- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Simple and Efficient Method for Concomitant Isolation and Culture of Enriched Astroglial and Microglial Cells from the Rat Spinal Cord
Published: Vol 10, Iss 2, Jan 20, 2020 DOI: 10.21769/BioProtoc.3501 Views: 5140
Reviewed by: LAXMI NARAYAN MISHRAFanny EhretAlexandros C Kokotos
Original research article
The authors used this protocol in:

Dec 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Investigations into glial biology have contributed substantially in understanding the physiology and pathology of the nervous system. However, intricacies of the neuron-glial and glial-glial interactions in vivo present significant challenges while delineating the individual cell-type contributions, thus making the in vitro techniques exceedingly relevant to study glial biology. However, obtaining optimal yield along with high purity has been challenging for microglial cultures. Here we present a simple protocol to establish enriched astroglial as well as microglial cultures from the neonatal rat spinal cord. This method results in highly enriched astroglial and microglial cultures with maximal yield.
Keywords: Primary culturesBackground
Astrocytes and microglial cells play a crucial role during the development, as well as in the normal physiology and pathology of the nervous system. These include structural, vascular, metabolic and neuro-regulatory functions like modulation of transmission by forming ‘quad-partite synapses’, release of gliotransmitters and trophic factors, and integration of neuronal-glial networks (Parpura et al., 1994; Araque et al., 1999; Schafer et al., 2013; Michell-Robinson et al., 2015; Rossi, 2015). Microglia, in addition to the canonical immune functions, also plays an important role in adult neurogenesis, differentiation, maturation and integration within the neuronal circuitry (Rezaie and Male, 2002; Polazzi and Monti, 2010; Michell-Robinson et al., 2015; Ransohoff and El Khoury, 2015). In response to a pathological insult, the glial cells react by undergoing morphological and physiological changes and rescue the vulnerable neurons from the insult (Ferraiuolo et al., 2011; Pekny et al., 2014; Mishra et al., 2016; Mishra et al., 2017). At the tissue level, astrocytes undergo morphological transformation from protoplasmic to fibrillary phenotype and move to the site of injury to create a physical barrier between damaged and healthy cells, or by forming glial scars in a process called reactive gliosis. In a similar manner, microglia undergo morphological transformation from the resting/ramified phenotype to the activated/amoeboid phenotype, via intermediate activation stages, (Rezaie and Male, 2002; Michell-Robinson et al., 2015; Ransohoff and El Khoury, 2015). Both the glial cell types thus promote neuronal repair through acute inflammation, which modulates the microenvironment by regulating production and release of trophic factors, gliotransmitters, cytokines and chemokines (Raivich et al., 1999; Parpura et al., 2012; Verkhratsky and Butt, 2013; Pekny et al., 2014). Reactive gliosis is reversible and beneficial during the acute insult. However, in response to prolonged insult or genetic dysregulation, the balance is disrupted and may lead to irreversible and erroneous activation resulting in chronic inflammation and neurodegeneration (Lobsiger and Cleveland, 2007). Therefore, delineating the precise glial response as well as their role in modulating neuronal physiology becomes relevant. However, an in vivo approach may present with a disadvantage owing to the complex neuron-glial and glial-glial interactions, which dynamically modulate these changes at the tissue level. Primary glial cultures help us to overcome this issue, by enabling observations in an isolated cell-type model system. In vitro experimentation also widens the scope for further comparing the effect of neuron-glial/glial-glial interactions through a co-culture model system.
Various approaches have been adopted to culture astrocytes and microglia from animal brains and spinal cords. These methods vary considerably not only in terms of purity and yield, but also with respect to the cell population targeted for isolation (Giulian and Baker, 1986; Saura et al., 2003; Floden and Combs, 2007; Scorisa et al., 2010; Kerstetter and Miller, 2012). For instance, the conventional repeated shaking methods for culturing microglia exploit the microglial layer growing atop astrocytes in the mixed glial cultures (Giulian and Baker, 1986; Scorisa et al., 2010), while the mild trypsinization method targets the ones growing beneath (Saura et al., 2003). We compared all the methods and standardized a procedure to establish exceedingly enriched astroglial and microglial cultures, while maximizing the yield. The current protocol generates enough microglial and astroglial cells per spinal cord to execute parallel experiments in both the cell types, thus minimizing the inter-population variability while comparing cell-specific responses.
