- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Estimating Cellular Abundances of Halo-tagged Proteins in Live Mammalian Cells by Flow Cytometry
Published: Vol 10, Iss 4, Feb 20, 2020 DOI: 10.21769/BioProtoc.3527 Views: 7300
Reviewed by: Chiara AmbrogioMaría Antonia Sánchez RomeroElif Karaca
Original research article
The authors used this protocol in:

Jun 2019


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Accurate abundance measurements of cellular proteins are required to achieve a quantitative and predictive understanding of any biological process inside the cell. Existing methods to determine absolute protein abundances are labor-intensive and/or require sophisticated experimental and computational infrastructure (e.g., fluorescence correlation spectroscopy (FCS)-calibrated imaging and quantitative mass spectrometry). Here we detail a straightforward flow cytometry-based method to measure the absolute abundance of any Halo-tagged protein in live cells that uses a standard mammalian cell line with a known number of Halo-CTCF proteins recently characterized in our lab. The protocol only comprises a few steps. First, a cell line expressing the Halo-tagged protein of interest is grown and labeled side-by-side with our standard line. Then, average fluorescence intensities are measured by conventional flow cytometry analysis and finally a simple calculation is applied to estimate the absolute number of the Halo-tagged protein of interest per cell. Once the protein of interest has been endogenously tagged with HaloTag, which we routinely achieve by Cas9-mediated genome editing, the presented protocol is fast, convenient, reproducible, cost-effective and readily accessible.
Keywords: HaloTagBackground
Assigning numbers to biological processes and to the molecules participating in them is of foremost importance to constrain models and attain a mechanistic and quantitative knowledge of biological phenomena (Ghaemmaghami et al., 2003). Proteins are macromolecules essential to cell, tissue, and organism structure, function and regulation. Although highly desirable, measuring absolute abundances of cellular proteins is a non-trivial undertaking. The few methods currently available are time-consuming, costly and technically challenging, and for the most part rely on complicated equipment and analytical tools not readily accessible to most laboratories. This is certainly the case for quantitative mass spectrometry, a powerful yet complex technology that allows high-throughput measurements of un-modified proteins (Bantscheff et al., 2007; Bantscheff et al., 2012; Holzmann et al., 2019). Quantitative FCS-calibrated imaging is a live cell method that can be streamlined to achieve high-throughput abundance measurement during dynamic cellular processes, while retaining single-cell resolution, and can be selective for different sub-cellular compartments (Cai et al., 2018; Politi et al., 2018; Walther et al., 2018; Holzmann et al., 2019). However, it again requires advanced microscopy equipment and computational infrastructure. Electrophoresis-based methods (e.g., traditional SDS-PAGE or more advanced capillary systems followed by staining/blotting (Chen et al., 2013; Bennett et al., 2017) are less complex, but still rely on accurate cell counting and/or depend on the availability of a pure recombinant protein to use as a calibrator.
We have recently developed a new flow cytometry-based absolute quantification method that can be easily applied to any Halo-tagged protein in the cell. This was part of a larger effort to quantify nuclear transcription factors and architectural 3D-genome regulators like CTCF and cohesin (Cattoglio et al., 2019), with the ultimate goal of deciphering the spatiotemporal regulation of transcription and its interplay with 3D genome organization. The method has several advantages over existing ones: it employs a human U2OS-derived cell line standard that is easy to culture; it only requires a flow cytometer, an instrument available in most institutions; it has single-cell resolution; it can be reproducibly repeated multiple times with little effort and cost; and, it involves very basic data analysis.
