- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High-throughput Site-directed Scanning Mutagenesis Using a Two-fragment PCR Approach



(*contributed equally to this work) Published: Vol 10, Iss 1, Jan 5, 2020 DOI: 10.21769/BioProtoc.3484 Views: 6733
Reviewed by: Pooja VermaVinay PanwarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Site-directed scanning mutagenesis is a useful tool applied in studying protein function and designing proteins with new properties, such as increased stability or enzymatic activity. Creating a systematic library of hundreds of site-directed mutants is still a demanding and expensive task. The established protocols for making such libraries include PCR amplification of the recombinant DNA using a pair of primers carrying a target mutation in the same PCR. Unfortunately, this approach is very often coupled with PCR artifacts which compromise overall efficiency of site-directed mutagenesis. To reduce the failure rate due to the PCR artifacts, we have set up a high-throughput mutagenesis protocol based on a two-fragment PCR approach. To this end, each mutation is introduced in two separate PCRs resulting in two linear fragments of the mutated plasmid. In the next steps, the PCR template is digested and the two matching plasmid fragments are joined together using Gibson assembly. Separating the corresponding mutagenic primers into two different PCRs decreases a number of artifacts and thus increases overall cloning efficiency. Furthermore, free software that we developed facilitates both high-throughput primer design and analysis of sequencing results. Overall, this protocol enabled us to efficiently produce several alanine-scanning libraries of 400 single-point mutations with complete coverage of the protein sequence.
Keywords: Scanning mutagenesisBackground
High-throughput cloning techniques are widely used for research involving expression and purification of proteins. Site-directed scanning mutagenesis techniques in particular are often used to generate mutants for functional studies or to generate stabilized proteins for biophysics and structural biology. Libraries necessary for these purposes may contain several hundred mutants and their generation can be expensive and time consuming. Many single-point mutant libraries have been created using single vector PCRs with overlapping primers, e.g. QuikChange (Agilent). Although this and similar mutagenesis techniques are well established and widely used (for a recent review see Ortega et al., 2019), they may lead to a number of different errors in the final vector (Liu and Naismith, 2008; Edelheit et al., 2009; Sun et al., 2013; Heydenreich et al., 2017) caused by inherent sequence overlap of primers. Such errors include additional mutations and primer repeats, which necessitates sequencing of