Materials and Reagents
- Animals
Wistar strain rat pups with post-natal Day 0-2 (P0-P2), housed and maintained in accordance with the institutional ethics guidelines - Culture products
- 0.22 µm membrane filter (Millipore, catalog number: GSWP04700)
- T-25 ml tissue culture flask (Corning, Sigma, catalog number: CLS430639)
- 24-well plate (Thermo Fisher Scientific, catalog number: 142475)
- Coverslips, 13 mm, circular (Thermo Fisher Scientific, catalog number: 12-519-21G)
- DNase-1 (Sigma-Aldrich, catalog number: 11 284 932 001)
- Hanks’ balanced salt solution (HCMF, Thermo Fisher Scientific, catalog number: 14170112)
- Poly-L-lysine hydrobromide (PLL, Sigma-Aldrich, catalog number: P5899-5G)
- Dulbecco’s modified Eagle’s medium/F-12 (DMEM/F-12) (GIBCO, Invitrogen, USA, catalog number: 12500062)
- L-Glutamine, 200 mM solution (Thermo Fisher Scientific, catalog number: 25030081)
- Fetal bovine serum-Heat inactivated (FBS-HI) (GIBCO, Invitrogen, USA, catalog number: 10082147)
- 0.25% trypsin-EDTA (GIBCO, Invitrogen, USA, catalog number: 25200)
- Penicillin-streptomycin (Pen/Strep; 100x solution; 10,000 units/ml each) (Thermo Fisher Scientific, catalog number: 15140122)
- HEPES (1 M) (Thermo Fisher Scientific, catalog number: 15630080)
- MilliQ water
- HCl (Sigma-Aldrich, catalog number: 7647-01-0)
- Ethanol (Sigma-Aldrich, catalog number: 64-17-5)
- HBSS
- Glucose powder
- NaCl
- KCl
- Na2HPO4
- KH2PO4
- PFA
- Growth medium (500 ml) (see Recipes)
- 50 ml FBS-HI (10% v/v) (see Recipes)
- Dissection medium (500 ml) (see Recipes)
- Dissociation medium (10 ml) (see Recipes)
- DMEM-trypsin-EDTA (1:4) (10 ml) (see Recipes)
- Immunostaining
- 0.1 M PBS, freshly prepared
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153)
- Anti-ChAT (rabbit polyclonal Abcam, catalog number: ab18736; 1:500)
- Anti-GFAP (mouse monoclonal, Abcam, catalog number: ab24345; 1:500)
- Anti-IBA1 (Rabbit polyclonal Abcam, catalog number: ab153696; 1:500)
- Anti-mouse IgG (Cy3-conjugated; 1:200, Sigma-Aldrich)
- Anti-rabbit IgG (FITC-conjugated; 1:200, Chemicon)
- PVA-DABCO anti-fade mounting medium (Sigma-Aldrich, catalog number: 10981)
- PBS 1x (pH-7.4) (see Recipes)
- PFA, 4% (see Recipes)
Equipment
- Fine forceps, Dumont #5 (2), curved (1) (Fine Science Tools)
- Scissors (1), 51/2 in., straight, operating (Fine Science Tools)
- Scissors (2), 4 in., straight, micro dissecting (Fine Science Tools)
- Dissecting microscope (Leica Microsystems)
- Phase contrast microscope (Leica Microsystems)
- Confocal laser-scanning microscope (Leica Microsystems)
- CO2 incubator (Eppendorf)
- Orbital Shaker (New Brunswick, Eppendorf)
- Centrifuge (Thermo Scientific)
- Fire polished glass pipette (Fisherbrand)
- Light source
- Laminar flow hood
- 4 °C refrigerator
- -20 °C freezer
- -80 °C freezer
- Hemocytometer
Software
- Photoshop CS2 (Adobe)
- Microsoft Office
Procedure
- Coating the coverslips
- Wash, dry and sterilize the coverslips by autoclaving and/or ethanol treatment (95% to absolute ethanol) prior to coating.
- Incubate the dried, sterile coverslips with PLL (0.1 mg/ml, final concentration in PBS) for 30 min, followed by rinsing twice for 30 s with PBS to prevent the toxicity associated with excess PLL.
- Let the coverslips dry completely before use. We recommend using freshly coated coverslips. Alternatively, the pre-coated coverslips can be stored at 4 °C for up to 3 weeks.
- Dissection of neonatal spinal cords
- Prepare ice-cold dissection medium (see Recipes).
- Carefully anesthetize the pups. Wipe the body with 70% ethanol and swiftly decapitate with the help of a scalpel.
- Make an incision in the lower back and expose the vertebral column. The skin and vertebral column tissue of the neonatal rats are soft and can be dissected out with relative ease when compared to the adults.
- With the help of spring scissors and fine forceps, carefully cut open the vertebral column to expose the spinal cord, moving from the caudal to rostral end. Gently but swiftly remove the spinal cord and place it into ice-cold dissection medium. For a description of the dissection process, we recommend the protocol previously published by Gal et al. (2016).
- Thoroughly separate the meninges from the spinal cords using the tips of fine forceps and minimizing tissue damage.