We focused on the HaloTag protein-fusion platform (Los et al., 2008) because of its popularity and versatility, with applications in a broad range of experimental systems (England et al., 2015), and since it is currently the preferred choice for live-cell single-molecule imaging (Presman et al., 2017). In fact, we first generated what is here used as a standard cell line with the intent of studying CTCF nuclear dynamics by single-molecule super-resolution microscopy (Hansen et al., 2017). This is a U2OS human clonal cell line (clone C32) with the endogenous CTCF locus homozygously engineered via Cas9-mediated genome editing to express a Halo-CTCF protein. As such, its expression level is stable and reflects endogenous CTCF. We realized that clone C32 could become a quantification standard for any Halo-tagged protein if we could estimate and cross-validate the absolute protein copy number per cell of Halo-CTCF in this U2OS cell line. We achieved this through a combination of in-gel fluorescence and FCS-calibrated imaging, obtaining consistent results across the two orthogonal methods performed in two different labs (~104,900 ± 14,600 and 114,600 ± 10,200 Halo-CTCF proteins per U2OS interphase cell, respectively) (Cattoglio et al., 2019). We took the mean of the two methods, 109,800 CTCF proteins per cell, as our best and final cross-validated estimate. This number can be now easily used to infer absolute abundances of other Halo-tagged proteins by simply growing, labeling and measuring fluorescence intensities of the cell line of interest side-by-side with our C32 standard, as we have previously demonstrated using Sox2 and TBP in mouse embryonic stem cells (Teves et al., 2016 and 2018; Cattoglio et al., 2019) (see also the data re-analysis in Table 1). After subtracting the background fluorescence of unstained cells, the mean fluorescence intensity of the cell line of interest is divided by the C32 standard fluorescence and multiplied by 109,800 to obtain the absolute abundance of the Halo-tagged protein under study (Figure 1). Although we focus exclusively on endogenously tagged cell lines here, the same pipeline can also be deployed to calculate abundances of exogenously expressed Halo-tagged proteins.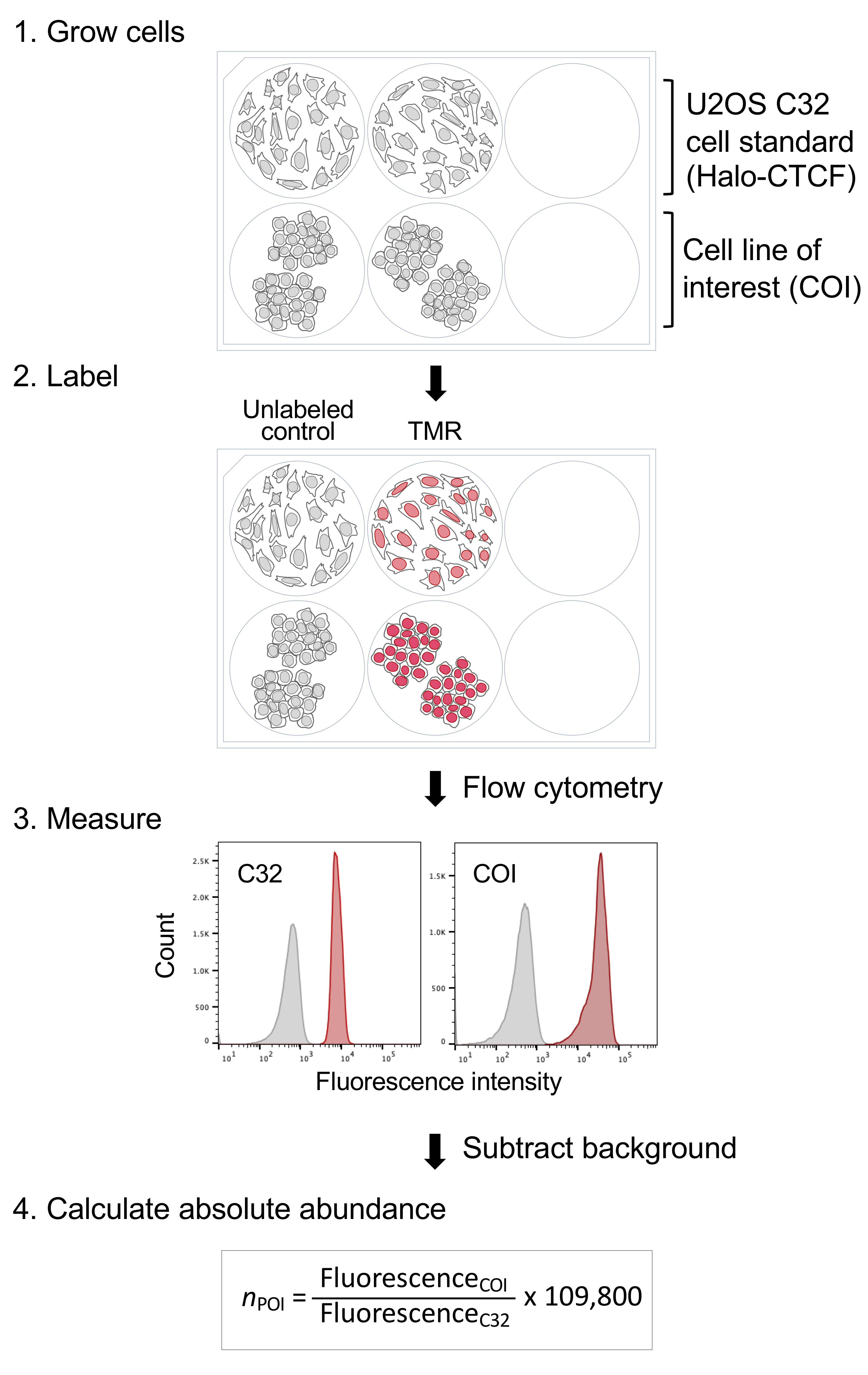
Figure 1. A streamlined method for absolute quantification of Halo-tagged proteins. (1) Grow your cell line of interest (COI), expressing the Halo-tagged protein to be quantified, side-by-side with the U2OS C32 Halo-CTCF cell standard. We usually perform the experiment in 6-well plates, but you can scale it up or down as required. (2) Label cells with the HaloTag ligand conjugated to a fluorophore (e.g., TMR) and (3) measure absolute fluorescence intensities by flow cytometry. (4) Subtract the background fluorescence of unlabeled controls and use the adjusted fluorescence values to calculate the absolute abundance (n) of the protein of interest (POI). 109,800 is the average number of Halo-CTCF molecules we estimated in the U2OS C32 cell standard.
The quantitative approach described here comes with a few limitations. It does not allow direct measurement of the concentration of the protein of interest, nor its quantification in different cellular compartments, it may not be robust for very lowly or very highly expressed proteins (below ~10,000 and above 10-20 million proteins/cell, respectively), and it is obviously restricted to Halo-tagged proteins. However, several streamlined protocols are now available to perform efficient genome editing with CRISPR-Cas9 in a variety of model organisms and cell types, and these are described elsewhere (Reference 2; Roberts et al., 2017; Aird et al., 2018; Haupt et al., 2018; Sharma et al., 2018; Tasan et al., 2018; Zhang et al., 2018; Liu et al., 2019).
Apart from these few drawbacks, our quantification protocol is simple and yet powerful. For instance, although we have only estimated average cellular protein numbers so far, the method can be used in principle to measure protein abundances in single cells, since the flow cytometer outputs fluorescence values for each individual cell. Another advantage of using flow cytometry as a readout is that the Halo-labeling can be multiplexed with other staining strategies (e.g., cell cycle analysis, surface and/or intracellular staining for the markers of interest) (Kovalenko et al., 2011) to determine protein abundances in different phases of the cell cycle and/or within different subpopulations of complex biological samples, like those obtained in differentiation and reprogramming experiments.
In summary, we here detail a rapid, straightforward and convenient method that will allow other researchers to estimate absolute cellular abundances of their Halo-tagged protein of interest with only a few hours of hands-on work.
Materials and Reagents
Note: With the exception of the U2OS C32 cell standard, you can replace all the listed reagents, equipment and software with equivalent alternatives.
