- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Expression and Purification of Adeno-associated Virus Virus-like Particles in a Baculovirus System and AAVR Ectodomain Constructs in E. coli
Published: Vol 10, Iss 3, Feb 5, 2020 DOI: 10.21769/BioProtoc.3513 Views: 8101
Reviewed by: Longping Victor TseBeatrice LiAkira Karasawa
Original research article
The authors used this protocol in:

May 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Adeno-associated virus (AAV) is a promising gene therapy vector and the biophysical characterization of its interactions with host proteins is a critical foundation for engineering tissue targeting and immune escape. Presented here are protocols for the production of: (a) the outer protein shells (virus-like particles or VLPs) for serotype 2 (AAV-2) and (b) two fragments from the binding ectodomain of AAV’s cellular receptor, AAVR. His6PKD1-2 comprises the first two polycystic kidney disease (PKD) domains, the minimal required for efficient binding of AAV, expressed with an N-terminal histidine tag. MBP-PKD1-5 is a fusion of the maltose binding protein with all five of the PKD domains of the AAVR receptor. Presented are the expression and purification of milligram quantities, ample for in vitro analyses. For AAV-2, the protocol offers an alternative to the use of (infectious) wild-type virus or transducing vectors. One of the methods for producing transducing vector is in Sf9 cells, and the production of VLPs is based on this. For AAVR, the protocols enable biochemical and biophysical characterization of virus-binding. The minimal two-domain construct allows more saturated binding to symmetry-equivalent sites on the virus, while the larger construct might be better expected to reflect the native receptor.
Keywords: VirusBackground
The human parvovirus Adeno-associated virus (AAV) has a 60-subunit protein capsid shell containing a single-stranded DNA genome (Xie et al., 2002; Chapman and Agbandje-McKenna 2006). Several recombinant AAV serotypes are employed as vectors in emerging clinical gene therapy applications (Gaudet et al., 2013; Russell et al., 2017). AAV infection starts with capsid attachment to serotype-specific extracellular glycans, followed by receptor-mediated endocytotic entry. The cellular receptor, AAVR, otherwise known as KIAA0319L, was implicated as key for entry of a panel of AAV serotypes into representative cell types and for transduction in mouse models (Pillay et al., 2016); although it is now known that the AAV-4 clade, comprising three related AAVs, enter cells independently of AAVR (Dudek et al., 2018). For the majority of AAVs, AAVR functions not only in cell entry, but in trafficking endosomally localized AAV towards the peri-nuclear trans Golgi network. This process likely mimics an uncharacterized host-cell function of the AAVR protein as a recycling receptor (Pillay et al., 2016). AAVR is an integral transmembrane glycoprotein but AAVR's function in binding AAV is independent of its glycosylation state, thereby opening several possibilities of heterologous AAVR expression for in vitro study of its interactions with AAV (Pillay et al., 2016 and 2017).
Potential relevance for improving gene therapy delivery is driving intense interest in AAV-receptor interactions. It is presumed that interactions with AAVR, or possibly other receptors, might be part-mediators of host-cell specificity that can differ among AAV serotypes. A detailed understanding of AAV-receptor interactions may, in turn, open a path towards engineering different tropism or higher efficiency.
Immune neutralization can be another limitation of in vivo viral vector delivery (Wang et al., 2011; Fitzpatrick et al., 2018). Strategies for developing immune-escape variants are currently being explored (Li et al., 2012; McCraw et al., 2012; Tseng and Agbandje-McKenna, 2014; Buning et al., 2015; Majowicz et al., 2017; Paulk et al., 2018). Therefore, a mapping of key AAV-receptor interactions is needed to understand the limits of capsid modification if viable transduction is to be retained (Meyer et al., 2019).
When structural studies of AAV were first attempted in the 1990s, methods were not available to produce the milligram levels of AAV needed for crystallization. Structural studies became possible with the optimization of methods to produce wild-type (i.e., infectious) AAV-2 by transfection of cultured human cells with virus-encoding bacterial plasmid (Samulski et al., 1982; Laughlin et al., 1983) in the presence of the adenovirus helper needed for AAV replication (Xie et al., 2004). The elucidation of the type-species structure, AAV-2 (Xie et al., 2002), paved the way for studies of immunologically distinct serotypes. Structural studies of several serotypes (e.g., AAV-3, AAV-4, AAV-5, AAV-6) used sample prepared in analogous ways (Kaludov et al., 2003; Walters et al., 2004; Xie et al., 2008; Lerch et al., 2010). More recently, biophysical studies have taken advantage of other preparative methods.
At first, it was not practical to use recombinant AAV (rAAV) vectors produced in human cells by double or triple transfection (Grimm et al., 1998; Matsushita et al., 1998), because the quantities, while sufficient for transduction experiments, were insufficient for biophysics. The higher-yield production of rAAV-2 vector in insect cells using baculoviral vectors (Urabe et al., 2002) inspired a different approach, that was used for the crystal structures of AAV8 and AAV9 (Nam et al., 2007, DiMattia et al., 2012). The same bacmids encoding capsid proteins are used, but bacmids encoding the AAV ITR vector genome and the replication protein are omitted. This generates empty capsid protein shells, or virus-like-particles (VLPs), instead of vectors (Lane et al., 2005). The same approach has been used for cryo-electron microscopy (EM), as exemplified by the engineered vector, AAV-DJ which has been solved at near atomic resolution (Lerch et al., 2012, Xie et al., 2017).
This protocol provides details for purifying milligram quantities of type species AAV-2 VLPs based on the AAV-DJ strategy summarized earlier (Lerch et al., 2012). We combine AAV8-like insect cell expression of VLPs with high-capacity purification steps used for the biophysics of wild-type viruses (Xie et al., 2004; Lerch et al., 2012) that can be more stringent than in rAAV productions designed for in vivo use. Repeated CsCl density gradient ultracentrifugation and desalting via dialysis is employed in contrast to the more common use of iodixanol for rAAV vectors (Hermens et al., 1999) and VLPs (Drouin et al., 2016). Furthermore, we take advantage of the heparin binding of AAV2-like viruses with an affinity purification step in lieu of ion exchange chromatography (Drouin et al., 2016).
The shift from crystallography to cryo-EM means that AAV structure is now possible with the smaller quantities available by rAAV triple transfection production methods (Zhang et al., 2019). (In our hands, preparation of a single EM grid uses ~1 μg, but for the optimization of conditions one would like > 25 μg.) The AAV-2 structure has now been determined by crystallography for infectious wild-type (wt) particles (Xie et al., 2002), by cryo-EM for VLPs (Drouin et al., 2016) and cryo-EM for rAAV vectors (Zhang et al., 2019). For most of the capsid protein, the structures superimpose within the experimental error at 2.8 to 3.8 Å resolution but some differences have been noted near the surface spikes (Drouin et al., 2016) where crystal packing contacts could influence structure. The three sources of material (wt, rAAV & VLP) can all support studies of atomic structure, but the highest resolution structures are coming from VLPs. There could be other explanations, but one possible reason is the purity achievable with the higher yield VLP protocol presented here.
The capsids of natural AAV are comprised mostly of viral protein (VP) 3, but about 10% of VP3s in the 60-subunit capsid are replaced by variant VP1, and another ~10% by VP2 (Berns, 1996). The three proteins are mostly the same, but VP1 and 2 have N-terminal extensions derived from alternate start codons. VLP constructs are designed to express variants in the right proportions (Urabe et al., 2002). However, it is only the 60-fold symmetric parts of AAV that have been resolved in structures. So, even though the properties of wt, rAAV and VLP appear similar, one cannot verify that the structures of the N-terminal extensions are configured exactly the same. In spite of this caveat, VLPs are considered to be a good proxy for wild-type and rAAV vectors, and are often chosen, because of the ease of producing large quantities of pure sample without biosafety level II containment.
The receptor protein, AAVR, is an integral membrane glycoprotein, that consists of the following domains, starting from the extracellular N-terminus: a signal peptide (SP), a MANEC domain (motif at the N-terminus with eight cysteines), five immunoglobulin-like polycystic kidney disease (PKD) domains (Ibraghimov-Beskrovnaya et al., 2000), a transmembrane region and a small cytoplasmic domain. A soluble fusion construct consisting of maltose binding protein (MBP), N-terminally fused to PKD1-5 (Pillay et al., 2016), and lacking SP, MANEC, TM and the C-terminal tail was pivotal in implicating the PKD domains in AAV-binding via inhibition of rAAV cell transduction. This soluble construct, herein denoted MBP-PKD1-5, was used to measure competitive inhibition of AAV cell-transduction by solubilized receptor ectodomain, to measure AAV-binding strength using ELISA and surface plasmon resonance, and also for low resolution cryo-Electron Tomographic imaging of an AAV-2/AAVR complex (Pillay et al., 2016, Meyer et al., 2019). The expression and purification of the MBP-PKD1-5 AAVR construct is described in this protocol.
Over-expression of “mini-AAVR” on the cell surface supports AAV transduction in an AAVR knock-out (KO) (Pillay et al., 2016). The mini-AAVR construct consists of the SP domain, omits the MANEC domain, and contains PKD domains 1-3 linked directly to the TM domain and the cytoplasmic tail. Additional AAVR domain deletion mutants, expressed in an AAVRKO, indicated that PKD domains 4 and 5 had little negative impact upon rAAV-2 transduction, and that deletion of PKD3-4 had much less of an effect than PKD1-2 (Pillay et al., 2016). These observations prompted the expression of a number of shorter constructs (Pillay et al., 2017), the first of which was an N-terminally histidine tagged PKD1-2, denoted His6PKD1-2. The expression and purification protocol for His6PKD1-2, which was used for the high resolution cryo-EM structure of the AAVR/AAV-2 complex (Meyer et al., 2019), is also presented here.
A number of single-PKD-domain constructs, created for binding and inhibition studies using analogous methods of expression and purification (Pillay et al., 2017), are not further detailed here. Also not covered is another PKD1-5 construct used for structural studies (Zhang et al., 2019). It is similar to our MBP-PKD1-5 (Pillay et al., 2016), except that the MBP fusion domain is replaced by an N-terminal histidine tag (Zhang et al., 2019) similar to the His6PKD1-2 construct detailed here (Pillay et al., 2017). All three types of construct can bind AAV, but higher saturation of symmetry-equivalent sites on the virus is achieved with the smaller constructs, likely because there is less steric occlusion (Meyer et al., 2019).
