming that each complete fusion event proceeds through hemifusion intermediates, the efficiency of local membrane merger events yielding either hemi- or complete fusion can be quantified as (Nhf + Ncf)/Nt, where Nhf is the number of hemifusion events, Ncf, the number of complete fusion events (estimated as the number of nuclei in the cells with at least 2 nuclei) and Nt, the total number of the nuclei in this field.
Notes
Modifications of the assay for different cell-cell fusion processes
- Lipid mixing assay has been successfully adapted for fusion of primary murine myoblasts (Gamage et al., 2017; Leikina et al., 2013 and 2018), HAP2-mediated fusion of BHK cells (Valansi et al., 2017) and osteoclast precursors (Verma et al., 2018). The required modifications mostly reflected the differences in characteristic rates of the cell fusion processes necessitating adjustment of the relative timing of labeling stages. For instance, primary murine myoblasts that differentiate and fuse faster than C2C12 cells, were labeled in PM rather than in DM (Leikina et al., 2013). Then differently labeled cells were co-plated in DM to start myogenic differentiation and fusion.
- In Valansi et al., 2017 and Leikina et al., 2018, we used a modification of the lipid mixing assay, in which we co-plated cells labeled with both lipid and content probe with unlabeled cells, instead of co-plating cells labeled with membrane probe and cells labeled with content probe. In this experimental design, cell fusion stalled at the hemifusion stage produces cells labeled with membrane, but not content probe. Application of this assay requires control experiments with cells labeled with both membrane and content probes cultured in the absence of unlabeled cells to verify that cells labeled with only membrane probe are generated by interactions between labeled and unlabeled cells but not by content probe leakage.
Concerns and controls
- An important concern in developing lipid mixing assays for very slow fusion processes is related to the ability of lipid probes to be transferred from membrane to membrane by lipid-exchanging proteins, lipid micelles or extracellular vesicles (reviewed in Merklinger et al., 2016), i.e., by mechanisms that do not depend on the cell fusion machinery. DiI, lipid probe used in our assay, has been developed for long-term labeling and, after careful optimization, DiI and similar lipid probes have been used for tracking individual cells in tissues for several days and even weeks (reviewed in Progatzky et al., 2013). Still, application of DiI-based lipid mixing assay for slow fusion processes requires a set of controls to assure that redistribution of lipid probes is due to membrane merger rather than to the fusion-independent transfer of the probes. If, under some conditions, fusion-independent redistribution of lipid probes dominates fusion-dependent redistribution, the conditions should be altered to reduce fusion-unrelated components of the lipid probe transfer between the membranes. To optimize the experimental procedure, we use cells lacking functional fusion machinery (proliferating satellite cells, differentiating myomaker-deficient myoblasts in DM and differentiating myoblasts in the presence of hemifusion inhibitor lysophosphatidylcholine [Leikina et al., 2013]). The main factors contributing to the fusion-independent lipid mixing are the details of cell labeling protocol (probe concentration and incubation time), number of washes, cell viability (to minimize cell debris), and fusion efficiency of the cells (to increase the relative contribution of the membrane-merger-dependent lipid mixing). In addition, too high confluency of cell monolayer can lead to undetected overlaps between green cells and red cells in fluorescence microscopy images.
- Myomaker-deficient C2C12 cells and Myomerger-deficient C2C12 cells provide convenient controls for the lipid mixing assay. As seen in Figure 6, Myomerger-deficient cells demonstrate much higher levels of lipid mixing than Myomaker-deficient cells. Importantly, we show that lipid probe transfer for Myomerger-deficient cells depends on the contacts between differently labeled cells rather than on the transfer through the medium (for instance, by extracellular vesicles), by incubating unlabeled differentiating cells in the conditioned medium from DiI- labeled differentiating cells (Figure 7).