- With the help of a scalpel, cut the tissue into small pieces and proceed with trituration in a Laminar Air Flow hood (LAF).
- Mixed glial cultures
- Transfer the tissue to 1 ml of the dissociation medium per spinal cord, at 37 °C for 15 min. Trypsin helps in dissociation of the tissue while addition of DNase aids by digesting the viscous DNA released from the damaged cells.
- Stop the reaction by adding pre-warmed growth media (DMEM/F-12 supplemented with 10% FBS-HI) and centrifuge at 400 x g for 3 min.
- Replace the supernatant with growth media, and mechanically triturate by passing through 1 ml fire polished glass pipette 8-10 times gently. Care should be taken to avoid bubbles.
- Allow the undissociated tissue to settle down and transfer the supernatant to another vial. Add fresh media and repeat the process of mechanical trituration 2 more times or till the tissue is dissociated. The optimal shear stress causes the tissues to dislodge into a single-cell suspension. However, harsh trituration may lead to loss of yield due to cell death.
- Pellet down the single-cell suspension thus obtained at 400 x g for 3 min.
- Resuspend in fresh media.
- Count the cells using a hemocytometer and plate with a seeding density of 2.5 x 104 cells/ml in T-25 tissue culture flask. Incubate at 37 °C in a sterile cell culture incubator with 5% CO2 and 95% humidity.
Note: Coating the flasks with PLL prior to seeding is not necessary, as glial cells adhere robustly. In our experience, seeding on uncoated flasks further ensured enrichment. - Change the media every day for 3-4 days to ensure the proper removal of residual debris. Afterward, replenish the media as per the requirement. By day in vitro (DIV) 7, the cultures start attaining confluence, and by DIV 12, a population of microglia with fringent, phase-bright soma can be seen growing on the top of astrocytes (Figure 1).
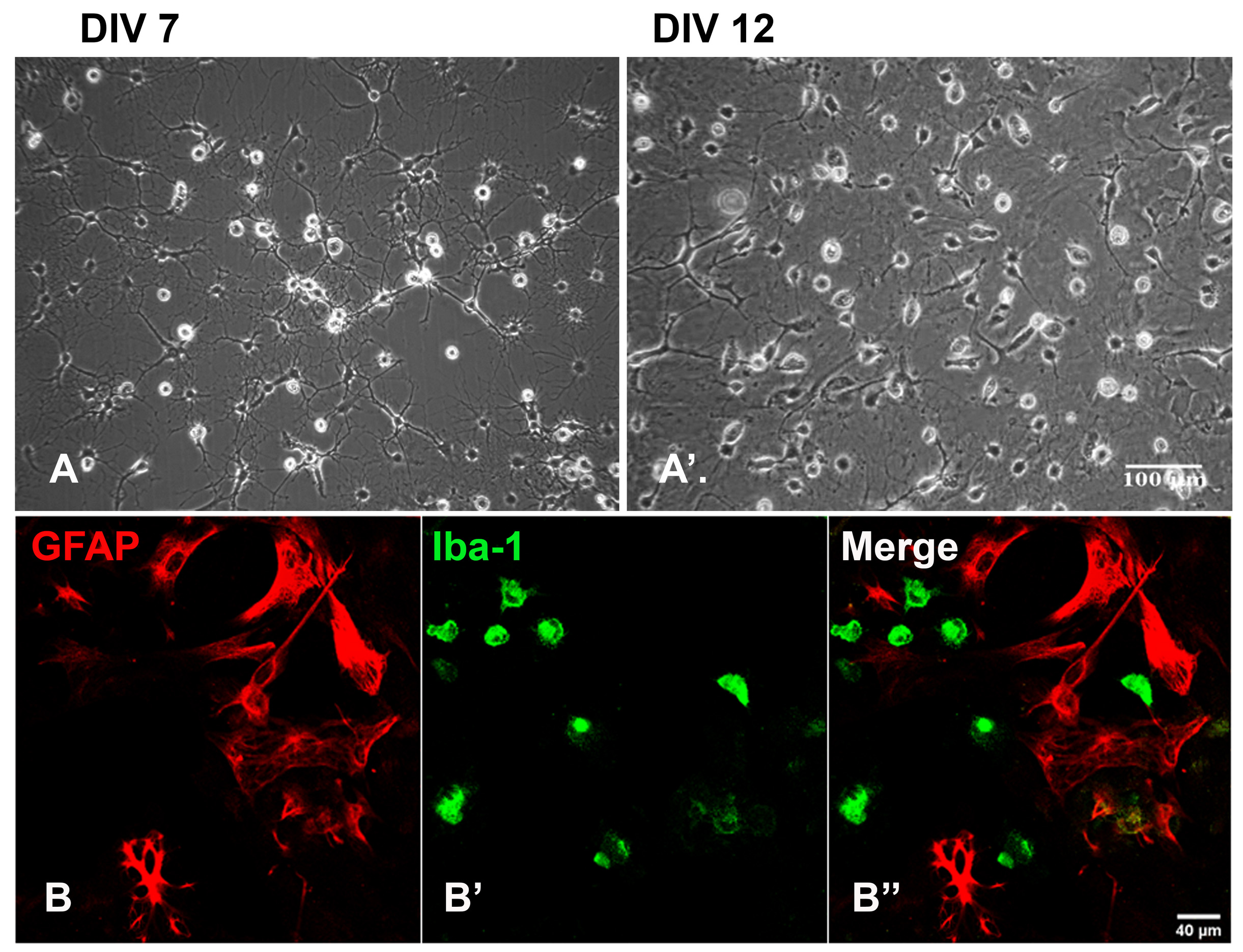
Figure 1. Mixed glial cultures isolated from the rat spinal cord. A and A’ represent the growing (DIV 7) and confluent (DIV 12) cultures, respectively. Panel B depicts the presence of distinct microglial marker, Iba-1 positive cells growing on top of the astroglial marker, GFAP labeled cells. Scale bars are indicated.