Materials and Reagents
- AAV-2 VLP expression in Sf9 cells
- Disposable standard transfer pipets, 3 ml (Fisher Scientific, catalog number: 13-680-50)
- Disposable fine-tipped transfer pipets, 3 ml (Fisher Scientific, catalog number: 02-708-018)
- Disposable serological pipets, 1, 10, 25, 50 ml (Thermo Fisher, catalog numbers: 13-678-11B, -11E, -11, -11F)
- Disposable polycarbonate Erlenmeyer flasks, 250 ml (Fisher Scientific, catalog number: 07-200-668)
- Sterile Petri dishes (Thermo Fisher, catalog number: FB0875712)
- 500 ml centrifuge bottles (Nalgene, catalog number: 05-564-3)
- 6-well tissue culture plate (Sarstedt, catalog number: 83.1839)
- CorningTM cell culture flasks, 25 cm2 surface area (Sigma-Aldrich, catalog number: CLS430639)
- CorningTM cell culture flasks, 75 cm2 surface area (Sigma-Aldrich, catalog number: CLS430641)
- pFBDVPm11 donor plasmid (donation of Rob Kotin) used to generate bacmid shuttle vector (Urabe, Ding et al. 2002)
- Chemically competent DH10Bac cells (Thermo Fisher, catalog number: 10361-012)
- Bluo-Gal (Gold Biotechnology, catalog number: B-673-1)
- IPTG (Gold Biotechnology, catalog number: I2481C25)
- Tetracycline (Gold Biotechnology, catalog number: T-101-25)
- Gentamicin (Gold Biotechnology, catalog number: G-400-5)
- Luria Broth (LB) Base (Thermo Fisher, catalog number: 12795027)
- Agar powder (Thermo Fisher, catalog number: BP1423-500)
- M13F and M13R primers (CCCAGTCACGACGTTGTAAAACG and AGCGGATAACAATTTCACACAGG, respectively)
- Qiagen Large-Construct kit (Qiagen, catalog number: 12462)
- Isopropanol (Thermo Fisher, catalog number: A520-4)
- GibcoTM Sf9 cells in Sf-900TM II SFM (Thermo Fisher, catalog number: 11496015, store at -80 °C)
- Grace’s insect medium, supplemented (Thermo Fisher, catalog number: 11605094, store at 4 °C)
- Grace’s insect medium, unsupplemented (Thermo Fisher, catalog number: 11595030, store at 4 °C)
- CellfectinTM II reagent (Thermo Fisher, catalog number: 10362100, store at 4 °C)
- HyCloneTM Fetal Bovine Serum (Fisher Scientific, catalog number: SH3007102, store at -20 °C)
- Sf-900TM II SFM cell medium (Thermo Fisher, catalog number: 10902187, store at 4 °C)
- Trypan blue, 0.4% (Gibco, catalog number: 15250-0601)
- BaculoQUANT All-in-One Virus extraction & titration Kit (Genway Biotech, Inc., catalog number GWB-100602)
- AAV-2 VLP purification
- Ultracentrifuge tubes, 13 x 51 mm (Beckman Coulter, catalog number: 344057)
- Pyrex storage bottles, 250, 500, 1,000 ml (Fisher Scientific, catalog numbers: 06-414-1B, -1C, -1D)
- Microcentrifuge tubes, 1.5 ml (Thermo Fisher, catalog number: 02-681-331)
- 0.45 μm syringe filter units (Millipore Sigma, catalog number: SLHV033RS)
- Dialysis tubing (50kDa MWCO), e.g., Repligen Spectra/Por® 6 Standard Regenerated Cellulose (RC) Dialysis Tubing, Pre-Wetted, catalog number: 888-11389)
- Slide-A-Lyzer dialysis cassette, 10 kDa MWCO (Thermo Fisher, catalog number: 66380)
- HiTrap Heparin HP column, 1 ml (GE, catalog number: 17-0406-01)
- Cesium chloride (Sigma-Aldrich, catalog number: C4036)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888)
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Millipore Sigma, catalog number: 1374248)
- Ethanol (Thermo Fisher, catalog number: S25307B)
- Benzonase (Millipore Sigma, catalog number: E1014-25kU)
- Bleach (Staples, catalog number: KIK8635042)
- CsCl gradient solution (see Recipes)
- Heparin column running buffer (Buffer A) (see Recipes)
- Heparin column elution buffer (Buffer B) (see Recipes)
- HM buffer (see Recipes)
- HN buffer (see Recipes)
- Phosphate buffered saline (PBS) (see Recipes)
- AAVR construct expression in E. coli
- Cell culture tubes (Thermo Fisher, catalog number: 14-956-6D)
- BL21(DE3) E. coli (NEB, catalog number: C2527I)
- pET11a vector (Novagen, catalog number: 69436-3)
- pMALTM Protein Fusion and Purification System (New England Biolabs, catalog number: E8200S), composed of pMAL-c5X, pMAL-p5X, amylose resin, factor Xa, anti-MBP monoclonal antibody, MBP5 protein, MBP5-paramyosin ∆Sal and E. coli ER2523
- NEB Express competent E. coli (NEB, catalog number: C2523I)
- IPTG (Gold Biotechnology, catalog number: I2481C25)
- Ampicillin (Gold Biotechnology, catalog number: A-301-5)
- Kanamycin (Gold Biotechnology, catalog number: K-120-5)
- Tris Base ((CH2OH)3CNH2) (Fisher Scientific, catalog number: BP152-1)
- LB-ampicillin medium (see Recipes)
- LB-ampicillin agar plates (see Recipes)
- AAVR construct purification
- MBPTrap HP column, 5 ml (GE, catalog number: 28-9187-79)
- HiLoad 16/600 Superdex 200 pg SE column (GE Healthcare, catalog number: 28989335)
- HiLoad 16/600 Superdex 75 pg SE column (GE Healthcare, catalog number: 28989333)
- Amicon Ultra-0.5 Centrifugal Filter Units, 3 kDa MWCO (Millipore Sigma, catalog number: UFC500324)
- Amicon Ultra-0.5 Centrifugal Filter Units, 30 kDa MWCO (Millipore Sigma, catalog number: UFC503024)
- 10 ml Luer lock syringes (e.g., Fisher Scientific, catalog number: 14-955-459)
- 0.45 μm syringe filter units (Millipore Sigma, catalog number: SLHV033RS)
- HALT Protease Inhibitor Cocktail (100x) (Thermo Fisher, catalog number: 78429)
- HiTrap Chelating HP (IMAC) column (GE, catalog number: 17-0409-01) charged with Co2+)
- Gel filtration standard (Bio-Rad, catalog number: 151-1901)
- Imidazole (Acros, catalog number: 12202-5000)
- Sodium phosphate dibasic (Na2HPO4) (Thermo Fisher, catalog number: S374-500)
- Potassium phosphate monobasic (KH2PO4) (Thermo Fisher, catalog number: P285-500)
- MBPTrap column running buffer (Buffer A) (see Recipes)
- MBPTrap column elution buffer (Buffer B) (see Recipes)
- IMAC column running buffer (Buffer A) (see Recipes)
- IMAC column elution buffer (Buffer B) (see Recipes)
Equipment
- Water bath capable of 42 °C (e.g., Thermo Fisher, model: Isotemp)
- Shaking incubator, 27 °C (e.g., Thermo Fisher, model: MaxQ 6000)
- Shaking incubator, 37 °C (e.g., New Brunswick Scientific, model: Excella E25)
- qPCR machine (e.g., Applied Biosystems, model: StepOne)
- French press for cell lysis (e.g., SLM Aminco, model: FA-078)
- Table-top microcentrifuge capable 16,000 x g (e.g., Thermo Fisher, model: accuSpin Micro)
- FPLC (e.g., GE Healthcare, model: AKTA chromatography system )
- FPLC system sample loops: 2 ml (GE Healthcare, catalog number: 18111402); 10 ml (superloop) (18-1113-81) (or 50 ml, 18-1113-82)
- SDS-PAGE equipment (e.g., Bio-Rad, model: Mini Protean 3 Cell)
- Thermocycler (e.g., MJ Research, model: PTC-200)
- Preparative centrifuge (e.g., Sorvall, model: RC5C)
- Centrifuge rotor, fixed angle, fitting 50 ml tubes (e.g., Beckman Coulter JA-20 or FiberLite F13-14x50cy)
- Centrifuge rotor, fixed angle, fitting 500 ml bottles (e.g., Beckman Coulter J-LITE® JLA-10.500)
- Ultracentrifuge (e.g., Beckman model: L7-55)
- Swinging-bucket rotor for CsCl, capable of 135,000 x g (e.g., Beckman SW50.1)
- Retort stand and clamp used for holding ultracentrifuge tubes
- pH meter (e.g.,Thermo Fisher, model: Acumet AB15)
- Pipets (e.g., Gilson Pipetman®, models: P1000, P200, P20)
- Stir plate (e.g., Fisher Scientific, catalog number: SP88854200)
- Magnetic stir bars (Nalgene, catalog number: 93-5521-5)
- Balance accurate to 0.01 g (e.g., Ohaus, model: Scout Pro SP402)
- UV-Spectrophotometer (e.g., Shimadzu, model: UV-1800)
- Custom-made dialysis buttons (parts purchased from McMaster-Carr, catalog number: 8546K15, and machined at the University of Missouri, Columbia; Figure 1 and Table 1 for assembly and measurements). An alternative might be the Slide-A-Lyzer MINI dialysis device, 20K MWCO (Thermo Fisher, catalog number: 69590 or 69555), see Procedure D below: “Desalting by dialysis”
- Refractometer (e.g., Abbe, model: C10)
- Hemocytometer (e.g., Thermo Fisher, catalog number: 02-671-6)
- Inverted microscope (for cell counting) (e.g., Nikon, model: Eclipse Ts2)

Figure 1. Custom dialysis button. Parts were machined from PTFE (McMaster-Carr) with threaded caps and chamber. Milled separately, the inner chamber is cemented into the outer chamber. The bore has a capacity of 50 μl (left) or 60 μl (right). Between the bottom screwcap and the cylinder is sandwiched a dialysis membrane, and at the top end is a microscope cover slip, providing a window. After assembling the bottom part, sample is loaded from the top, before the cover slip is added and the top sealed. The button is hung membrane-side down from the side of a beaker in exchange buffer. Air that is trapped in the pocket under the bottom cap must be removed to ensure contact between membrane and buffer.