- FalconTM standard 100-mm tissue culture dishes (CorningTM 353000, Fisher Scientific, catalog number: 08-772-E)
- FalconTM polystyrene 6-well microplates (CorningTM 353046, Fisher Scientific, catalog number: 08-772-1B)
- Cryovials (CorningTM 430487, Fisher Scientific, catalog number: 03-374-20)
- EppendorfTM snap-cap microcentrifuge safe-lockTM tubes, 1.5 ml (Fisher Scientific, catalog number: 05-402)
- Test tube with cell strainer snap cap (CorningTM 352235, Fisher Scientific, catalog number: 08-771-23)
- Eye protection
- Mask
- U2OS C32 Halo-CTCF standard cell line [first described in Hansen et al. (2017), available upon request]. Thaw the contents of one cryovial and plate onto one 100-mm cell culture plate
- User-provided cell line(s) expressing the Halo-tagged protein(s) of interest
- DMEM, low glucose, GlutaMAXTM Supplement, pyruvate (Fisher Scientific, catalog number: 10567014). Storage temperature: 4 °C. Shelf-life: 12 months from date of manufacture
- Fetal bovine serum (FBS) (HyClone, catalog number: SH30910.03). Not fully-defined FBS will also work. Store in 50 ml aliquots at -20 °C. Shelf-life: specified by the manufacturer
- 50x Penicillin-streptomycin mixture, 5,000 units potassium penicillin and 5,000 μg streptomycin sulfate per ml in 0.85% saline (LonzaTM 17-603E, Fisher Scientific, catalog number: BW17-603E). Store in 5 ml aliquots at -20 °C. Shelf-life: 12 months from date of manufacture
Note: The working concentration is 100 units of potassium penicillin and 100 μg of streptomycin sulfate per 1 ml of culture media. - TMR-HaloTag® ligand (Promega, catalog number: G8251). Storage temperature: -20 °C. Shelf-life: specified by the manufacturer
Note: If you need spectral properties different from those of TMR (555Ex/585Em), Promega offers a few alternatives, including HaloTag® ligands conjugated to Janelia Fluor® dyes. - Dimethyl sulfoxide, DMSO (Millipore Sigma, catalog number: D2650-5X5ML). Store at room temperature. Shelf-life: specified by the manufacturer. We recommend using a freshly opened ampule to dilute fluorescent HaloTag® ligands
- HCl (Fisher Scientific, catalog number: A144-50)
- NaCl (Fisher Scientific, catalog number: S271-10)
- KCl (Fisher Scientific, catalog number: P217-10)
- Na2HPO4 (Fisher Scientific, catalog number: P285-500)
- KH2PO4 (Fisher Scientific, catalog number: S375-212)
- NaOH pellets (Fisher Scientific, catalog number: S318-100), store at room temperature. Shelf-life: specified by the manufacturer
- 2.5% Trypsin (Fisher Scientific, Gibco, catalog number: 15090046). Storage temperature: -20 °C. Shelf-life: 24 months from date of manufacture for the 2.5% stock; 12 months from date of dilution for the diluted aliquots
- EDTA (C10H18N2Na2O10, Fisher Scientific, catalog number: S311-500). Storage temperature: room temperature. Shelf-life: specified by the manufacturer
- 10x PBS, pH 7.4 (1 L) (see Recipes)
- 1x PBS, pH 7.4 (see Recipes)
- 0.1 M EDTA pH 8.0 (see Recipes)
- 0.05% Trypsin (see Recipes)
- U2OS media (see Recipes)
- U2OS freezing media (see Recipes)
Equipment
- Cell culture incubator (Sanyo copper alloy IncuSafe humidified incubator, model number: MCO-18AIC(UV)). Set at 37 °C and 5.5% CO2
- Biosafety cabinet (Baker Sterligard III, model: SG403A)
- Water bath (Thermo Fisher Scientific Precision, model: 185 (2843))
- Microscope (Nikon Diaphot inverted phase-contrast microscope)
- Tabletop centrifuge (Eppendorf, model: 5702)
- Microfuge (Eppendorf, model: 5418)
- Vortex mixer (Scientific Industries, Inc., model: Vortex-Genie 2)
- BD LSRFortessaTM flow cytometer
Software
- BD FACSDivaTM 6.2 (BD, https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software)
- FlowJo 10.6.1 (BD, https://www.flowjo.com)
- Excel 16.30 or equivalent (Microsoft Corporation, https://www.microsoft.com/Microsoft/Excel)
Procedure
- Cell culture
Note: Perform all cell culture procedures in a biosafety cabinet to maintain sterility. General guidelines to perform cell and tissue culture are available from different organizations/companies [Abcam (Reference 1); ATCC (Reference 4); ECACC (Reference 11); LifeTechnologies (Reference 18)].- Culture your cell line(s) of interest side-by-side with the U2OS C32 cell standard you received from our lab. To thaw U2OS cells:
- Warm up U2OS media (Recipe 4) in a water bath set to 37 °C.
- Add 9 ml of U2OS media to a 15 ml conical tube.
- Put the cryovial in the 37 °C water bath and gently swirl until almost completely thawed.
- With a P1000 pipet, transfer the content of the cryovial (1 ml or less) to the 15 ml conical tube and invert the tube a few times.
- Spin cells down in a tabletop centrifuge (200 x g, 5 min, at room temperature).
- Aspirate the supernatant, flick the tube to resuspend the cells and add 10 ml of U2OS media.
- Plate to a 100-mm tissue culture dish and put cells overnight in the incubator set at 37 °C and 5.5% CO2.
- When cells are ready (60-80% confluent) split them to 6-well plates
Note: We normally perform the labeling experiment in 6-well plates, but you can scale it up or down as required.- Warm up U2OS media and 1x PBS in a water bath set to 37 °C, and trypsin at room temperature.
- Take the 100-mm plate out of the incubator.
- Aspirate media and wash with 10 ml of 1x PBS.
- Aspirate PBS and add 1.5 ml of trypsin.
- Move plate to the incubator for 1-2 min or until the cells look round and partially detached under a microscope (hitting the side of the plate with your hand helps detaching most of the cells).
- Add 10.5 ml of U2OS media and pipet extensively up and down to achieve a single-cell suspension.
- Distribute the cell suspension to 6-well microplates. Seed at least 2 wells per cell type: one to be left unlabeled to measure background fluorescence and the other to be stained with TMR. One ml of cells per well will give you enough cells to perform staining and flow cytometry the day after seeding (1 ml will contain ~250,000-500,000 U2OS cells when starting from a 60-80% confluent 100-mm plate). Use less if you need to delay the experiment (e.g., 0.5 ml to wait one additional day or 0.25 ml to wait a couple of days).
Note: We suggest you create your own stocks of the U2OS C32 cell standard at this point. To freeze U2OS cells, spin down all the cells you spare in a 15 ml conical tube. Resuspend the pellet in freezing media (Recipe 5) and aliquot to cryovials (we typically prepare 2 vials starting from a confluent 100-mm plate, resuspending the pellet in 500 μl to 1 ml of freezing media per vial). - Add media to 2 ml final volume and put plates back in the incubator.
- Culture your cell line(s) of interest side-by-side with the U2OS C32 cell standard you received from our lab. To thaw U2OS cells:
- TMR labeling
Note: When working with fluorophores, try and minimize light exposure (e.g., keep the hood’s lights off, cover plates and tubes with foil).- Pre-warm 1x PBS, U2OS media and any other media required for your specific cell line(s) at 37 °C, and trypsin at room temperature. You will need ~5 ml of media per each well of the 6-well plate.
Note: If your cell line of interest grows in suspension, spin cells down to replace media or to perform PBS washes. Because the cells will not need trypsinization, after the 5 min incubation to wash off unreacted ligand you can perform a PBS wash and directly resuspend cells in fresh media for flow cytometry. - Thaw the 5 mM TMR-HaloTag® ligand provided by Promega.
- Dilute 1:10 in DMSO to create a 1,000x stock solution (500 μM) and make smaller aliquots to be stored at -20 °C (e.g., dilute 10 μl to 90 μl DMSO and prepare 10x 10 μl aliquots). Avoid freeze-thawing the 1,000x stock if you can, although a few freeze-thawing cycles should not be an issue.