- Astroglial cultures
- Establish and allow the mixed cultures to attain confluence (8-10 DIV). Replace the media every alternate day. On the 11th DIV, place the culture flasks on an orbital shaker at 200 rpm for 3-4 h maintained at 37 °C. Aspirate the resultant media that contains the less adherent microglial cells dislodged from the top of astrocytes.
- The media containing microglial cells obtained at this stage can be further centrifuged at 400 x g for 3 min to pellet microglia. This microglial population can be resuspended in DMEM/F-12 and plated for future experiments in appropriate culture dishes with the desired plating density. However, care must be taken to avoid the dislodged non-microglial cells, including astrocytes, as it can easily lead to cross-contamination. Moreover, we adopted another, more efficient method to culture microglia (Saura et al., 2003), that provided better yield and reduced contamination, as described later.
- Thoroughly wash the intact astroglial layer with PBS, followed by incubation in 0.25% trypsin-EDTA for 1-2 min, to ensure proper detachment of the cells.
- Monitor the rounding of cells and stop the trypsinization with an equal amount of growth media when the detachment is complete.
- Pellet the cells at 400 x g for 3 min and resuspend in fresh growth media. Replate at a density of 2.5 x 104 cells/ml on the circular coverslips in a 24-well plate and proceed with the experiments (Figure 2).
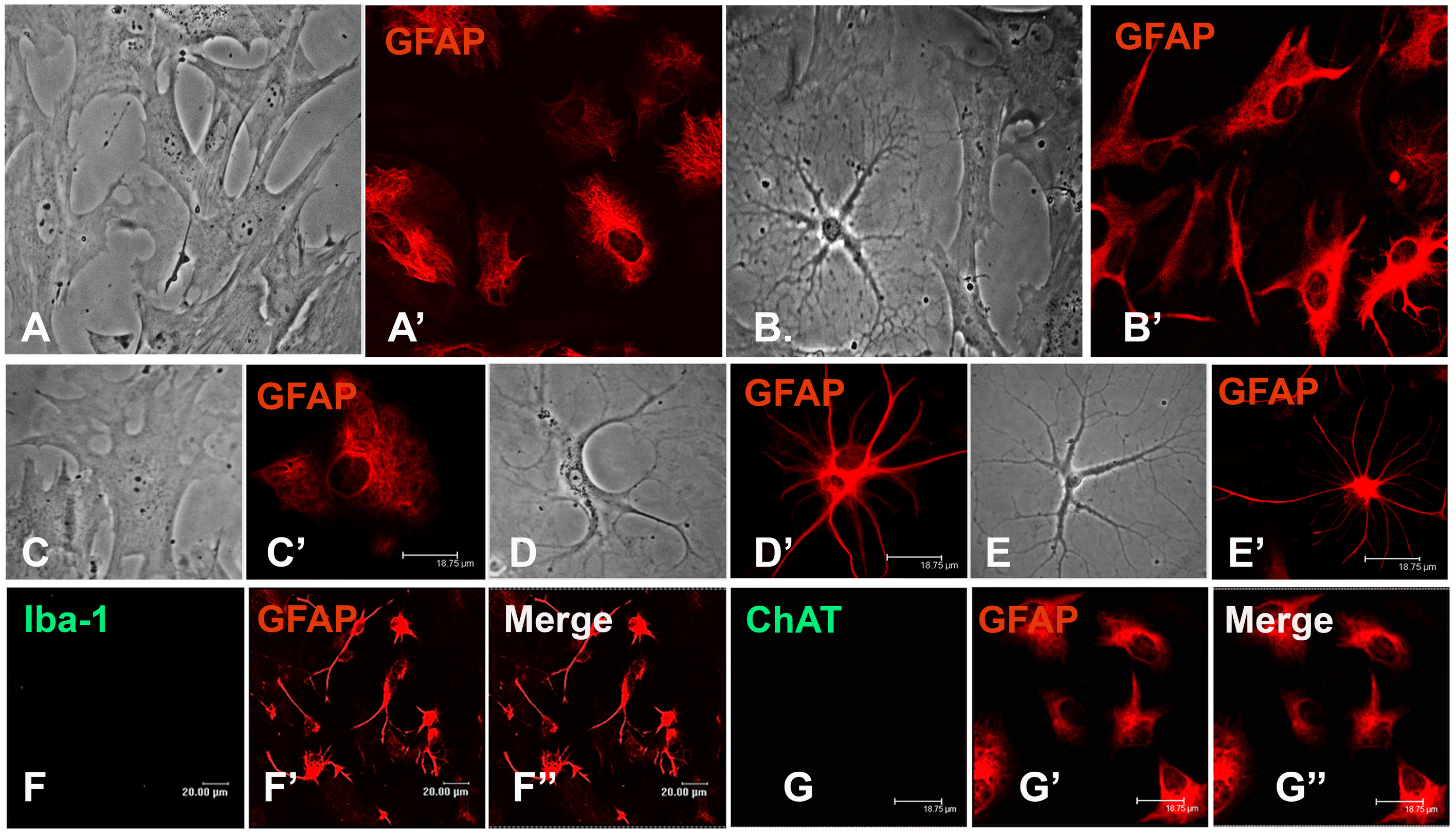