Table 1. Dimensions of dialysis buttonsDialysis button part Outer diam (mm) Inner hole diam (mm) Height (mm) Top cap 19 3 8.5 Outer chamber 19 13 18 Inner chamber 13 3 6.5 Bottom cap 19 7.5 9.5
Procedure
- Baculoviral expression of AAV-2 VLPs in Sf9 cells
This consists of five steps: (1) starting and maintaining a culture of Sf9 cells, (2) construction and purification of the bacmid DNA containing the AAV-2 capsid gene, (3) transfection of Sf9 cells with the prepared bacmid DNA, (4) collection and amplification of baculovirus stock, and (5) baculoviral infection of Sf9 cells for production of VLPs.- Growth of Sf9 cells
- Warm frozen Sf9 stock at 37 °C until nearly thawed.
- Aseptically transfer the cells into a 250 ml Erlenmeyer flask containing 25 ml of pre-warmed (25-30 °C) Sf-900TM II SFM.
- Incubate at 28 °C while shaking at 135 rpm, protected from light (as suggested by the manufacturer).
Note: A culture from frozen stock can take a couple of weeks to grow to a suitable density. - Count cells daily when culture shows signs of growth. To count, mix 1 ml of culture with 100 µl of trypan blue and mix gently by flicking the tube. Wait 2 min, then remix and count with a hemocytometer. (It can also usually be assumed that cells will have propagated sufficiently, and can be counted at 48 h, and split immediately, as described in the next paragraph.)
- When cells reach a density of 4 x 106-6 x 106 cells/ml, passage to 0.5 x 106-1.0 x 106 cells/ml in a final volume of 50 ml. Healthy cultures double in ~24 h and should be passaged every 2-4 days. Continue passaging as needed to maintain an appropriate cell density. Cultures may show decreasing viability after passage 30 and a new culture may need to be started.
Note: Starting a culture with a density < 0.5 x 106 cells/ml or allowing a culture to exceed 8 x 106 cells/ml is highly discouraged as it leads to longer growth lags and decreased viability.
- Generation and purification of bacmid DNA
- From the pFBDVPm11 donor plasmid, generate bacmid shuttle vector in DH10Bac cells following the Bac-to-BacTM protocol (Thermo Fisher, publ. MAN0000414).
- Purify bacmid using Qiagen’s endotoxin-free large construct kit.
- Transfection of bacmid DNA into Sf9 cells according to the Bac-to-BacTM protocol (see above).
- Isolation of P1 baculovirus stock according to the Bac-to-BacTM protocol. Modifications to the Bac-to-BacTM procedure include:
- Quantifying the virus titer by qPCR with the BaculoQUANT All-In-One Extraction & Titration Kit to estimate readiness for virus harvest.
- Optionally, instead of harvesting when the Sf9 cells are 20-30% viable, harvest when the baculovirus titer is at least 106 viral genomes (vg)/ml.
Note: This tends to take about 4-5 days post transfection.
- Amplification of baculovirus stock (generation of P2 stock)
This continues to follow the Bac-to-BacTM, modified as follows:
Quantifying the P2 virus titer by qPCR.
Note: This tends to take about 3-4 days post infection. - Sf9 infection for VLP production
- Scale up a Sf9 culture to 600-800 ml and a density of 2 x 106-4 x 106 cells/ml, as described in Step A1, above.
- Infect the culture with the P2 baculovirus stock. Add P2 to a MOI of between 5 and 10.
- Incubate the culture as before and check cell density and viability daily. When viability starts to decrease (at 40-60 h), check culture more frequently and harvest when the viability is between 80-60% (typically between 55-80 h).
- Transfer the culture into two 500 ml centrifuge bottles, balance and centrifuge at 5,500 x g for 30 min at 20 °C.
- Resuspend each pellet in 10 ml of HM buffer and pool into a 50 ml centrifuge tube. Collect any additional cells with a single aliquot of 5 ml of additional HM buffer.
- Store the ~25 ml of resuspended cell pellet at -80 °C until ready for VLP purification.
- Growth of Sf9 cells
- AAV-2 VLP purification
AAV-2 VLP purification consists of four steps: (1) lysis of the cell pellet by freeze-thaw cycles, (2) clarification of the VLP product from the remaining soluble components, (3) affinity column purification and (4) desalting by dialysis. First is an initial lysis that extracts VLPs from the cells. The second step relies upon ultracentrifugation of the sample in a cesium chloride gradient to separate the VLP from the other soluble components using differential density centrifugation. Following three rounds of CsCl gradient centrifugation, the sample is 50-70% pure. For greater purity, the sample is run through a heparin affinity column followed by one additional CsCl centrifugation to concentrate the sample. [For serotypes that do not have high affinity for heparin, one can substitute ion exchange for affinity chromatography (Kaludov et al., 2002), or Cs-gradient ultracentrifugation can be performed with higher stringency, further repeating, or accepting only a narrower fraction near the peak of the band (Xie et al., 2004)]. The yield for a VLP preparation is typically 2-4 mg/ml in 100 µl. Lastly, since the sample contains a high concentration of CsCl which would interfere with electron microscopy visualization, the sample is dialyzed using a custom dialysis device (dialysis button) designed for 40-60 µl volumes. Due to the relative stability of the VLP sample in 3.3 M CsCl and the instability in low-salt solution, the last dialysis step is performed on small volumes of purified sample that will be used within 1-2 days. (AAV-2 is more prone to aggregation at low salt than other serotypes, many of which are stable at 4 °C for up to 4 weeks.)- Lysis of the Sf9 cells and collection of soluble proteins
- Thaw the VLP-containing cell suspension in 37 °C water bath for 30 min.
- Refreeze the cell suspension at -80 °C for at least 2 h.
- Repeat Steps B1a and B1b 4 more times (5 cycles of thaw and freeze).
- Thaw the cell suspension in 37 °C water bath for 20 min, add 0.25 µl benzonase per ml of cell suspension, mix and continue incubating in 37 °C water bath for 20 min.
Note: Digestion of the nucleic acids reduces the viscosity. - Add 5 M sodium chloride solution to increase sodium chloride in cell suspension by 100 mM (e.g., add 0.6 ml to a 30 ml suspension) and freeze at -80 °C for at least 2 h.
Note: Addition of sodium chloride deactivates benzonase. - Thaw the suspension a final time in a 37 °C bath and dispense 2-ml aliquots into fifteen 2-ml tubes and spin at 16,000 x g for 10 min.
- Pool the supernatant from all tubes and filter through a 0.45 µm filter.
- CsCl gradient purification of AAV-2 VLP
- Add CsCl to the pooled supernatant (above) until the refractive index (RI) is 1.3690 (± 0.0005).
Note: This will be approximately 0.5 g per ml of sample and brings it to ~3.3 M CsCl. - Make an extra 50 ml of HM buffer plus CsCl, to the same RI, for balancing (Recipe 11).
- Transfer ~5 ml of sample into each of 6 ultracentrifuge tubes and balance each pair to within 0.05 g, using the solution made above (Step C2b) to top off tubes as needed. Fill each tube to within 1 mm of the top to avoid collapse during centrifugation.
- Centrifuge the samples at 135,000 x g with the SW50.1 rotor (or similar) in an ultracentrifuge at 20 °C for 20-24 h.
- Gently transport the tubes to your working area to preserve gradient separation. Wipe the outside of each tube with ethanol and visualize the AAV band using a bright halogen lamp (see Figure 2). As a guide, mark the side of the tube about 2 mm above and below the AAV-2 band (e.g., with a Sharpie pen).

Figure 2. Side-illumination of the centrifuge tube after separation of AAV-2 by CsCl gradient centrifugation. The cloudy white band in the middle is AAV-2 VLP. - Use a standard disposable transfer pipette to remove the liquid down to the mark 2 mm above the AAV-2 band and discard this into a 10% bleach solution.
- Use a fine-tipped disposable transfer pipette to gently collect the sample containing AAV-2 (take 2 mm above to 2 mm below the band), and transfer it to a new ultracentrifuge tube. Collect the AAV-2 band from all six tubes similarly and pool into a single ultracentrifuge tube. Check and record the refractive index (RI).
- Balance and centrifuge the tube containing the pooled AAV-2 samples as before (Steps C2c-C2e, above) (second spin).
- Repeat the AAV-2 band collection and ultracentrifugation step one more time (third spin).
- Add CsCl to the pooled supernatant (above) until the refractive index (RI) is 1.3690 (± 0.0005).
- Collect the sample after the third ultracentrifugation and inject into a Slide-A-Lyzer dialysis cassette with the 1 ml syringe and 18 gauge needle provided, as per product instructions.
- Float the dialysis cassette in 2 L of phosphate buffered saline (PBS) and stir for 2 h at 4 °C. Replace with 2 L of fresh PBS and allow to dialyze overnight at 4 °C.
- Transfer the dialyzed sample from the Slide-A-Lyzer cassette to a low protein-binding microcentrifuge tube; store at 4 °C until loading onto heparin affinity column.
- Lysis of the Sf9 cells and collection of soluble proteins
- Heparin affinity column purification of AAV-2 VLP
- Prepare AKTA system and heparin affinity column
- Verify the 2 ml sample loop is connected to the FPLC system.
- Purge the FPLC system lines A and B with ultra-pure deionized water.
- Purge the FPLC system lines with heparin affinity buffers: line A with the running buffer A and line B with the elution buffer B (Recipe 6).
- With buffer A flowing at 0.5 ml/min connect the Heparin HiTrap column.
- Equilibrate the column with 20 ml of 100% buffer A at 1 ml/min.
- Wash the column with 10 ml of 100% buffer B at 1 ml/min.
- Wash the column with at least 10 ml of 100% buffer A.
- Once UV and conductivity stabilize, end the flow of buffer A.
- Run sample on heparin affinity column
- Spin the dialyzed sample at 16,000 x g for 10 min to remove aggregates.
- Set the system to “Load” and flush the loop with 10 ml of PBS using a Luer lock syringe.
- Set the system to “Inject” and remove the syringe.
- Load a 1 ml syringe with your VLP sample and connect it to the loop.
- Set the system back to “Load” and inject your sample into the loop, being careful not to inject air.
- Set the flow to 1 ml/min and introduce your sample to the column by setting the system to “Inject” and flushing the loop with 3 ml of 100% buffer A.
- Set back to “Load” and continue to run 100% buffer A at 1 ml/min for 5 min.
- Run a linear gradient from 0-85% buffer B over 25 min, collecting 0.5 ml fractions.
- Examine the UV chromatogram and set aside the eluate fractions containing the AAV-2 peak for evaluation by SDS-PAGE.
- Run a sample from each AAV-2 peak-containing fraction on an SDS-PAGE gel, following standard protocols, to verify the presence of VLP.
- Pool the AAV-2-containing fractions and run a final CsCl gradient ultracentrifugation as before (see Steps C2c-C2e above). This gradient serves to concentrate the VLPs.