- Prepare 1.5 ml of labeling media per each well to be stained, diluting the 1,000x TMR-HaloTag® ligand stock in cell culture media (1:1,000 dilution: 1.5 μl in each 1.5 ml of media). Remember to leave 1 well unlabeled for each cell line to be measured.
- Take out the 6-well plates you prepared the previous day(s) from the incubator.
- Aspirate the regular cell culture media and add 1.5 ml of either labeling media (labeled wells) or regular media (unlabeled wells for background measurement).
- Transfer the plates back to the 37 °C incubator for 30 min.
- Aspirate the supernatant and wash with 4 ml of 1x PBS.
- Aspirate the PBS and add 2 ml of regular media.
- Transfer the plates back to the 37 °C incubator for 5 min to remove unreacted ligand.
- Pre-warm 1x PBS, U2OS media and any other media required for your specific cell line(s) at 37 °C, and trypsin at room temperature. You will need ~5 ml of media per each well of the 6-well plate.
- Collect cells for flow cytometry
- Trypsinize cells. For each well:
- Wash with 2 ml 1x PBS.
- Aspirate PBS and add 300 μl of trypsin.
- Move the plates back to the 37 °C incubator as long as needed to detach the cells.
- Inactivate trypsin by adding 900 μl of cell culture media.
- Resuspend each well up and down a few times with a P1000 and transfer the contents to a 1.5 ml Eppendorf tube.
- Spin cells down in a microfuge (200 x g, 5 min, at room temperature).
- Resuspend cell pellets in 500 μl of fresh media.
- Transfer cells to flow cytometry test tubes passing them through the tubes’ cell strainer snap cap.
- Store tubes on ice, protected from light, and proceed immediately to flow cytometry.
- Trypsinize cells. For each well:
- Measure fluorescence intensities by flow cytometry
Most flow cytometers and software will work, but here we illustrate the protocol using a BD LSRFortessaTM instrument and the associated BD FACSDivaTM acquisition software.
Note: If you are using a different flow cytometer and/or a different acquisition software, refer to their operation manuals. If you are not familiar with flow cytometry, we advise running at least the first flow analysis with an experienced user. Also, we suggest to set up the flow cytometry experiment and adjust all the required settings in advance, so that the stained cells only sit on ice for a limited time (~20 min).- Turn on the BD LSRFortessaTM flow cytometer and the computer connected to it.
- Open the BD FACSDivaTM acquisition software and wait for the cytometer to connect. If you are not familiar with the software, you can find the complete reference manual for the 6.0 version at https://www.bu.edu/flow-cytometry/files/2010/10/BDFACSDivaSoftwareReferenceManual.pdf.
- If a “CST Mismatch” window appears, click on “Use CST Settings”.
- In the upper menu bar, click “View” and make sure the following items are checked, and thus the corresponding window open: Toolbar, Status Bar, Browser, Cytometer, Inspector, Worksheet and Acquisition Dashboard.
- In the upper menu bar, click “Experiment” and choose “New Experiment” from the dropdown menu.
- Select a blank template. Your experiment will now appear as an opened book in the Browser window. Right-click on the experiment name and click “Rename” to assign a different name.
- Below the experiment icon in the Browser window, click on “Cytometer Settings”.
- Move to the Inspector window to setup the Cytometer Settings. In the “Parameters” tab, select and delete everything but FSC (forward scatter), SSC (side scatter) and PE-Tx-Red-YG (for TMR staining; the instrument will use a 561-nm laser as an excitation source and read the emission through a 610/20 band pass filter or similar (TMR emission max is ~585 nm). If you are using a different fluorophore, modify accordingly. Check “A” and “H” for the FSC and SSC parameters to record the signal area and height (this will allow you to distinguish single cells from doublets during analysis). For the PE-Tx-Red-YG parameter, check “A” and “Log”, for the data to be in logarithmic scale.
- Move back to the upper menu bar, click “Experiment” and choose “New Specimen” from the dropdown menu. A Panel Template window will appear. Click “OK”. A new specimen will be created (syringe icon) as a dependent of your open experiment in the Browser window. Right-click on the specimen name and change it to today’s date (YYYYMMDD).
- In the Browser window, select the Global Worksheets and then move to the Global Worksheet window.
- Draw a dot plot (5th button from the left in the window toolbar, click and drag anywhere in the sheet; the dot plot will appear as you release the mouse).
- Create a polygon gate (14th button from the left) by positioning the cursor inside the dot plot and clicking several times to draw your shape of choice. You will adjust the gate to select live cells later, when acquiring your sample.
- Create a histogram plot (7th button from the left) and, if needed, click on the x-axis to change the displayed parameter to PE-Tx-Red-YG.
- With the histogram plot selected, move to the Inspector window and check “P1”. This will only display the TMR signal from the live cells that you gated in 12.
- Move back to the Browser window.
- Expand the specimen icon by clicking on the “+” sign to expose the “Tube” icon. Right-click on it and rename it with your sample name (U2OS_C32_unlabeled).
- Click on the left grey pointer to select the sample you have just created.
- Take your first sample (U2OS_C32_unlabeled), vortex briefly, remove the cap and load it on the flow cytometer.
- Set the flow rate to low using the “LO” button in the instrument control panel.
- Hit the “RUN” button in the instrument control panel.
- Go back to the Diva software, move to the Acquisition Dashboard and hit “Acquire” to visualize your cells in the Worksheet window.
- In the Cytometer window, adjust FSC and SSC voltages to distinguish dead from live cells in the Worksheet dot plot (for U2OS we use ~220 for the FSC and ~280 for the SSC). In the Acquisition Dashboard window, hit “Restart” to only visualize the cells acquired with the adjusted voltages. Now you can adjust your P1 gate to only include live cells.
Note: Forward and side scatter values vary from cell to cell depending on cellular size (FSC) and internal complexity (SSC). Live cells have larger FSC and SSC values than dead cells or cell debris, and normally constitute the majority of the population. Cell doublets/aggregates will have higher FSC and SSC values than single cells but should not represent the majority of cells, if you have thoroughly resuspended the samples (you will exclude doublets at the analysis stage. See Step E5 below). - Back to the Inspector window, adjust the PE-Tx-Red-YG voltage to move the Worksheet histogram to the leftmost part of the x-axis while still displaying all the events (for U2OS we use ~520).
- In the Acquisition Dashboard window, hit “Stop Acquiring”, and in the instrument control panel hit “STNDBY” to pause acquisition.
- Quickly check other unlabeled samples to make sure that the voltages you chose for U2OS cells are reasonable for the other cell types as well. If not, adjust them as necessary.
Notes:- We usually run water between one sample and the other, to avoid cross-sample contamination.