Figure 2. Enriched astroglial cultures derived from the mixed cultures. A, A’, C, C’. The cultures mainly consist of a flat/protoplasmic morphology. B, D-E’. Fibrous morphology can be seen occasionally in unstimulated cultures but frequently in cultures provided with inflammatory stimulus. B, B’. Astrocytes further subcultured (20-25 DIV) do not show a significant change in their viability or reactive ability. The cultures stain negative for microglia as shown by Iba-1 staining (F-F”) and neurons, as shown by ChAT staining (G-G”). Scale bars are indicated.
- Microglial cultures
- Derive the mixed glial cultures from the Wistar rat pups as described above and proceed for establishing microglial cultures using mild trypsinization method.
- After the cultures reach confluence, allow the cultures to mature for a few more days while constantly replenishing one-half of the media with fresh medium every alternate day. Don’t completely remove the old media conditioned with glial factors, as these factors allow for the optimal growth of microglia.
- After the intact astroglial monolayer has matured for a while, subject the glial cultures to mild trypsin-EDTA treatment. Add 4-6 ml of Trypsin EDTA-DMEM/F-12, in a ratio of 1:4 per flask, enough to cover the cells. The mild trypsinization step removes the astroglial monolayer, revealing the microglial layer growing beneath, while DMEM/F-12 provides nourishment throughout the process. The process usually takes 10-15 min. Periodically check for the detachment of the monolayer under a phase contrast microscope and standardize the time for optimal detachment.
- Once the astroglial layer is fully detached, discard the media containing the astroglial layer and wash the culture dish thoroughly with DMEM/F-12 to remove traces of non-microglial cells.
- Check the cultures for purity. Afterward, use the cells directly, or replate on the circular coverslips in a 24-well plate, or as per the requirement of the experiment (Figure 3).