- Store the collected sample in a low protein-binding microcentrifuge tube.
- Clean the column by running 10 column volumes of elution buffer B, ultra-pure DI water, and 20% ethanol, sequentially. Store the column in 20% ethanol.
- Prepare AKTA system and heparin affinity column
- Desalting by dialysis
Our protocol continues to use a custom mini-dialysis apparatus that we designed for low volume and high recovery before somewhat similar devices became available commercially. It is likely that the Slide-A-Lyzer MINI dialysis device (see Materials) could be substituted, following the manufacturer’s directions, rejoining the protocol at Step D3i below, “Measure the absorbance…”.We continue the protocol based on the custom device:- Prepare the dialysis membrane, according to the manufacturer’s instructions, then:
Using a 12 mm circular glass cover side as a template, cut small circles out of dialysis membrane; store at 4 °C in HM buffer plus 0.02% sodium azide until use. - Prepare the dialysis button
Assemble the custom dialysis button according to Figure 1, ensuring a smooth fit of the membrane between the bottom inner chamber and the plastic washer inside the bottom cap. - If fully assembled, remove the top cap, cover slip and rubber washer
- Aliquot a sample (30-60 µl) of the purified VLP (from Procedure D) into the inner hole of the inner chamber, taking care to avoid introducing air bubbles onto the surface of the dialysis membrane.
- Place the glass coverslip, rubber washer, and top cap in the outer chamber and tighten carefully to seal.
- Use a twist-tie to suspend the dialysis button partially in a 50 ml beaker with the glass coverslip facing up.
- Add HN buffer so that the lower portion of the dialysis button is submerged while the top is above the surface.
- Use a Pasteur pipette that has been melted (to form a 180°-hairpin) to remove air bubbles present on the bottom surface of the dialysis membrane in the bottom cap of the dialysis button.
Note: The presence of air bubbles will occlude the liquid portion from allowing dialysis of the salts from the sample and may harm protein structure. - Dialyze overnight with stirring at 4 °C.
- Remove the dialysis button from the beaker and gently dry the area near the dialysis membrane by wicking away with strips of filter paper.
- Use a 10 µl tip to carefully puncture the dialysis membrane and pipette the sample into a sterile, low protein-binding microcentrifuge tube.
- Measure the absorbance of the sample at 280 nm and determined the protein concentration using an extinction coefficient of 17.3 (g/100 ml)-1/cm, otherwise known as
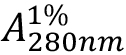 , or E1%).
, or E1%).
- Prepare the dialysis membrane, according to the manufacturer’s instructions, then:
- Expression of MBP-PKD1-5 and His6PKD1-2 AAVR constructs in E. coli
- Subclone desired PKD1-5 fragments from the KIAA0319L cDNA clone (clone ID 3843301; GE Dharmacon; Pillay et al., 2016). PKD1-5 is subcloned into the pMAL-c5X vector as a fusion with MBP whereas PKD1-2 is subcloned into the pET-11a vector with an N-terminal His-tag (Figure 3).
- Transform pMAL-c5x plasmids (1 pg) into NEB Express cells and pET-11a plasmids (1 pg) into BL21(DE3) cells using standard techniques and plate on LB-ampicillin agar plates (100 µg/ml).
Note: Both constructs can be expressed using the procedure delineated below (Steps E3-E5). Transformation efficiencies did not vary significantly from the advertised values of 8 x 108 CFU/µg for NEB Express and 3 x 107 CFU/µg for BL21(DE3). - Inoculate a single transformed BL21(DE3) colony from a freshly streaked LB-ampicillin agar plate into a volume of LB-ampicillin (100 µg/ml) media that is 5% of the final total desired culture volume (e.g., 50 ml overnight culture into 1,000 ml final volume). Grow the starter culture overnight at 37 °C with shaking at 210 rpm.
- The next day, transfer the starter culture into the final media volume and incubate at 37 °C with shaking at 210 rpm until the OD600 is ~1.0. Remove a 1 ml aliquot (pre-induction sample) and place into a 1.5 ml tube. Spin this aliquot for 1 min at 6,000 x g and discard the supernatant into a 10% bleach solution. Store the bacterial pellet at -20 °C for post-expression analysis by SDS-PAGE, if desired.
- Add IPTG to a concentration of 1 mM and incubate the culture at 37 °C with shaking at 210 rpm for an additional 4-5 h. After incubation, remove a 1 ml aliquot (post-induction sample) and pellet these cells as described in Step E4 for the pre-induction sample. Store this pellet also at -20 °C for post-expression analysis. Centrifuge the remaining culture at 6,000 x g, discard the supernatant into a 10% bleach solution, and store the pellets at -20 °C until lysis and purification steps.
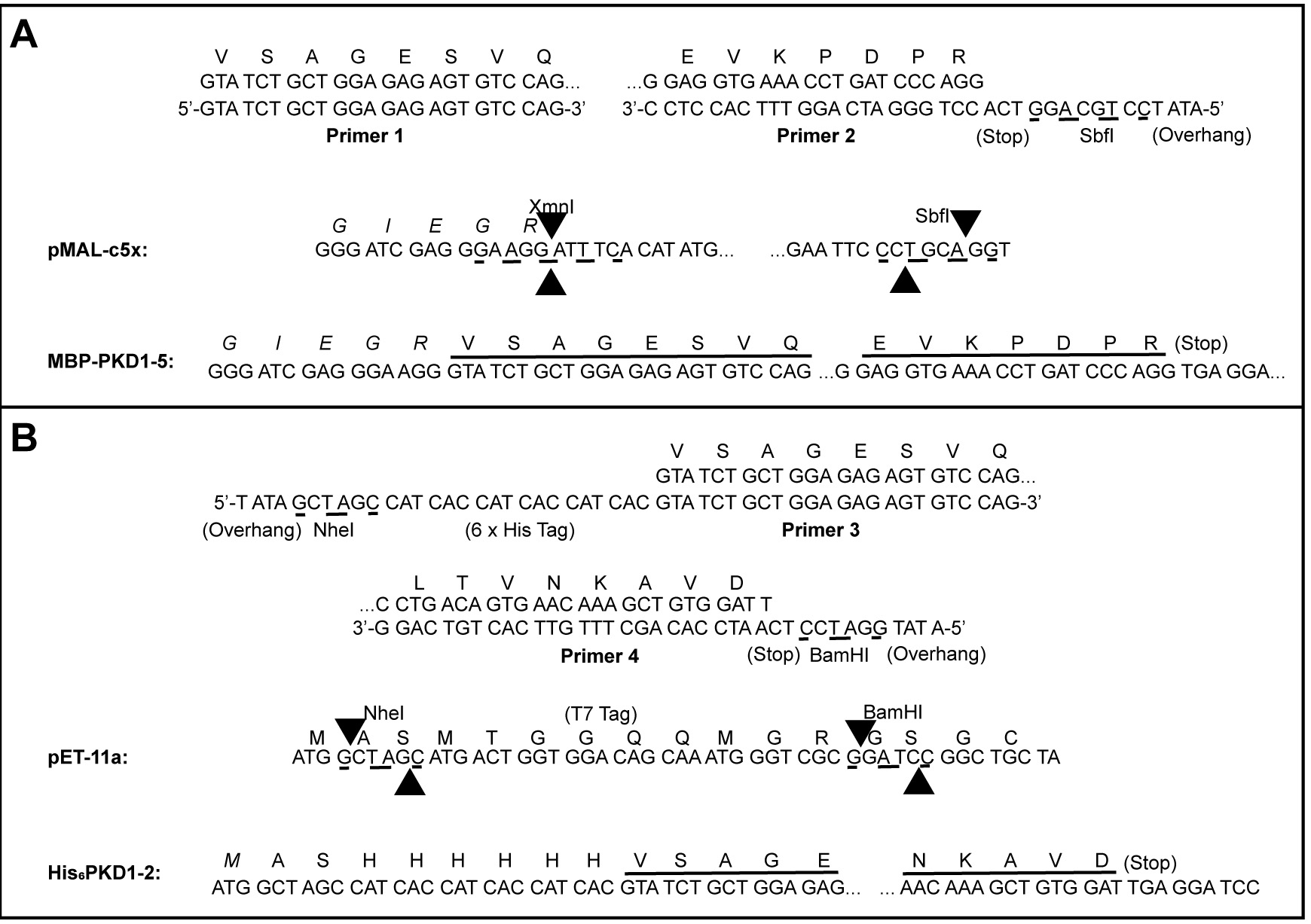
Figure 3. Subcloning of MBP-PKD1-5 and His6PKD1-2. A. Use the KIAA0319L cDNA clone as a template for PCR amplification using primers 1 and 2. Cut the PCR product with SbfI then phosphorylate using T4 Polynucleotide Kinase. Cut pMAL-c5x with XmnI and SbfI. Ligate the cut/phosphorylated PCR product and the cut pMAL-c5x vector together to make MBP-PKD1-5. B. Use the same DNA as a template for PCR using primers 3 and 4. Cut the PCR fragment with NheI and BamHI. Cut pET-11a with NheI and BamHI. Ligate the cut PCR product and the cut pET-11a vector together to make His6PKD1-2.
- Purification of AAVR constructs
- MBP-PKD1-5 purification
- Cell lysis (do on ice)
- Resuspend frozen pellets in 25 ml of MBPTrap running buffer containing 100 µl HALT Protease Inhibitor Cocktail 100x.
- Load cell slurry into a French press cell and pass through two complete rounds of lysis at 1,000 psi using a slow flow rate to collect the lysate into a 50 ml centrifuge tube.
- Centrifuge lysate at 4 °C, 16,000 x g, for 45 min in a F13-14x50cy rotor (or similar). While spinning, prepare the AKTA system.
- MBP affinity chromatography on the AKTA system
- Washing and equilibrating the MBPTrap column1)Prior to attaching the column, place line A of the AKTA into MBPTrap running buffer and line B into MBPTrap elution buffer.2)Inject 10 ml of MBPTrap running buffer into the superloop (valve setting = Load).3)Start flow at 5 ml/min and 50%B, bypassing superloop (valve setting = Load). Run for about 5 min.4)Switch to buffer A (0%B), and run for 5 more min.5)Reduce flow to 2 ml/min and flow through superloop (valve setting = Inject) for 10 min or until the superloop is purged completely.6)Reduce flow to 1 ml/min, switch valve back to “Load”, and attach the MBPTrap HP column, avoiding introduction of air.7)Equilibrate the column with 5 column volumes of elution buffer B at 1 ml/min and until UV and conductivity baselines stabilize.8)Switch to running buffer A and wash until the baseline stabilizes, at least 10 column volumes.