- When working with different cell types you might need to move the P1 gate between one cell type and the other. However, you must find voltage values that are acceptable for all cell types and keep them constant during acquisition of all samples.
- Go back to your original sample. Repeat Steps D18-D21. If no further voltage adjustments are required, in the Acquisition Dashboard window modify the Acquisition Setup to decide which Stopping Gate to use (choose P1) and how many Events to Record (choose 50,000). In case you need to re-adjust voltages, go back to Step D25.
- Click the “Record Data” button to save data to an FCS file. Increase the flow rate to MED or HIGH to speed up the acquisition if samples are too diluted. Once the specified number of events are recorded, on the instrument control panel hit “STNDBY” to pause acquisition.
- In the Acquisition Dashboard window click on “Next Tube” to create the next sample. You can rename it either right-clicking on it in the Browser window or from the “Tube” tab in the Inspector window.
- Take the next sample and repeat Steps D18-D21 and D27-D28, until you have acquired and recorded all your samples.
- Perform any recommended cleaning procedure for your flow cytometer.
- In the Browser window, export your data by right-clicking on the specimen name. Select “Export”, “FCS files” and leave the default FCS3.0 format. Click “OK”, when prompted choose a directory where to save your data and click “Save”.
- Quit the BD FACSDIVATM software (from the top menu click first “File” and then “Quit”).
- Analyze flow cytometry data and obtain mean fluorescence intensities using the FlowJo software (see docs.flowjo.com/d2/ for full software documentation)
Note: You can use the BD FACSDivaTM software for both data acquisition and analysis, instead of FlowJo. A license-free alternative is the Matlab code we used to analyze the data in our original manuscript (Cattoglio et al., 2019), available at https://gitlab.com/tjian-darzacq-lab/cattoglio_et_al_absoluteabundance_2019.- Open the FlowJo program.
- Drag and drop your FCS 3.0 files into the lower workspace window.
- Select the U2OS C32 unlabeled control.
- Create a gate on live cells.
- Double-click on the file name; this opens a dot plot of forward (FSC-A) and side scatter (SSC-A) values.
- Create a free-polygon gate (5th button from the top left) to enclose live cells (name it “Live Cells”).
- In the workspace window select the “Live Cells” gate and apply it to all samples (simply drag and drop it on the above window on the “All Samples” group).
- Go back to the forward and side-scatter plot; move from one sample to the next using the arrow at the top-right of the window and adjust the “Live Cells” gate as needed, dragging it around. If necessary, you can also modify the gate shape (Figure 2).
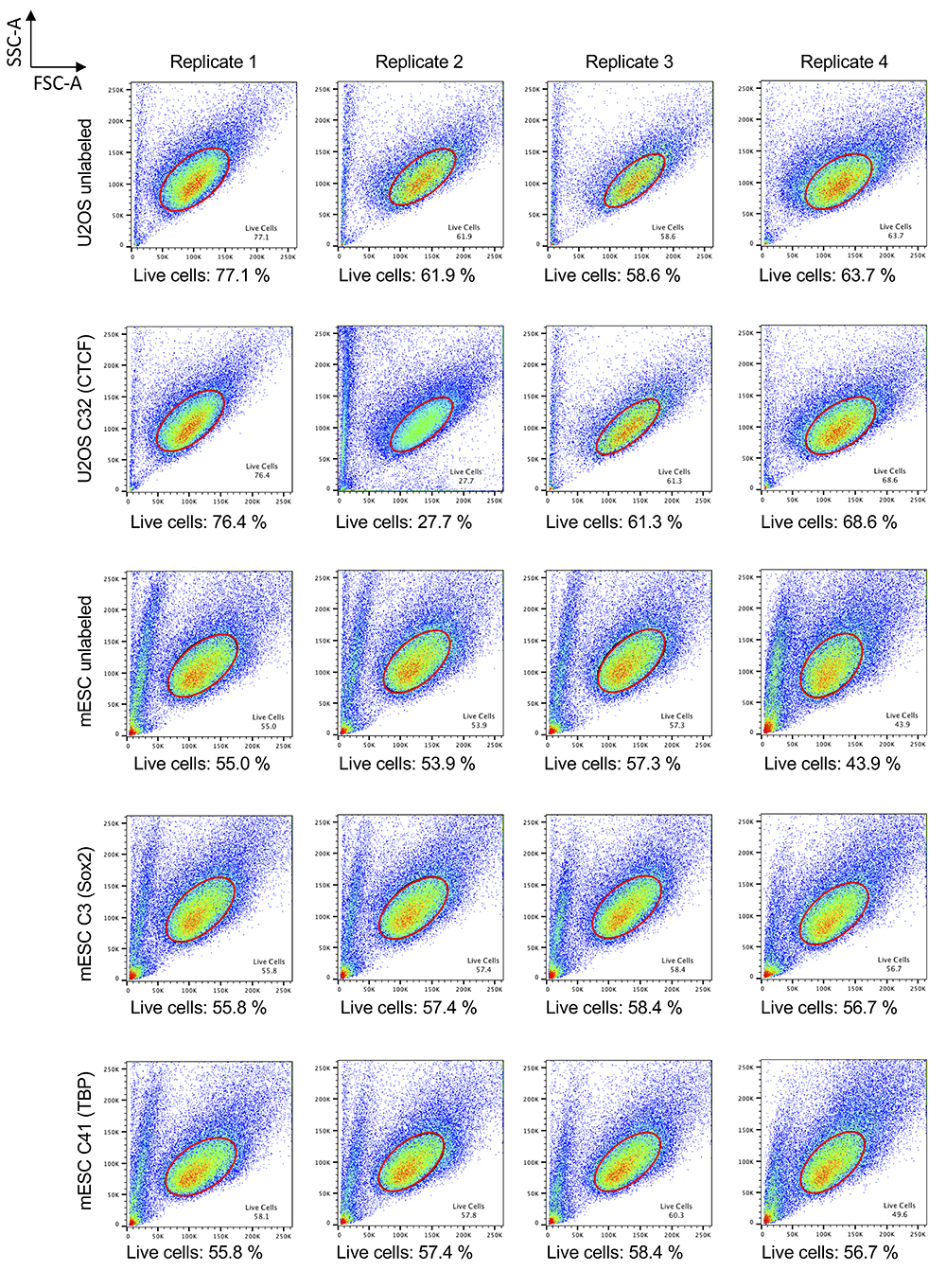
Figure 2. Live-cell gating in FlowJo. Forward- and side-scatter plots generated by FlowJo that illustrate the “Live Cells” gating applied to the data re-analyzed here. The gating choice is likely to affect your absolute quantifications, since cell size correlates with protein abundances.