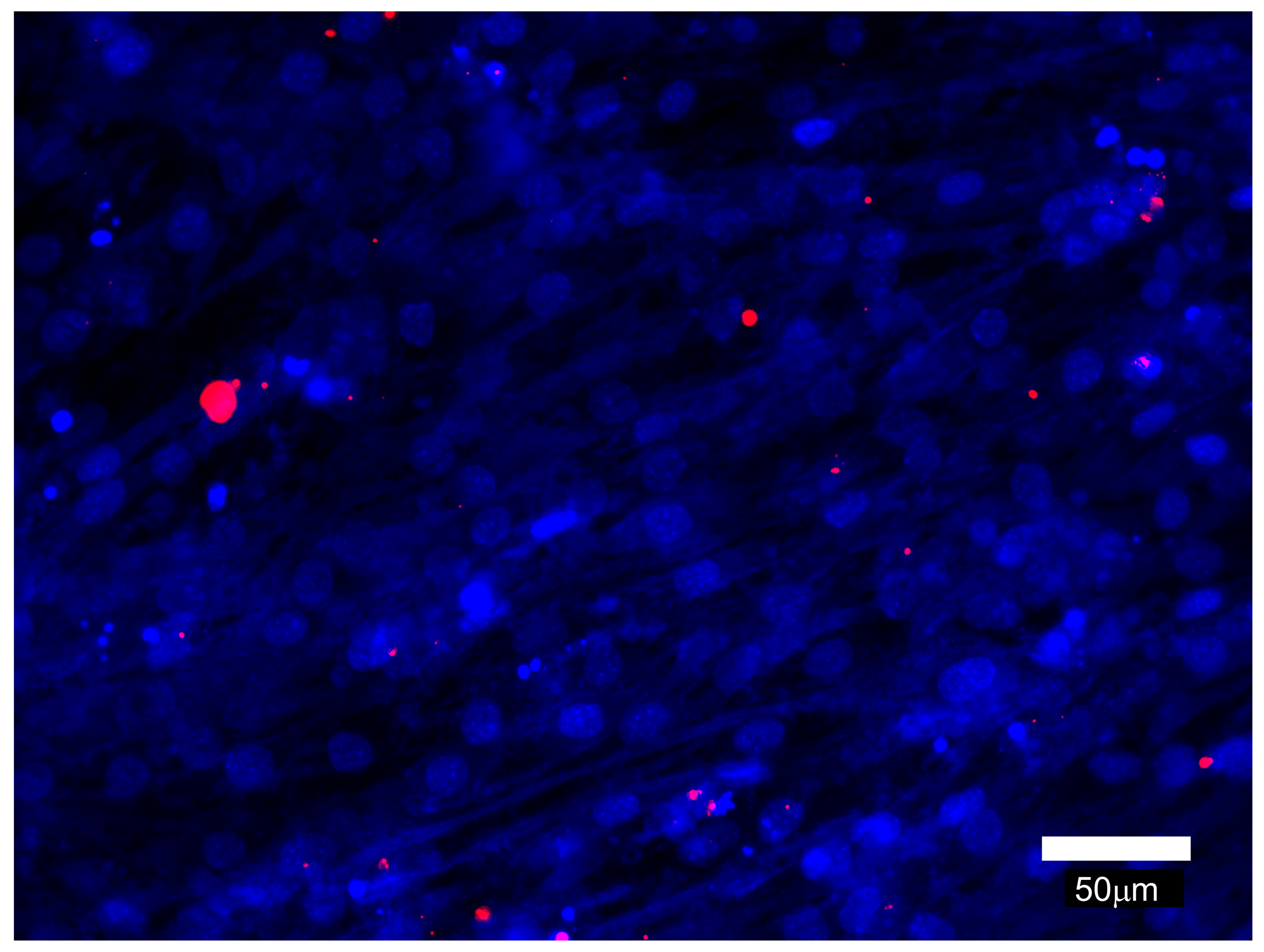
Figure 7. Representative fluorescence microscopy image that illustrates cell-cell fusion independent acquisition of Dil by unlabeled cells incubated with conditioned medium from Dil-labeled cells. The conditioned medium from Myomerger-deficient cells committed to differentiation and labeled with Dil as described above was collected at t = 72 h post differentiation. In another dish, unlabeled differentiating Myomerger-deficient cells were placed into this conditioned medium at t = 24 h. At t = 72 h, these cells were fixed and analyzed. Scale bar = 50 μm.
As expected, in these experiments we find only few cells that acquired lipid probe and no cells containing more than 3 distinct perinuclearly located DiI puncta. In this experimental design, finding significant numbers of DiI-labeled cells would suggest problems with DiI labeling. - Identification of hemifusion phenotype in lipid mixing assay can be further validated by independent experimental approaches that do not utilize lipid mixing assays. These two approaches (short-term application of hypotonic shock or chlorpromazine) reveal hemifusion intermediates by converting them into easy-to-detect complete fusion (Melikyan et al., 1997; Chernomordik et al., 1998). Hypotonic osmotic shock generates membrane tension that breaks hemifusion structures, and chlorpromazine preferentially partitions to inner monolayers of plasma membranes and destabilizes hemifusion intermediate formed by these monolayers. Neither of the treatments induces complete fusion if applied to tightly bound rather than hemifused cells.