- Loading sample onto MBPTrap column1)Remove the tube containing cell lysate from the centrifuge and transfer the supernatant to a new 50 ml centrifuge tube.2)Filter through a 0.45 µm filter using a 10 ml Luer lock syringe. It may take multiple filters if membranes become fouled.3)With the valve setting set to “Load”, inject 10 ml of lysate into the superloop.4)Set flow rate to 0.5 ml/min, fraction size to 10 ml, and the valve to “Inject”.5)Flow through the superloop into the column until the superloop is almost empty.6)At this point, pause the FPLC, switch the valve to “Load”, inject another 10 ml into superloop, switch back to “Inject”, and resume flow. Repeat this process until entire lysate volume is loaded onto the column (with a high capacity superloop, the entire sample could be loaded in a single step).
- Elution of MBP-PKD1-51)After loading, wash the column by switching the valve to “Load” and increasing the flow of running buffer to 1 ml/min.2)Once baseline is stabilized–at least 10 column volumes–reduce the flow to 0.5 ml/min and the fraction size to 2 ml.3)Flow 100% elution buffer B for 45 min to elute MBP-PKD-1-5.
- Processing MBPTrap column fractions1)Run peak fractions (Figure 4) on SDS-PAGE to verify the presence of MBP-PKD-1-5 and pool the appropriate fractions.2)Concentrate the pooled sample to ~1 ml using a 30 kDa Amicon Ultra centrifugal filter unit.
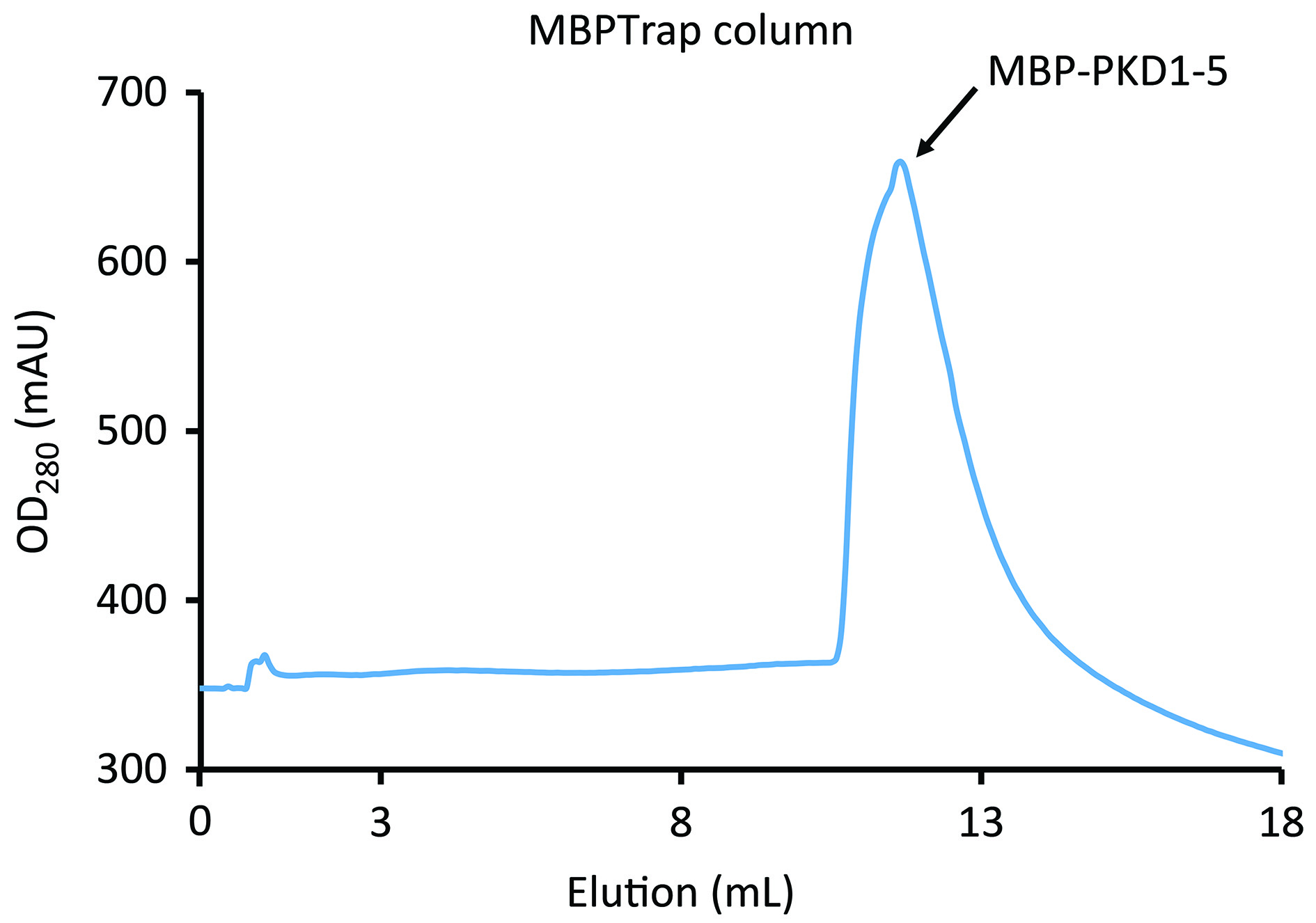
Figure 4. Elution of MBP-PKD1-5 from MBPTrap HP column. The flow was switched from 100% A to 100% elution buffer B at elution volume 0.
- Washing and equilibrating the MBPTrap column
- Superdex 200 SEC on the AKTA system
Note: MBP-PKD1-5 oligomerizes slowly (over a ~30 day period at 4 °C) (Meyer et al., 2019). This SEC step allows dimers to be selected over higher order oligomers shortly before use, and must be repeated, if this separation is desired, after the sample has been stored at 4 °C for an extended time.- Washing and equilibrating the Superdex 2001)Prior to attaching the column, place line A of the AKTA into HN buffer and flush line A by running 0%B at 5 ml/min for 5 min.2)Inject 10 ml of HN buffer into superloop (ensure valve setting = Load).3)Set flow to 2 ml/min and flow through superloop (valve setting = Inject) for 10 min or until the superloop is purged completely.4)Reduce the flow rate to 1 ml/min and attach a pre-calibrated Superdex 200 size exclusion column, avoiding introduction of air. To equilibrate, flow HN buffer through the column at 1-1.5 ml/min until the baseline stabilizes.
- Loading sample onto the Superdex 2001)Pause the AKTA and inject into the superloop a sample volume that is no more than 1% of the column volume (valve setting = Load).2)Set flow rate to 1 ml/min and resume flow (valve setting = Inject).3)Collect the fractions (Figure 6) that correspond to the molecular weight of the MBP-PKD1-5 dimer (190 kDa) based on your gel filtration d calibration results and Meyer et al., 2019. Figure 5 provides an example of what a standard calibration may look like.

Figure 5. Calibration of Superdex 200 using the BioRad Gel Filtration Standards. The blue line tracks elution of standards (left to right): bovine thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa) and vitamin B12 (1.35 kDa). Chromatography of the standards should be performed under the same conditions (flow rates, etc.) as will be used for sample (see Step F1c and Figure 7).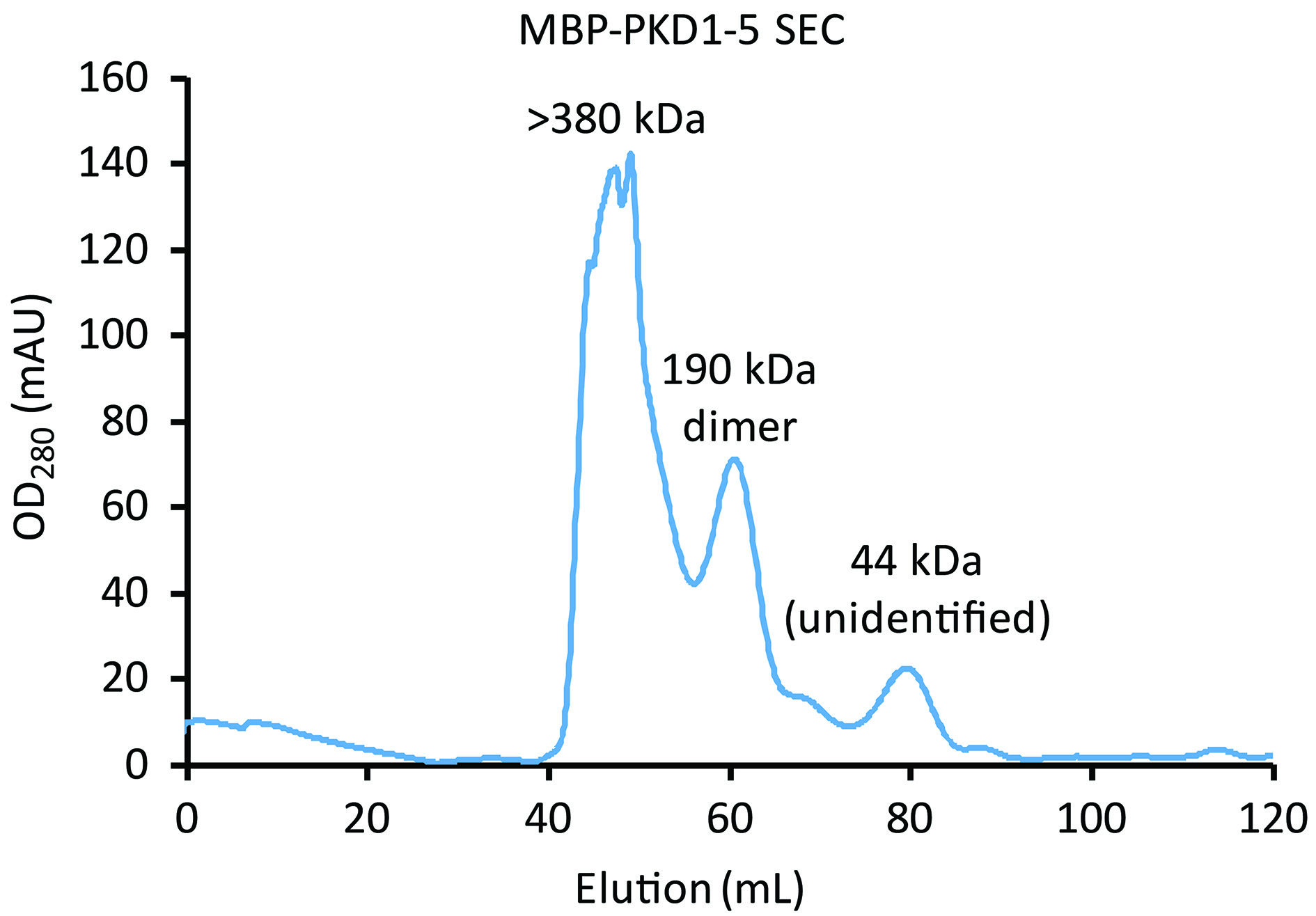
Figure 6. MBP-PKD1-5 elution from Superdex 200 SEC. Comparison of the elution times with Figure 5 indicates that the larger left peak corresponds to higher order oligomers of > 380 kDa. The smaller right peak, corresponding to dimers of MBP-PKD1-5 (190 kDa), is collected.