- Exclude cell doublets
Note: In the original flow cytometry experiments published in Cattoglio et al. (2019) we did not record FSC-H and SSC-H values. Thus, in the re-analysis of Table 1 below we could not exclude cell doublets as recommended in Step E5.- Go back to the U2OS C32 unlabeled control’s scatter plot and double-click anywhere inside the “Live Cells” gate. This will open a new scatterplot that only displays gated cells.
- Set the y-axis to FSC-H and the x-axis to FSC-A to exclude doublets. Single cells localize along the same diagonal, while doublets lie underneath (Figure 3).
- Create another free-polygon gate to only include cells along the top diagonal. Click “OK” to name it “Single Cells”.
- In the workspace window select the “Single Cells” gate and apply it to all samples (simply drag and drop it on the above window on the “Live Cells” subgroup).
- Adjust the “Single Cells” gate to each sample individually as you did in Step E4d for the “Live Cells” gate.
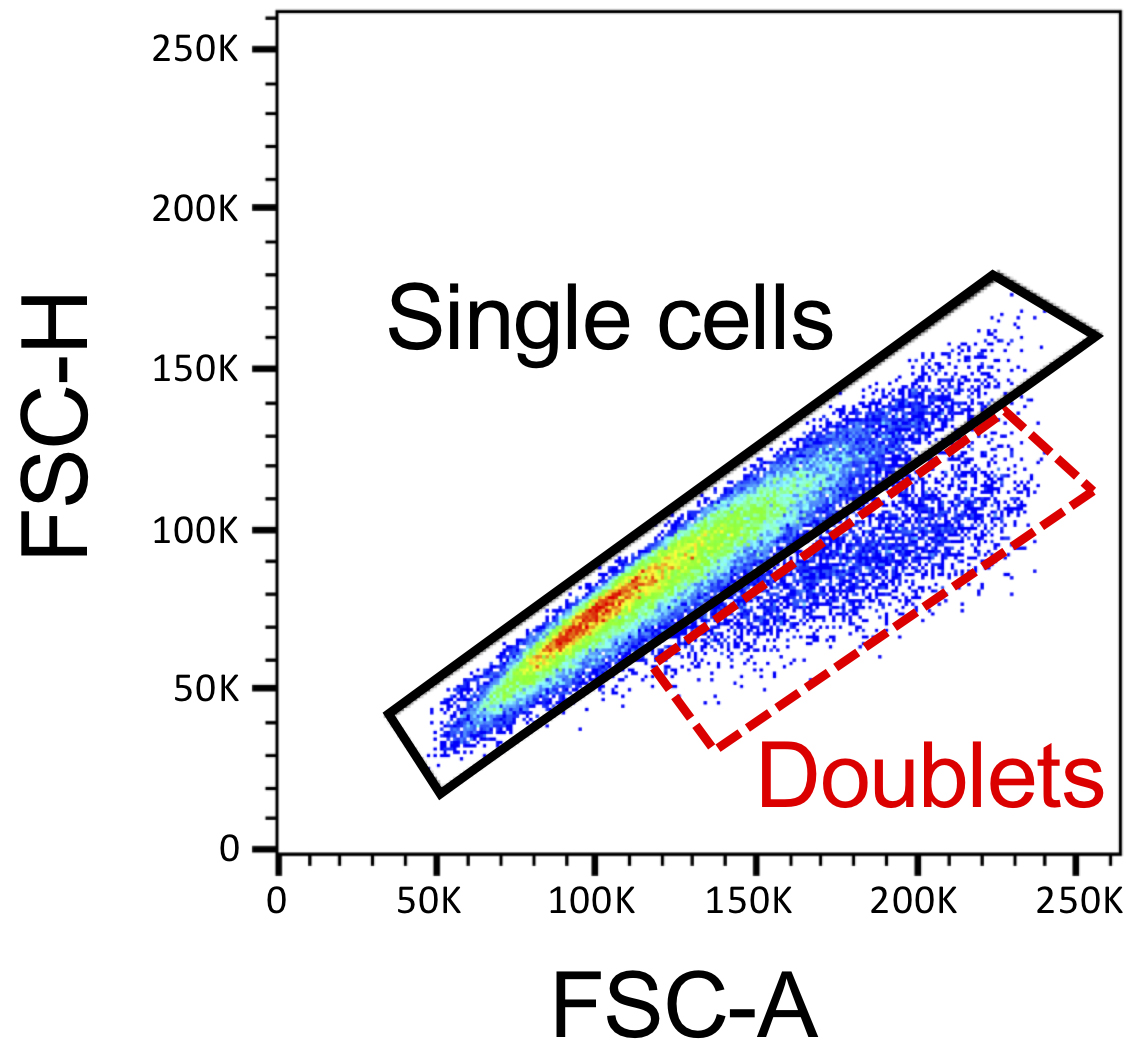
Figure 3. Doublets exclusion in FlowJo. FSC-H and FSC-A scatter plot generated by FlowJo that illustrate single cells lying on the main diagonal (black polygon gate) and cell doublets just underneath it (red dashed gate).
- Obtain a mean fluorescence intensity (MFI) table
- Back to the workspace, select the Table Editor and click the “Edit” tab. Click “Add Column” and select “Mean” on the left, and the “Single Cells” Population and the “PE-Tx-Red-YG-A” Parameter from the dropdown menus on the right.
- Click OK and go back to the “Table Editor” tab. Click on “Create Table”. This generates a table with the mean fluorescence intensities for all the samples and the unlabeled controls. To download the table as an Excel file, from the dropdown menus to the right of the “Create Table button” choose “To File” and “Excel”. Choose a “Destination” folder and hit “Create Table” again.
- Quit FlowJo saving the workspace.
- Obtain the absolute amount of your protein of interest
- Open the table generated by FlowJo in Microsoft Excel.
- For each TMR-labeled cell line, subtract the MFI of the unlabeled sample (background fluorescence, bkg) from the MFI of each TMR-labeled sample to obtain the adjusted MFI as follows:MFIadj = MFITMR - MFIbkg
- Use the MFIadj values to calculate the absolute abundance of the protein of interest (nPOI) as follows:
- Average nPOI obtained over several biological replicates to get mean and standard deviation values.