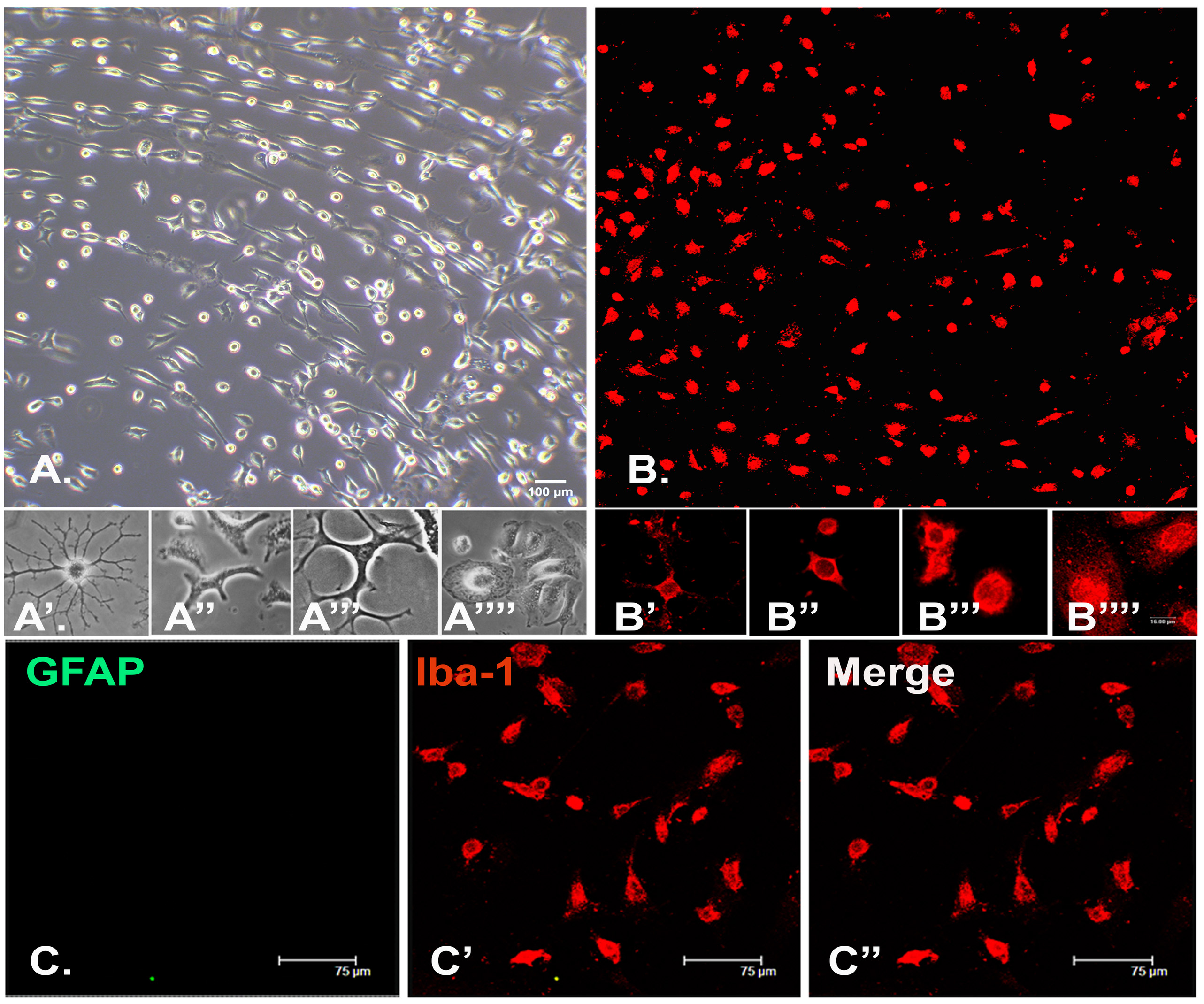
Figure 3. Enriched microglial cultures derived from the mixed cultures. A-A’’, B-B’’. Healthy cultures predominantly display process bearing morphology in the unstimulated state. However, intermediate (A’’’, B’’’) and reactive, phagocytotic (A’’’’, B’’’’) stages can be observed as the microglial response to external stimuli. The cultures stain negative for astroglial contamination (C-C’’). Scale bars are indicated.
- Fixation and immunostaining
- Wash the coverslips containing cells with sterile PBS and follow with fixation using 4% PFA for 15 min at RT. Aspirate PFA and thoroughly wash with PBS before proceeding for immunostaining.
- Antigen retrieval step is optional and should be standardized depending upon the guidelines for each antibody. For the antibodies used in the present study, antigen retrieval was neither needed nor performed.
- Proceed for blocking with 3% BSA for an hour, followed by the overnight incubation at 4 °C with the primary antibody of interest. After primary incubation, subject the coverslips to 3 washes of 5 min each before adding appropriate, fluorescently labeled secondary antibodies. After incubation for 2 h at RT, wash the coverslips with PBS thrice for 5 min each and mount them on a clean slide with PVA-DABCO anti-fade mounting medium. Once the slide has dried completely, proceed for imaging with a confocal microscope.
Data analysis
The mixed glial cultures, as well as the primary astroglial and microglial cultures obtained using these protocols were investigated and compared for their purity and individual responses through a series of assays. GFAP, Iba-1 and DAPI staining demonstrated > 98% enrichment of both the astroglial as well as microglial cultures (Mishra et al., 2016 and 2017). However, the analysis is beyond the scope of this protocol and has already been published. Therefore, the data analysis has not been discussed.
Notes
- Supplementation with muscle extract from the gastrocnemius muscles of the wistar rats promotes the growth of motor neurons at the Step D7, and should be done if motor neuronal survival is desired in the mixed cultures (Shobha et al., 2007).
- The mixed cultures can be further subcultured by trypsinization prior to isolation of astrocytes and /or microglia, to further enhance the yield. In that case, the cultures should be standardized and characterized prior to planning the experiments. Moreover, all the experiments must be conducted and replicated at the same passage status to avoid inducing any phenotypic variability that might arise due to subculturing.