- Washing and equilibrating the Superdex 200
- Cell lysis (do on ice)
- Purification of His6PKD1-2
- Cell lysis (do on ice)
- Resuspend frozen pellets in 25 ml of IMAC running buffer containing 100 µl HALT Protease Inhibitor Cocktail 100x.
- Load the cell slurry into a French press cell and pass through 2 complete rounds of lysis at 1,000 psi using a flow rate slow enough to avoid foaming, collecting the lysate into a 50 ml centrifuge tube.
- Centrifuge lysate at 4 °C, 16,000 x g, for 45 min in a F13-14x50cy rotor (or similar). While spinning, prepare the AKTA system.
- IMAC affinity chromatography on the AKTA system
- Washing and equilibrating the IMAC column1)Place line A of the AKTA in the IMAC running buffer and line B in the IMAC elution buffer. The column should not be attached to the AKTA at this point unless the valve is set in the bypass mode.2)Inject 10 ml of IMAC running buffer into the superloop (ensure valve setting = Load).3)Start the flow at 5 ml/min and 50%B, bypassing the superloop (valve setting = Load). Run for about 5 min.4)Switch the buffer to A (i.e., 0%B), and run for an additional 5 min.5)Reduce the flow to 2 ml/min and flow through the superloop (valve setting = Inject) for 10 min or until the superloop is purged completely.6)Reduce the flow to 1 ml/min, switch valve back to “Load”, and attach the HiTrap HP (IMAC) column, avoiding introduction of air.7)Equilibrate the column by flowing elution buffer for 5 column volumes at 1 ml/min and until UV and conductivity baselines stabilize.8)Switch to running buffer and flow until the baseline stabilizes, at least 10 column volumes.
- Loading sample onto the IMAC column1)Remove the tube containing the cell lysate from the centrifuge and transfer the supernatant to a new 50 ml centrifuge tube.2)Filter through 0.45 µm filter. It may take multiple filters if membranes become fouled.3)With the FPLC valve setting set to load, inject 10 ml of lysate into the superloop.4)Set the flow rate to 0.5 ml/min, fraction size to 10 ml, and valve to inject.5)Flow through the superloop into the column until the superloop is almost empty.6)At this point, pause the FPLC, switch the valve to load, inject another 10 ml into the superloop, switch the valve to inject, and continue running the FPLC. Repeat this process until the entire lysate is loaded onto column.
- Elution of His6PKD1-21)Wash the column by switching the injection valve to “Load” and increasing the flow to 1 ml/min.2)Once the baseline stabilizes–at least 10 column volumes–reduce the flow rate to 0.5 ml/min and the fraction size to 2 ml.3)Run a linear gradient from 0%-100% elution buffer over 45 min.
- Processing IMAC column fractions1)Run the peak fractions (Figure 7) on SDS-PAGE and pool the appropriate fractions.2)Concentrate pooled sample to ~1 ml using a 3 kDa MWCO Amicon Ultra centrifugal filter unit.
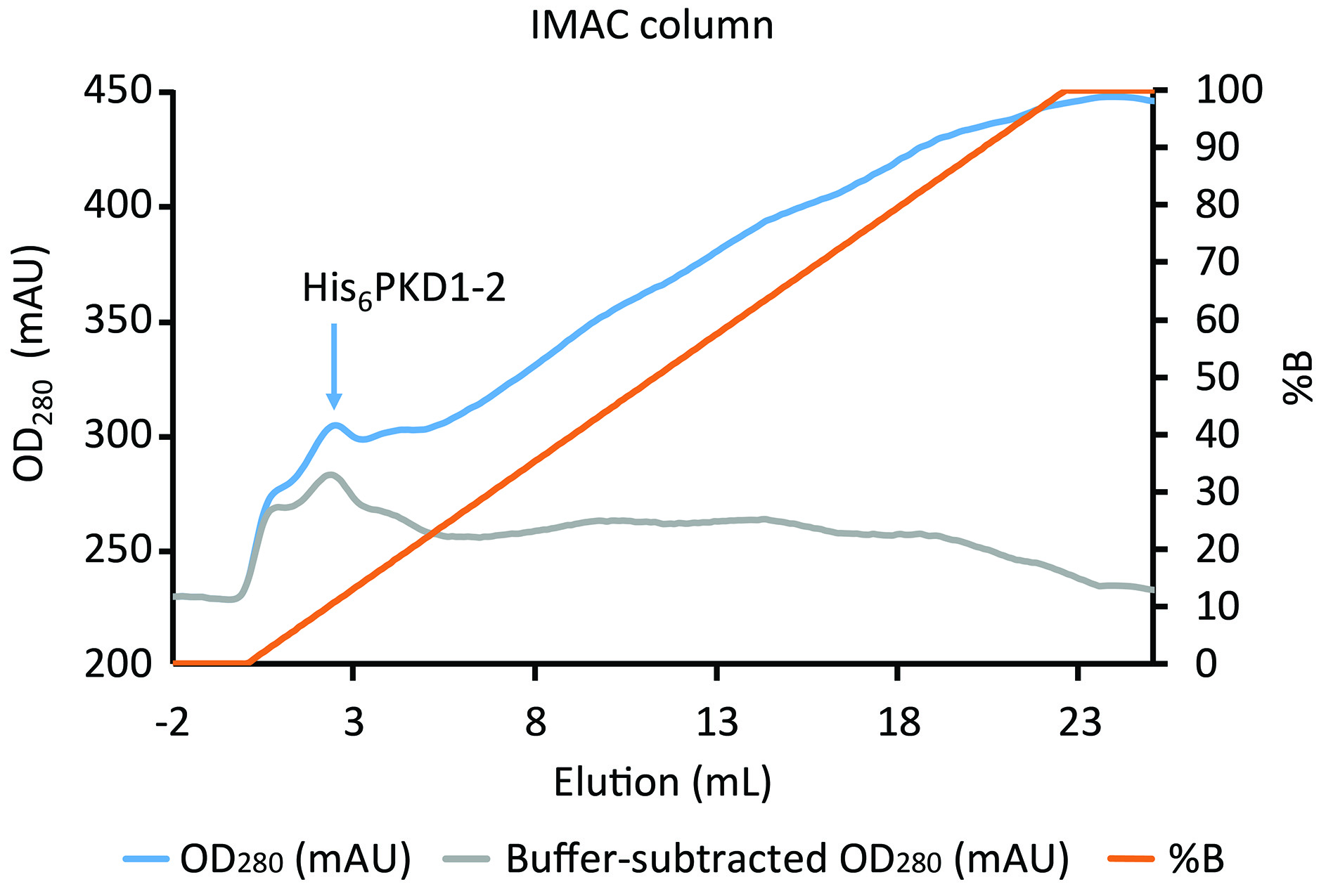
Figure 7. Elution of His6PKD1-2 from the IMAC column. The blue line shows the elution of the His-tagged protein peaking in the first 3 ml over a large background. Background, coming from absorption of the imidazole in buffer B (orange line), is approximately removed in the gray line through subtraction of a value proportional to %B.
- Washing and equilibrating the IMAC column
- Superdex 75 SEC on the AKTA system
Note: As with MBP-PKD1-5, His6PKD-1-2 oligomerizes slowly at 4 °C (Meyer et al., 2019). This SEC step allows dimers to be selected over higher order oligomers shortly before use, and must be repeated, if this separation is desired, after the sample has been stored at 4 °C for an extended time.- Washing and equilibrating the Superdex 75
Wash and equilibrate the Superdex 75 column with HN buffer exactly as described for the Superdex 200 (Step F1c, above). - Loading sample onto the Superdex 751)After equilibration, pause the AKTA and, as with the Superdex 200, inject a sample volume that is no more than 1% of the column volume (set valve to “Load”).2)Set flow rate to 1 ml/min and resume flow (valve setting = Inject).Note: Loading and elution procedures, including flow rates, for the Superdex 75 SEC are the same as for the Superdex 200 SEC (Step F1c), but samples have different retention times. The expected retention time of the His6PKD-1-2 dimer can be estimated by gel filtration standard calibration results (see Figure 8 for an example) and Meyer et al., 2019.3)Collect the fractions that correspond to the molecular weight of the His6PKD1-2 dimer (44 kDa) based on your gel filtration standard calibration results and Meyer et al., 2019.