- Table 1 details a FlowJo re-analysis of the original data published in Cattoglio et al. (2019). You can download the raw FCS 3.0 files from github at https://gitlab.com/tjian-darzacq-lab/cattoglio_et_al_absoluteabundance_2019/tree/master/Data. Note that FSC/SSC gating can affect final absolute estimates, because cells of different size will also have different protein amounts (the gating applied in FlowJo for these samples is in Figure 2). The numbers we report here for mouse embryonic stem cells thus vary from those we initially published. Specifically, we now estimate ~539,000 ± 36,500 Sox2 molecules (vs. ~460,500 ± 25,600 in Cattoglio et al., 2019) and ~111,000 ± 32,000 TBP molecules (vs. ~99,100 ± 29,100 in Cattoglio et al., 2019)
Table 1. Absolute quantification of Sox2 and TBP in mouse embryonic stem cells. For each cell line, where a different protein of interest (POI) is endogenously Halo-tagged, the table reports flow cytometry fluorescence values (MFI, mean fluorescence intensity) obtained in 4 biologically independent replicates, separated by commas. We then subtract the background fluorescence of the unlabeled sample to obtain adjusted MFI values (MFIadj). We finally use these values to calculate absolute abundances of Sox2 and TBP in the mouse embryonic stem cell (mESC) lines C3 and C41, as detailed in Steps F2 to F4. The nPOI we report in the last column is the absolute quantification averaged across the 4 replicates ± standard deviation.A.U.: fluorescence arbitrary unitsCell line POI MFI (A.U.) MFIadj nPOI U2OS_unlabeled 574, 1071, 1216, 513 Subtracted from C32 U2OS_C32_TMR CTCF 7761, 10883, 10978, 5285 7187, 9812, 9762, 4772 109,800; standard mESC_unlabeled 451, 616, 640, 309 Subtracted from C3 and C41 mESC_C3_TMR Sox2 32260, 49995, 49373, 25052 31809, 49379, 48733, 24743 538,996 ± 36,512 mESC_C41_TMR TBP 6434, 9036, 9562, 7175 5983, 8420, 8922, 6866 110,991 ± 31,549
nPOI = (MFIadj,COI) ÷ (MFIadj,C32) × 109,800
where MFIadj,COI is the adjusted mean fluorescence intensity of the cell line of interest (COI) that expresses the Halo-tagged protein under investigation, MFIadj,C32 is the background-subtracted mean fluorescence intensity of the C32 cell standard and 109,800 is the absolute amount of Halo-CTCF we estimated in our standard U2OS C32 cell line.
Data analysis
Data analysis only entails the calculations detailed above (protocol Steps F2 to F4). Table 1 shows intermediary and final data after re-analysis of a subset of the TMR-labeling experiments first described in Cattoglio et al. (2019). The raw files are available for download at https://gitlab.com/tjian-darzacq-lab/cattoglio_et_al_absoluteabundance_2019/tree/master/Data.
Recipes
- 10x PBS, pH 7.4 (1 liter)
- To 800 ml of double-distilled water (ddH2O) add NaCl (80 g), KCl (2 g), Na2HPO4 (14.4 g) and KH2PO4 (2.4 g). Stir to dissolve
- Wear eye protection and adjust the pH to 7.4 with 6 N HCl (Thermo Fisher Scientific To prepare a 6 N HCl solution dilute the concentrated acid 1:1 with ddH2O)
- Bring the volume up to 1 liter with ddH2O and dispense 100 ml into autoclavable glass bottles
- Autoclave 30 min on liquid cycle
- 1x PBS, pH 7.4
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
2 mM KH2PO4
Dilute from a 10x autoclaved solution (Recipe 1) and re-autoclave
Note: You can also purchase ready-to-use, sterile 1x PBS. Storage temperature: 15-30 °C. Shelf-life: 12 months from date of preparation. - 0.1 M EDTA, pH 8.0 (1 L)
- To 700 ml of ddH2O add 37.22 g of EDTA and stir (EDTA will not dissolve until the pH is near to 8.0)
- Adjust the pH to 8.0 with NaOH pellets (wear eye protection and mask)
- Bring the solution up to volume with ddH2O
- Autoclave 30 minutes on liquid cycle
- 0.05% Trypsin
- To 1 liter of 1x PBS add 10 ml 0.1 M EDTA, pH 8.0
- Autoclave for 30 min, prepare 50 ml aliquots and add 1 ml of Gibco → 2.5% trypsin stock solution to each aliquot
- Store aliquots at -20 °C
- U2OS media
To a 500-ml low-glucose DMEM bottle add 50 ml of FBS (~10% final) and 5 ml of pen/strep mixture - U2OS freezing media
- To 1 ml of DMSO add 9 ml U2OS media (10% DMSO final). Open the DMSO in the biosafety cabinet to ensure sterility and aliquot the remaining DMSO to 15 ml conical tubes (1 ml aliquots)
- Store the freezing media in the fridge for up to 1 month and the DMSO aliquots at -20 °C for 12 months
Acknowledgments
ASH acknowledges support from a Siebel Stem Cell Institute post-doctoral fellowship and NIH NIGMS K99 Pathway to Independence Award K99GM130896. The Tjian-Darzacq lab is supported by NIH Common Fund 4D Nucleome Program U01-EB021236 and U54-DK107980 (XD), the California Institute of Regenerative Medicine grant LA1-08013 (XD), and by the Howard Hughes Medical Institute (003061, RT). The protocol detailed here was first described in Cattoglio et al. (2019) and it used Halo-tagged Sox2 and TBP mESC lines previously generated by Sheila Teves (Teves et al., 2016 and 2018). We thank Elena Slobodyanyuk for testing this protocol and Nike Walther for providing comments to this manuscript.
Competing interests
The authors declare no competing financial/non-financial interests.
References
- Abcam (2019). Cell culture guidelines. Accessed 24 October 2019.
- Addgene. CRISPR Guide. Accessed 24 October 2019.
- Aird, E. J., Lovendahl, K. N., St Martin, A., Harris, R. S. and Gordon, W. R. (2018). Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun Biol 1: 54.
- ATCC. Animal cell culture guide. Accessed 24 October 2019.
- Bantscheff, M., Lemeer, S., Savitski, M. M. and Kuster, B. (2012). Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem 404(4): 939-965.
- Bantscheff, M., Schirle, M., Sweetman, G., Rick, J. and Kuster, B. (2007). Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem 389(4): 1017-1031.
- Bennett, R. J., Simpson, D. M., Holman, S. W., Ryan, S., Brownridge, P., Eyers, C. E., Colyer, J. and Beynon, R. J. (2017). DOSCATs: Double standards for protein quantification. Sci Rep 7: 45570.
- Cai, Y., Hossain, M. J., Hériché, J. K., Politi, A. Z., Walther, N., Koch, B., Wachsmuth, M., Nijmeijer, B., Kueblbeck, M., Martinic-Kavur, M., Ladurner, R., Alexander, S., Peters, J. M. and Ellenberg, J. (2018). Experimental and computational framework for a dynamic protein atlas of human cell division. Nature 561(7723): 411-415.