Acknowledgments
The research in L.V.C.’s laboratory was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, and by Grant Number 2013151 from the United States-Israel Binational Science Foundation (BSF). The protocol described here has been developed in Leikina et al., 2013 and 2018.
Competing interests
The authors declare no competing financial or non-financial interests.
References
- Brukman, N. G., Uygur, B., Podbilewicz, B. and Chernomordik, L. V. (2019). How cells fuse. J Cell Biol 218(5): 1436-1451.
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P. and Zimmerberg, J. (1998). The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol 140(6): 1369-1382.
- Edelstein, A. D., Tsuchida, M. A., Amodaj, N., Pinkard, H., Vale, R. D. and Stuurman, N. (2014). Advanced methods of microscope control using muManager software. J Biol Methods 1(2).
- Gamage, D. G., Leikina, E., Quinn, M. E., Ratinov, A., Chernomordik, L. V. and Millay, D. P. (2017). Insights into the localization and function of myomaker during myoblast fusion. J Biol Chem 292(42): 17272-17289.
- Leikina, E., Gamage, D. G., Prasad, V., Goykhberg, J., Crowe, M., Diao, J., Kozlov, M. M., Chernomordik, L. V. and Millay, D. P. (2018). Myomaker and myomerger work independently to control distinct steps of membrane remodeling during myoblast fusion. Dev Cell 46(6): 767-780 e767.
- Leikina, E., Melikov, K., Sanyal, S., Verma, S. K., Eun, B., Gebert, C., Pfeifer, K., Lizunov, V. A., Kozlov, M. M. and Chernomordik, L. V. (2013). Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J Cell Biol 200(1): 109-123.
- Melikyan, G. B., Brener, S. A., Ok, D. C. and Cohen, F. S. (1997). Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J Cell Biol 136(5): 995-1005.
- Merklinger, E., Schloetel, J. G., Spitta, L., Thiele, C. and Lang, T. (2016). No evidence for spontaneous lipid transfer at ER-PM membrane contact sites. Journal of Membrane Biology 249(1-2): 41-56.
- Millay, D. P., Gamage, D. G., Quinn, M. E., Min, Y. L., Mitani, Y., Bassel-Duby, R. and Olson, E. N. (2016). Structure-function analysis of myomaker domains required for myoblast fusion. Proc Natl Acad Sci U S A 113(8): 2116-2121.
- Progatzky, F., Dallman, M. J. and Lo Celso, C. (2013). From seeing to believing: labelling strategies for in vivo cell-tracking experiments. Interface Focus 3(3): 20130001.
- Quinn, M. E., Goh, Q., Kurosaka, M., Gamage, D. G., Petrany, M. J., Prasad, V. and Millay, D. P. (2017). Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat Commun 8: 15665.
- Sampath, S. C., Sampath, S. C. and Millay, D. P. (2018). Myoblast fusion confusion: the resolution begins. Skelet Muscle 8(1): 3.
- Valansi, C., Moi, D., Leikina, E., Matveev, E., Grana, M., Chernomordik, L. V., Romero, H., Aguilar, P. S. and Podbilewicz, B. (2017). Arabidopsis HAP2/GCS1 is a gamete fusion protein homologous to somatic and viral fusogens. J Cell Biol 216(3): 571-581.
- Verma, S. K., Leikina, E., Melikov, K., Gebert, C., Kram, V., Young, M. F., Uygur, B. and Chernomordik, L. V. (2018). Cell-surface phosphatidylserine regulates osteoclast precursor fusion. J Biol Chem 293(1): 254-270.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Leikina, E., Melikov, K., Rabinovich, A. G., Millay, D. P. and Chernomordik, L. V. (2020). Lipid Mixing Assay for Murine Myoblast Fusion and Other Slow Cell-cell Fusion Processes. Bio-protocol 10(5): e3544. DOI: 10.21769/BioProtoc.3544.
Category
Developmental Biology > Cell growth and fate > Myofiber
Biochemistry > Lipid > Membrane lipid
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.