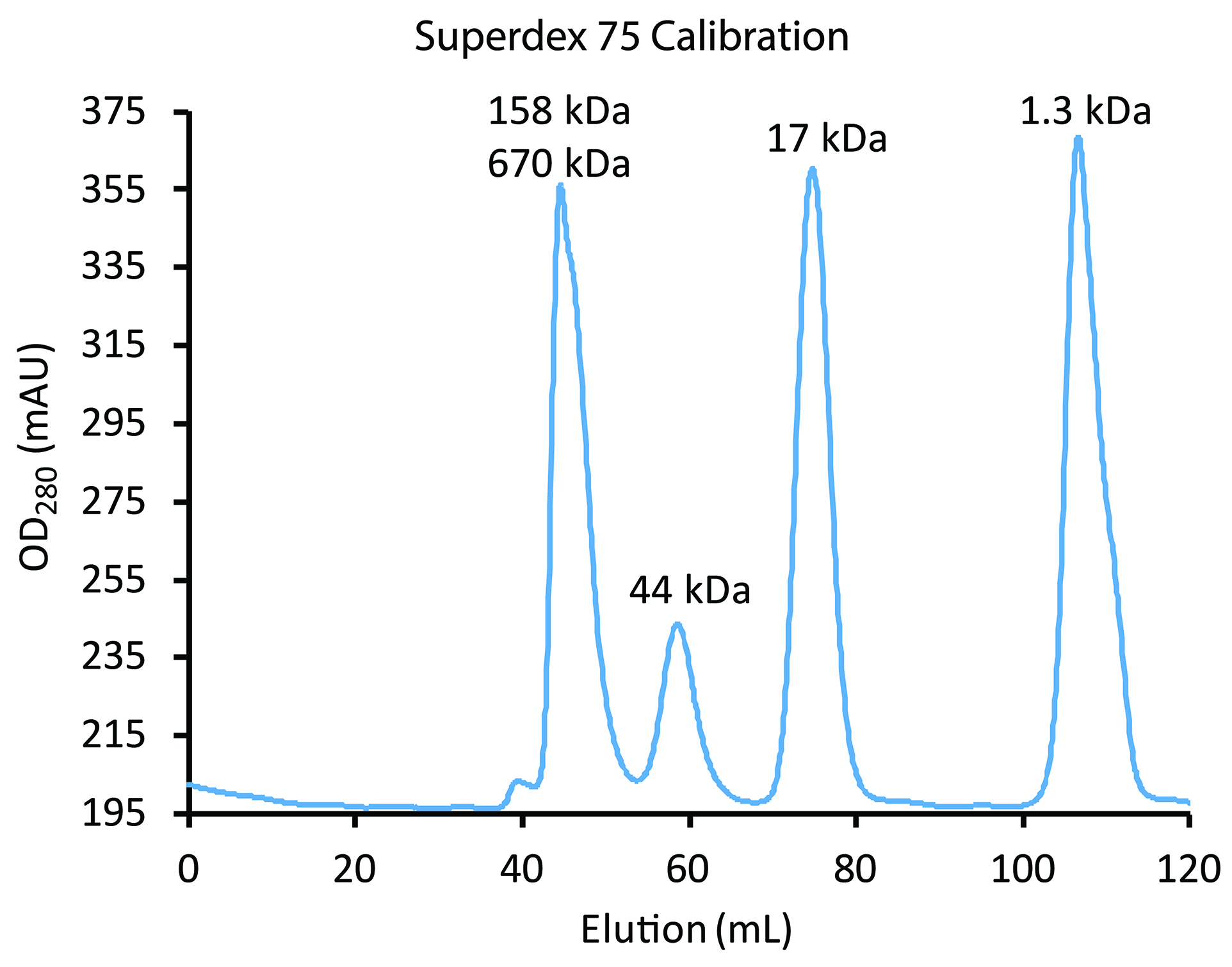
Figure 8. Calibration of Superdex 75 using the BioRad Gel Filtration Standards. The blue line tracks elution of standards (left to right): (a) the first major peak has a shoulder indicative that it is unresolved bovine thyroglobulin (670 kDa) and bovine γ-globulin (158 kDa); (b) chicken ovalbumin (44 kDa), (c) horse myoglobin (17 kDa); and (d) vitamin B12 (1.35 kDa). Chromatography of the standards should be performed under the same conditions (flow rates, etc.) as will be used for sample (see Step F2ci and Figure 9). Monomers of His6PKD1-2 (22 kDa) would be expected at about 70 ml and dimers at about 60 ml, the same elution time as ovalbumin.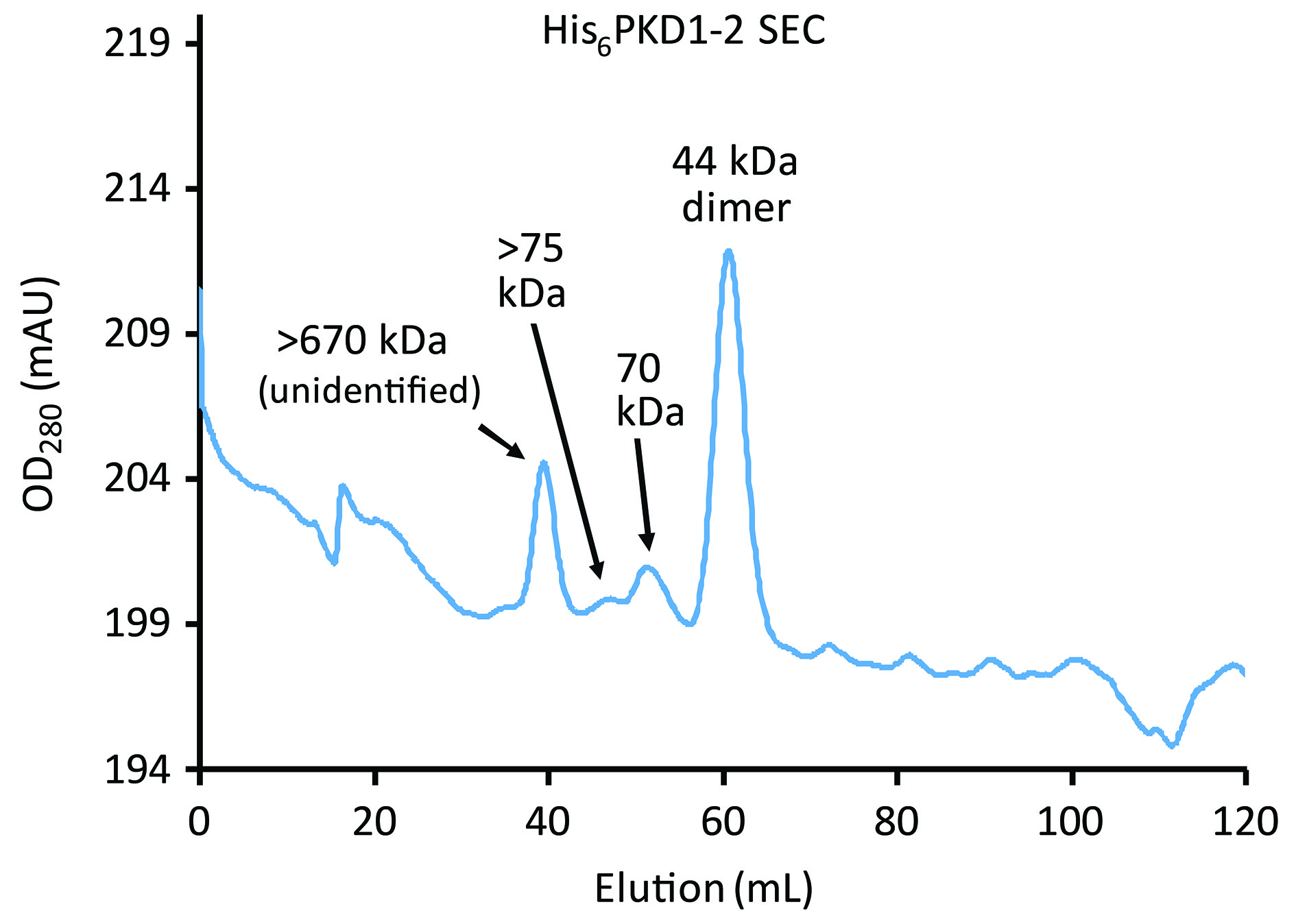
Figure 9. His6PKD1-2 elution from Superdex 75 SEC. Comparison of the elution times with Figure 8 indicates that the larger left composite peak corresponds to higher order oligomers exceeding > 75 kDa and approximately 670 kDa. The largest peak, corresponding to dimers of His6PKD1-2, is used, and there is only the faintest hint of something approximating a monomer eluting later.
- Washing and equilibrating the Superdex 75
- Cell lysis (do on ice)
- MBP-PKD1-5 purification
Recipes
- LB-ampicillin medium
20 g/L LB Broth Base
Autoclave at 121 °C, 2.6 bar for 30 min to sterilize
Cool to ~60 °C in a water bath, then add ampicillin to 100 µg/ml - LB-ampicillin agar plates
20 g/L LB Broth Base
15 g/L agar powder
Autoclave at 121 °C, 2.6 bar for 30 min to sterilize
Cool to ~60 °C in a water bath, then add ampicillin to 100 μg/ml and pour into sterile Petri dishes
Can be stored at 2-8 °C for 2-3 weeks - CsCl gradient solution (for balancing of ultracentrifuge tubes)
HM buffer (see Recipe 11)
Approx. 0.5 g/ml CsCl
Note: Add CsCl to 50 ml HM buffer until the RI is 1.3690 (± 0.0005); gives a solution of ~3.3 M CsCl. - Heparin affinity column running buffer (Buffer A)
Phosphate buffered saline (PBS) (see Recipe 12)
Filter sterilize - Heparin affinity column elution buffer (Buffer B)
PBS (see Recipe 12)
1 M NaCl
Filter sterilize - MBPTrap column running buffer (Buffer A)
20 mM Tris
200 mM NaCl
pH to 7.4 - MBPTrap column elution buffer (Buffer B)
20 mM Tris
200 mM NaCl
10 mM maltose
pH to 7.4 - IMAC column running buffer (Buffer A)
20 mM Na2HPO4
500 mM NaCl
40 mM imidazole
pH to 7.4 - IMAC column elution buffer (Buffer B)
20mM Na2HPO4
500 mM NaCl
500 mM imidazole
pH to 7.4 - HM buffer
25 mM HEPES
150 mM NaCl
50 mM MgCl2
pH to 7.4 - HN buffer
25 mM HEPES
150 mM NaCl
pH to 7.4 - Phosphate buffered saline (PBS)
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4 (dibasic)
2 mM KH2PO4 (monobasic)
pH to 7.4
Acknowledgments
The research was funded the National Institutes of Health: R01-GM066875 (MSC) and R35-GM122564 (MSC). The protocols for the AAVR constructs were summarized in Meyer et al. (2019) which adapts protocols for AAV VLP preparation in Lerch et al. (2012) and for AAV purification in Xie et al. (2004), all listed below.
Competing interests
The authors declare no conflicts of interest.
References
- Berns, K. I. (1996). Parvoviridae: The viruses and their replication. Virology. Fields, B. N. D., Knipe, M. and Howley, P. M.. Philadelphia, Raven: 1017-1041.
- Buning, H., Huber, A., Zhang, L., Meumann, N. and Hacker, U. (2015). Engineering the AAV capsid to optimize vector-host-interactions. Curr Opin Pharmacol 24: 94-104.
- Chapman, M. S. and M. Agbandje-McKenna (2006). Atomic structure of viral particles. Parvoviruses. Kerr, J. R., Cotmore, S. F., Bloom, M. E., Linden, R. M. and Parrish, C. R. London, Hodder Arnold, Ltd. 107-123.