- Cattoglio, C., Pustova, I., Walther, N., Ho, J. J., Hantsche-Grininger, M., Inouye, C. J., Hossain, M. J., Dailey, G. M., Ellenberg, J., Darzacq, X., Tjian, R. and Hansen, A. S. (2019). Determining cellular CTCF and cohesin abundances to constrain 3D genome models. Elife 8: e40164.
- Chen, J. Q., Heldman, M. R., Herrmann, M. A., Kedei, N., Woo, W., Blumberg, P. M. and Goldsmith, P. K. (2013). Absolute quantitation of endogenous proteins with precision and accuracy using a capillary Western system. Anal Biochem 442(1): 97-103.
- ECACC. (2019). Fundamental techniques in cell culture Fundamental techniques in cell culture. Accessed 24 October 2019.
- England, C. G., Luo, H. and Cai, W. (2015). HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem 26(6): 975-986.
- Ghaemmaghami, S., Huh, W. K., Bower, K., Howson, R. W., Belle, A., Dephoure, N., O'Shea, E. K. and Weissman, J. S. (2003). Global analysis of protein expression in yeast. Nature 425(6959): 737-741.
- Hansen, A. S., Pustova, I., Cattoglio, C., Tjian, R. and Darzacq, X. (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6: e25776.
- Haupt, A., Grancharova, T., Arakaki, J., Fuqua, M. A., Roberts, B. and Gunawardane, R. N. (2018). Endogenous protein tagging in human induced pluripotent stem cells using CRISPR/Cas9. J Vis Exp(138). doi: 10.3791/58130.
- Holzmann, J., Politi, A. Z., Nagasaka, K., Hantsche-Grininger, M., Walther, N., Koch, B., Fuchs, J., Dürnberger, G., Tang, W., Ladurner, R., Stocsits, R. R., Busslinger, G. A., Novak, B., Mechtler, K., Davidson, I. F., Ellenberg, J. and Peters, J. M. (2019). Absolute quantification of cohesin, CTCF and their regulators in human cells. Elife 8: e46269.
- Kovalenko, E. I., Ranjbar, S., Jasenosky, L. D., Goldfeld, A. E., Vorobjev, I. A. and Barteneva, N. S. (2011). The use of HaloTag-based technology in flow and laser scanning cytometry analysis of live and fixed cells. BMC Res Notes 4: 340.
- LifeTechnologies. (2019). Cell culture basics. Accessed on 24 October 2019.
- Liu, P., Li, Y., Lei, J. and Dong, L. (2019). Creating knockin alleles in mouse embryonic stem cells by CRISPR/Cas9-mediated homologous recombination without drug selection. Methods Mol Biol 1874: 115-137.
- Los, G. V., Encell, L. P., McDougall, M. G., Hartzell, D. D., Karassina, N., Zimprich, C., Wood, M. G., Learish, R., Ohana, R. F., Urh, M., Simpson, D., Mendez, J., Zimmerman, K., Otto, P., Vidugiris, G., Zhu, J., Darzins, A., Klaubert, D. H., Bulleit, R. F. and Wood, K. V. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3(6): 373-382.
- Politi, A. Z., Cai, Y., Walther, N., Hossain, M. J., Koch, B., Wachsmuth, M. and Ellenberg, J. (2018). Quantitative mapping of fluorescently tagged cellular proteins using FCS-calibrated four-dimensional imaging. Nat Protoc 13(6): 1445-1464.
- Presman, D. M., Ball, D. A., Paakinaho, V., Grimm, J. B., Lavis, L. D., Karpova, T. S. and Hager, G. L. (2017). Quantifying transcription factor binding dynamics at the single-molecule level in live cells. Methods 123: 76-88.
- Roberts, B., Haupt, A., Tucker, A., Grancharova, T., Arakaki, J., Fuqua, M. A., Nelson, A., Hookway, C., Ludmann, S. A., Mueller, I. A., Yang, R., Horwitz, R., Rafelski, S. M. and Gunawardane, R. N. (2017). Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol Biol Cell 28(21): 2854-2874.
- Sharma, A., Toepfer, C. N., Ward, T., Wasson, L., Agarwal, R., Conner, D. A., Hu, J. H. and Seidman, C. E. (2018). CRISPR/Cas9-mediated fluorescent tagging of endogenous proteins in human pluripotent stem cells. Curr Protoc Hum Genet 96: 21 11 21-21 11 20.
- Tasan, I., Sustackova, G., Zhang, L., Kim, J., Sivaguru, M., HamediRad, M., Wang, Y., Genova, J., Ma, J., Belmont, A. S. and Zhao, H. (2018). CRISPR/Cas9-mediated knock-in of an optimized TetO repeat for live cell imaging of endogenous loci. Nucleic Acids Res 46(17): e100.
- Teves, S. S., An, L., Bhargava-Shah, A., Xie, L., Darzacq, X. and Tjian, R. (2018). A stable mode of bookmarking by TBP recruits RNA polymerase II to mitotic chromosomes. Elife 7: e35621.
- Teves, S. S., An, L., Hansen, A. S., Xie, L., Darzacq, X. and Tjian, R. (2016). A dynamic mode of mitotic bookmarking by transcription factors. Elife 5: e22280.
- Walther, N., Hossain, M. J., Politi, A. Z., Koch, B., Kueblbeck, M., Odegard-Fougner, O., Lampe, M. and Ellenberg, J. (2018). A quantitative map of human Condensins provides new insights into mitotic chromosome architecture. J Cell Biol 217(7): 2309-2328.
- Zhang, X. R., He, J. B., Wang, Y. Z. and Du, L. L. (2018). A cloning-free method for CRISPR/Cas9-mediated genome editing in fission yeast. G3 (Bethesda) 8(6): 2067-2077.
Article Information
Copyright
![]() Cattoglio et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Cattoglio et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Cattoglio, C., Darzacq, X., Tjian, R. and Hansen, A. S. (2020). Estimating Cellular Abundances of Halo-tagged Proteins in Live Mammalian Cells by Flow Cytometry. Bio-protocol 10(4): e3527. DOI: 10.21769/BioProtoc.3527.
- Cattoglio, C., Pustova, I., Walther, N., Ho, J. J., Hantsche-Grininger, M., Inouye, C. J., Hossain, M. J., Dailey, G. M., Ellenberg, J., Darzacq, X., Tjian, R. and Hansen, A. S. (2019). Determining cellular CTCF and cohesin abundances to constrain 3D genome models. Elife 8: e40164.
Category
Cell Biology > Cell-based analysis > Flow cytometry
Biochemistry > Protein > Quantification
Molecular Biology > Protein > Flow Cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.