- Subculturing further ensures the purity of the cultures. The astroglial cultures can be subcultured 3-4 times without significant loss of characteristics. Plating at a high density further obliterates cross contamination.
- At the Step F2 it is crucial that the astroglial monolayer is allowed to mature and remain intact. An immature layer may not detach efficiently, resulting in improper isolation and/or cross-contamination of microglial cultures. Usually 5-7 days after the cultures have attained confluence, it is optimal to proceed with mild trypsinization.
- In our hands the trypsin-EDTA-DMEM/F-12 ratio of 1:4 used at the Step F3 efficiently removed the astroglial monolayer for 15-20 DIV cultures. However, the ratio can be further increased to 1:3 to 1:2 depending on the culture conditions and must be standardized. Moreover, EDTA is essential to ensure the selective removal of astrocytes, while leaving the microglial layer intact. For further insights, we recommend reading the article by Saura et al. (2003), where the protocol was first described.
- In view of achieving purity, the washing step is critical at the Step F4, as the astrocytes remaining in the culture dish can efficiently reattach and proliferate, leading to astroglial contamination.
- The microglia thus obtained can be propagated overnight in serum free DMEM/F-12 and plated for the experiment in the following day. Microglia could be grown in mixed glial/astroglial conditioned media for a longer duration. However, microglial cells are dynamic and very quickly respond to the slightest changes in their environment in vitro. Therefore, for the best, unbiased results, we recommend using the cultures at the earliest.
- The astroglial monolayer detached during the Step F3 can be further subcultured. However, since the confluent astroglial layer has a high nutritional demand, the media must be frequently replenished to ensure a healthy subculture. One way of determining the health of the cultures is to monitor the detachment of the layer, as unhealthy or starved astroglial layer detaches very quickly with or without mild trypsinization. We recommend standardizing and checking the integrity of the subcultured astrocytes before using them for further experimentation. In our hands, a second subculture yielded viable, protoplasmic astrocytes that actively responded to inflammatory stimulus (Figures 1B-B’). Further, replating the cultures at a high density ensured healthy and enriched astroglial cultures. Unlike astrocytes, microglial cells cannot be subcultured.
Recipes
Note: The media preparation should be done under a laminar flow hood to ensure sterility.
- Media preparation
DMEM/F-12 powdered media package into 800 ml of autoclaved MilliQ
0.11% HEPES and sodium bicarbonate, with pH set to 7.4 using 1 N HCl
10 ml of 1x antibiotic mixture (100,000 IU penicillin, 0.05% streptomycin and 0.002% amphotericin B)
Make the final volume up to 1 L and Sterile-filter the media using a 0.22 µm membrane filter store at 4 °C until further use - Growth medium (500 ml)
450 ml DMEM/F-12
50 ml FBS-HI (10% v/v)
Filter and store at 4 °C - Dissection medium (500 ml)
487 ml 1x HBSS
5 ml 1 M HEPES (0.1 M solution)
3 g glucose powder (6 mg/ml)
5 ml Pen/Strep solution (100 U/ml)
Filter and store at 4 °C - Dissociation medium (10 ml)
HBSS 10 ml
0.25% trypsin-EDTA
DNase 100 μg/ml - DMEM-trypsin-EDTA (1:4) (10 ml)
2 ml of 2.5% trypsin EDTA dissolved in 8 ml of DMEM/F-12, freshly prepared - PBS 1x (pH-7.4)
NaCl 8 g (0.137 M)
KCl 200 mg (0.0027 M)
Na2HPO4 1.44 g (0.01 M)
KH2PO4 240 mg (0.0018 M)
Make up the volume to 1 L with milliQ water, adjust the pH - PFA 4% (50 ml)
- 2 mg PFA in 20 ml Double Distilled water
- Adjust the pH to 7.4
- Make the volume 50 ml using water and 25 ml 2x PBS
Acknowledgments
The protocols were standardized with the help of grants from DBT and ICMR, govt. of India, respectively. PM was a Junior/senior research fellow under UGC/CSIR fellowship program by govt. of India. The protocols were used in two separate studies published by Mishra et al. (2016 and 2017). The authors are further grateful to Dr. Josep Saura and Dr. Kumarasamy Murali for troubleshooting and standardization of the enriched microglial cultures.
Competing interests
The authors declare no competing interest.
Ethics
All the animal procedures used in establishing the protocols were approved by the Institutional Animal Ethics Committee (AEC/44/264/NP and AEC/55/343/NP).
References
- Araque, A., Sanzgiri, R. P., Parpura, V. and Haydon, P. G. (1999). Astrocyte-induced modulation of synaptic transmission. Can J Physiol Pharmacol 77(9): 699-706.
- Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M. and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7(11): 616-630.
- Floden, A. M. and Combs, C. K. (2007). Microglia repetitively isolated from in vitro mixed glial cultures retain their initial phenotype. J Neurosci Methods 164(2): 218-224.
- Giulian, D. and Baker, T. J. (1986). Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 6(8): 2163-2178.
- Kerstetter, A. E. and Miller, R. H. (2012). Isolation and culture of spinal cord astrocytes. Methods Mol Biol 814: 93-104.
- Le Gal, J. P., Juvin, L., Cardoit, L. and Morin, D. (2016). Bimodal respiratory-locomotor neurons in the neonatal rat spinal cord. J Neurosci 36(3): 926-937.
- Lobsiger, C. S. and Cleveland, D. W. (2007). Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci 10(11): 1355-1360.
- Michell-Robinson, M. A., Touil, H., Healy, L. M., Owen, D. R., Durafourt, B. A., Bar-Or, A., Antel, J. P. and Moore, C. S. (2015). Roles of microglia in brain development, tissue maintenance and repair. Brain 138(Pt 5): 1138-1159.
- Mishra, P. S., Dhull, D. K., Nalini, A., Vijayalakshmi, K., Sathyaprabha, T. N., Alladi, P. A. and Raju, T. R. (2016). Astroglia acquires a toxic neuroinflammatory role in response to the cerebrospinal fluid from amyotrophic lateral sclerosis patients. J Neuroinflammation 13(1): 212.
- Mishra, P.-S., Vijayalakshmi, K., Nalini, A., Sathyaprabha, T., Kramer, B., Alladi, P. A. and Raju, T. J. J. o. n. (2017). Etiogenic factors present in the cerebrospinal fluid from amyotrophic lateral sclerosis patients induce predominantly pro-inflammatory responses in microglia. J Neuroinflammation 14(1): 251.
- Parpura, V., Basarsky, T. A., Liu, F., Jeftinija, K., Jeftinija, S. and Haydon, P. G. (1994). Glutamate-mediated astrocyte-neuron signalling. Nature 369(6483): 744-747.
- Parpura, V., Heneka, M. T., Montana, V., Oliet, S. H. R., Schousboe, A., Haydon, P. G., Stout, R. F., Spray, D. C., Reichenbach, A., Pannicke, T., Pekny, M., Pekna, M., Zorec, R. and Verkhratsky, A. (2012). Glial cells in (patho)physiology. J Neurochem 121(1): 4-27.
- Pekny, M., Wilhelmsson, U. and Pekna, M. (2014). The dual role of astrocyte activation and reactive gliosis. Neurosci Lett 565: 30-38.
- Polazzi, E. and Monti, B. (2010). Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 92(3): 293-315.
- Raivich, G., Bohatschek, M., Kloss, C. U., Werner, A., Jones, L. L. and Kreutzberg, G. W. (1999). Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev 30(1): 77-105.
- Ransohoff, R. M. and El Khoury, J. (2015). Microglia in health and disease. Cold Spring Harb Perspect Biol 8(1): a020560.
- Rezaie, P. and Male, D. (2002). Mesoglia & microglia--a historical review of the concept of mononuclear phagocytes within the central nervous system. J Hist Neurosci 11(4): 325-374.
- Rossi, D. (2015). Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol 130: 86-120.
- Saura, J., Tusell, J. M. and Serratosa, J. (2003). High-yield isolation of murine microglia by mild trypsinization. Glia 44(3): 183-189.
- Schafer, D. P., Lehrman, E. K. and Stevens, B. (2013). The "quad-partite" synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61(1): 24-36.
- Scorisa, J. M., Duobles, T., Oliveira, G. P., Maximino, J. R. and Chadi, G. (2010). The review of the methods to obtain non-neuronal cells to study glial influence on Amyotrophic Lateral Sclerosis pathophysiology at molecular level in vitro. Acta Cir Bras 25(3): 281-289.
- Shobha, K., Vijayalakshmi, K., Alladi, P. A., Nalini, A., Sathyaprabha, T. N. and Raju, T. R. (2007). Altered in-vitro and in vivo expression of glial glutamate transporter-1 following exposure to cerebrospinal fluid of amyotrophic lateral sclerosis patients. J Neurol Sci 254(1-2): 9-16.
- Verkhratsky, A., and Butt, A. M. (2013). Glial physiology and pathophysiology. John Wiley & Sons.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mishra, P. S. and Raju, T. R. (2020). A Simple and Efficient Method for Concomitant Isolation and Culture of Enriched Astroglial and Microglial Cells from the Rat Spinal Cord. Bio-protocol 10(2): e3501. DOI: 10.21769/BioProtoc.3501.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Monolayer culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.