- DiMattia, M. A., Nam, H. J., Van Vliet, K., Mitchell, M., Bennett, A., Gurda, B. L., McKenna, R., Olson, N. H., Sinkovits, R. S., Potter, M., Byrne, B. J., Aslanidi, G., Zolotukhin, S., Muzyczka, N., Baker, T. S. and Agbandje-McKenna, M. (2012). Structural insight into the unique properties of adeno-associated virus serotype 9. J Virol 86(12): 6947-6958.
- Drouin, L. M., Lins, B., Janssen, M., Bennett, A., Chipman, P., McKenna, R., Chen, W., Muzyczka, N., Cardone, G., Baker, T. S. and Agbandje-McKenna, M. (2016). Cryo-electron microscopy reconstruction and stability studies of the wild type and the R432A variant of adeno-associated virus Type 2 reveal that capsid structural stability is a major factor in genome packaging. J Virol 90(19): 8542-8551.
- Dudek, A. M., Pillay, S., Puschnik, A. S., Nagamine, C. M., Cheng, F., Qiu, J., Carette, J. E. and Vandenberghe, L. H. (2018). An Alternate Route for Adeno-associated Virus (AAV) Entry Independent of AAV Receptor. J Virol 92(7).
- Fitzpatrick, Z., Leborgne, C., Barbon, E., Masat, E., Ronzitti, G., van Wittenberghe, L., Vignaud, A., Collaud, F., Charles, S., Simon Sola, M., Jouen, F., Boyer, O. and Mingozzi, F. (2018). Influence of pre-existing anti-capsid neutralizing and binding antibodies on AAV vector transduction. Mol Ther Methods Clin Dev 9: 119-129.
- Gaudet, D., Methot, J., Dery, S., Brisson, D., Essiembre, C., Tremblay, G., Tremblay, K., de Wal, J., Twisk, J., van den Bulk, N., Sier-Ferreira, V. and van Deventer, S. (2013). Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther 20(4): 361-369.
- Grimm, D., Kern, A., Rittner, K. and Kleinschmidt, J. A. (1998). Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther 9(18): 2745-2760.
- Hermens, W. T., ter Brake, O., Dijkhuizen, P. A., Sonnemans, M. A., Grimm, D., Kleinschmidt, J. A. and Verhaagen, J. (1999). Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum Gene Ther 10(11): 1885-1891.
- Ibraghimov-Beskrovnaya, O., Bukanov, N. O., Donohue, L. C., Dackowski, W. R., Klinger K. W. and Landes, G. M. (2000). Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum Mol Genet 9: 1641-1649.
- Kaludov, N., Handelman, B. and Chiorini, J. A. (2002). Scalable purification of adeno-associated virus type 2, 4, or 5 using ion-exchange chromatography. Hum Gene Ther 13(10): 1235-1243.
- Kaludov, N., Padron, E., Govindasamy, L., McKenna, R., Chiorini, J. A. and Agbandje-McKenna, M. (2003). Production, purification and preliminary X-ray crystallographic studies of adeno-associated virus serotype 4. Virology 306(1): 1-6.
- Lane, M. D., Nam, H. J., Padron, E., Gurda-Whitaker, B., Kohlbrenner, E., Aslanidi, G., Byrne, B., McKenna, R., Muzyczka, N., Zolotukhin, S. and Agbandje-McKenna, M. (2005). Production, purification, crystallization and preliminary X-ray analysis of adeno-associated virus serotype 8. Acta Crystallogr Sect F Struct Biol Cryst Commun 61(Pt 6): 558-561.
- Laughlin, C. A., Tratschin, J. D., Coon, H. and Carter, B. J. (1983). Cloning of infectious adeno-associated virus genomes in bacterial plasmids. Gene 23(1): 65-73.
- Lerch, Thomas F., O'Donnell, Jason K., Meyer, Nancy L., Xie, Q., Taylor, Kenneth A., Stagg, Scott M. and Chapman, Michael S. (2012). Structure of AAV-DJ, a retargeted gene therapy vector: Cryo-electron microscopy at 4.5 Å resolution. Structure 20(8): 1310-1320.
- Lerch, T. F., Xie, Q. and Chapman, M. S. (2010). The structure of adeno-associated virus serotype 3B (AAV-3B): insights into receptor binding and immune evasion. Virology 403(1): 26-36.
- Li, C., Diprimio, N., Bowles, D. E., Hirsch, M. L., Monahan, P. E., Asokan, A., Rabinowitz, J., Agbandje-McKenna, M. and Samulski, R. J. (2012). Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol 86(15): 7752-7759.
- Majowicz, A., Salas, D., Zabaleta, N., Rodriguez-Garcia, E., Gonzalez-Aseguinolaza, G., Petry, H. and Ferreira, V. (2017). Successful repeated hepatic gene delivery in Mice and Non-human primates achieved by sequential administration of AAV5ch and AAV1. Mol Ther 25(8): 1831-1842.
- Matsushita, T., Elliger, S., Elliger, C., Podsakoff, G., Villarreal, L., Kurtzman, G. J., Iwaki, Y. and Colosi, P. (1998). Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 5(7): 938-945.
- McCraw, D. M., O’Donnell, J. K., Taylor, K. A., Stagg, S. M. and Chapman, M. S. (2012). Structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology 431(1–2): 40-49.
- Meyer, N. L., Hu, G., Davulcu, O., Xie, Q., Noble, A. J., Yoshioka, C., Gingerich, D. S., Trzynka, A., David, L., Stagg, S. M. and Chapman, M. S. (2019). Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. eLife 8: e44707.
- Nam, H. J., Lane, M. D., Padron, E., Gurda, B., McKenna, R., Kohlbrenner, E., Aslanidi, G., Byrne, B., Muzyczka, N., Zolotukhin, S. and Agbandje-McKenna, M. (2007). Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol 81(22): 12260-12271.
- Paulk, N. K., Pekrun, K., Zhu, E., Nygaard, S., Li, B., Xu, J., Chu, K., Leborgne, C., Dane, A. P., Haft, A., Zhang, Y., Zhang, F., Morton, C., Valentine, M. B., Davidoff, A. M., Nathwani, A. C., Mingozzi, F., Grompe, M., Alexander, I. E., Lisowski, L. and Kay, M. A. (2018). Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol Ther 26(1): 289-303.
- Pillay, S., Meyer, N. L., Puschnik, A. S., Davulcu, O., Diep, J., Ishikawa, Y., Jae, L. T., Wosen, J. E., Nagamine, C. M., Chapman, M. S. and Carette, J. E. (2016). An essential receptor for adeno-associated virus infection. Nature 530: 108-112.
- Pillay, S., Zou, W., Cheng, F., Puschnik, A. S., Meyer, N. L., Ganaie, S. S., Deng, X., Wosen, J. E., Davulcu, O., Yan, Z., Engelhardt, J. F., Brown, K. E., Chapman, M. S., Qiu, J. and Carette, J. E. (2017). AAV serotypes have distinctive interactions with domains of the cellular receptor AAVR. J Virol 91(18): e00391-00317.
- Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z. F., Tillman, A., Wittes, J., Pappas, J., Elci, O., McCague, S., Cross, D., Marshall, K. A., Walshire, J., Kehoe, T. L., Reichert, H., Davis, M., Raffini, L., George, L. A., Hudson, F. P., Dingfield, L., Zhu, X., Haller, J. A., Sohn, E. H., Mahajan, V. B., Pfeifer, W., Weckmann, M., Johnson, C., Gewaily, D., Drack, A., Stone, E., Wachtel, K., Simonelli, F., Leroy, B. P., Wright, J. F., High, K. A. and Maguire, A. M. (2017). Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390(10097): 849-860.
- Samulski, R. J., Berns, K. I., Tan, M. and Muzyczka, N. (1982). Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. 79: 2077-2081.
- Tseng, Y. S. and Agbandje-McKenna, M. (2014). Mapping the AAV Capsid host antibody response toward the Development of Second Generation Gene Delivery Vectors. Front Immunol 5: 9.
- Urabe, M., Ding, C. and Kotin, R. M. (2002). Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther 13(16): 1935-1943.
- Walters, R. W., Agbandje-McKenna, M., Bowman, V. D., Moninger, T. O., Olson, N. H., Seiler, M., Chiorini, J. A., Baker, T. S. and Zabner, J. (2004). Structure of Adeno-associated virus serotype 5. J Virol 78(7): 3361-3371.
- Wang, L., Calcedo, R., Bell, P., Lin, J., Grant, R. L., Siegel, D. L. and Wilson, J. M. (2011). Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum Gene Ther 22(11): 1389-1401.
- Xie, Q., Bu, W., Bhatia, S., Hare, J., Somasundaram, T., Azzi, A. and Chapman, M. S. (2002). The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A 99(16): 10405-10410.
- Xie, Q., Hare, J., Turnigan, J. and Chapman, M. S. (2004). Large-scale production, purification and crystallization of wild-type adeno-associated virus-2. J Virol Methods 122(1): 17-27.
- Xie, Q., Ongley, H. M., Hare, J. and Chapman, M. S. (2008). Crystallization and preliminary X-ray structural studies of adeno-associated virus serotype 6. Acta Crystallogr Sect F Struct Biol Cryst Commun 64(Pt 11): 1074-1078.
- Xie, Q., Spear, J. M., Noble, A. J., Sousa, D. R., Meyer, N. L., Davulcu, O., Zhang, F., Linhardt, R. J., Stagg, S. M. and Chapman, M. S. (2017). The 2.8 Å Electron Microscopy Structure of Adeno-Associated Virus-DJ Bound by a Heparinoid Pentasaccharide. Mol Ther Methods Clin Dev 5: 1-12.
- Zhang, R., Cao, L., Cui, M., Sun, Z., Hu, M., Zhang, R., Stuart, W., Zhao, X., Yang, Z., Li, X., Sun, Y., Li, S., Ding, W., Lou, Z. and Rao, Z. (2019). Adeno-associated virus 2 bound to its cellular receptor AAVR. Nat Microbiol 4(4): 675-682.
Article Information
Copyright
![]() Meyer et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Meyer et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Meyer, N., Davulcu, O., Xie, Q., Silveria, M., Zane, G. M., Large, E. and Chapman, M. S. (2020). Expression and Purification of Adeno-associated Virus Virus-like Particles in a Baculovirus System and AAVR Ectodomain Constructs in E. coli. Bio-protocol 10(3): e3513. DOI: 10.21769/BioProtoc.3513.
- Meyer, N. L., Hu, G., Davulcu, O., Xie, Q., Noble, A. J., Yoshioka, C., Gingerich, D. S., Trzynka, A., David, L., Stagg, S. M. and Chapman, M. S. (2019). Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. eLife 8: e44707.
Category
Microbiology > Microbe-host interactions > Virus
Biochemistry > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.