multiple colonies or additional rounds of mutagenesis. The protocol presented here is based on a two-fragment PCR mutagenesis, where the primers used in each PCR reaction have no sequence complementarity. The two halves of a plasmid to be cloned are amplified in two separate PCRs, followed by in vitro circularisation using Gibson assembly reaction (Figure 1). As a result, the number of successful mutagenesis reactions after sequencing only one clone per mutant is increased, allowing us to generate multiple alanine-scanning libraries of complete proteins of ca. 400 amino acids in length within approx. 6 weeks each (Heydenreich et al., 2017). In addition, our protocol can also be used to insert multiple point mutations in one round of cloning and for sequence modification (e.g., addition of purification tags, small insertions and deletions) with increased efficiency.
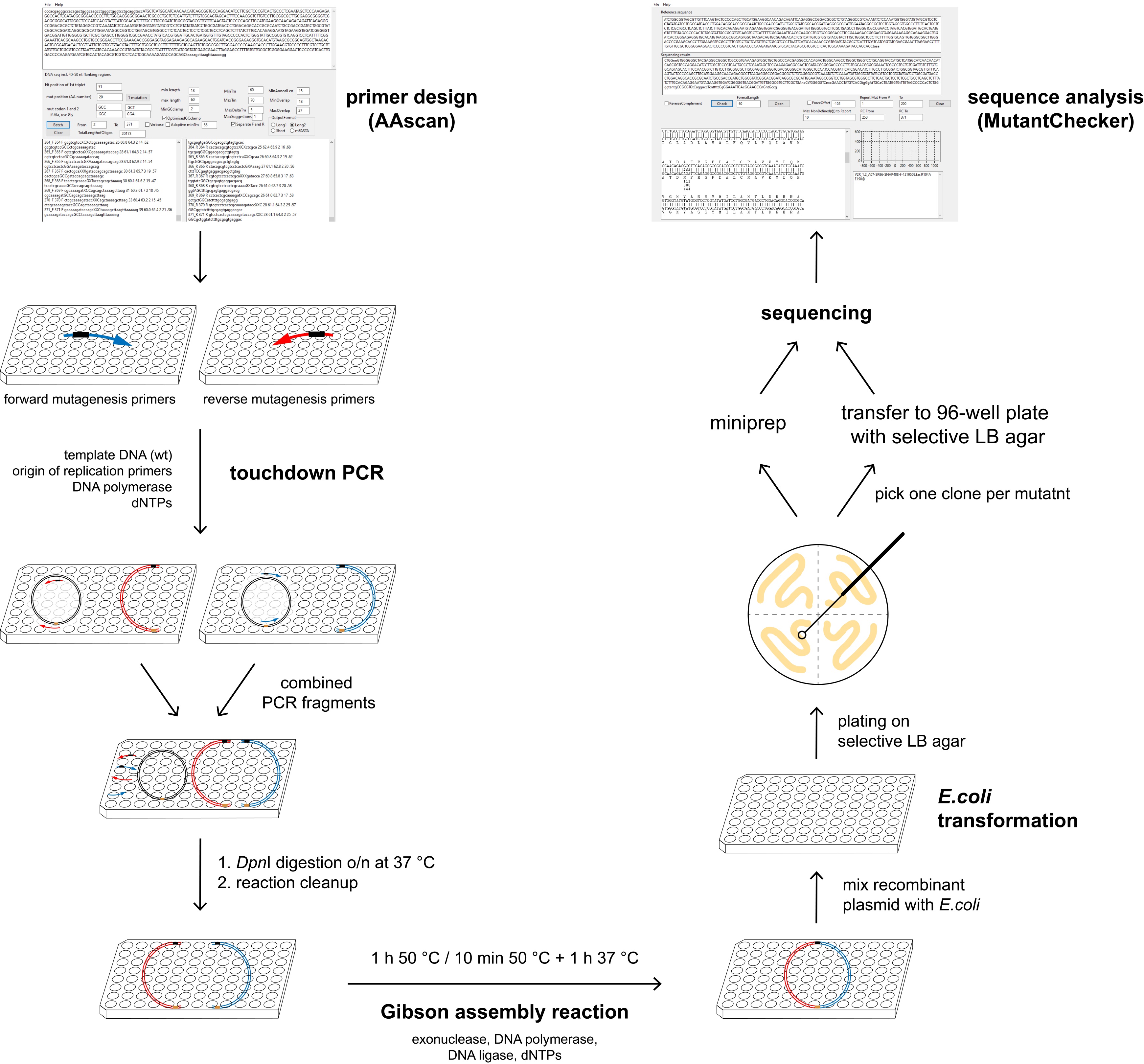
Figure 1. Overview of the mutagenesis technique. AAscan software (Sun et al., 2013) is used to design mutagenic primers for the high-throughput site-directed scanning mutagenesis. Two PCR reactions are done per mutant, in each of them approximately half of the vector is amplified. Two fragments containing one mutation are combined, followed by DpnI digestion at 37 °C overnight. Reaction clean-up is performed to purify DNA fragments, which are then assembled by Gibson assembly reaction. Bacteria are transformed with the resulting circular plasmid and plated on selective LB agar plates. One clone per mutant is sent for sequencing either on a selective LB agar 96-well plate or as purified DNA. All steps excluding the plating of the bacteria are done in 96-well plates. Sequencing results are analyzed in a high-throughput manner using MutantChecker program (Sun et al., 2013).
Materials and Reagents
Note: All reagents must be of analytical grade or higher and stored according to recommendations from the manufacturer. All plasticware must be DNase-free.
- LB-agar 96-well plates with the appropriate antibiotic(s) (can be ordered from a sequencing company; if not, see Recipes)
- Pipette tips, 10 μl, 200 μl and 1,250 μl (VWR, European catalog numbers: 613-1093, 613-1092 and 613-1087)
- (Optional) Sterile toothpicks
- Polypropylene conical tubes 15 ml and 50 ml (Falcon, catalog numbers: 352096 and 352070)
- Microcentrifuge tubes 1.5 ml (Simport, catalog number: T330-7G)
- Pipetting reservoir (Diversified Biotech, catalog number: REBP-3000)
- 96-well PCR plates, 0.2 ml (VWR, European catalog number: 211-0297)
- Microseal ’B’ PCR plate seals (Bio-Rad, catalog number: MSB1001)
- Deep-well plates 96/2,000 μl (Eppendorf, catalog number: 651033405)
- Adhesive gas-permeable film for culture plates (VWR, European catalog number: 391-1261)
- Sterile Petri dishes (e.g., ThermoScientific NuclonTM Delta Surface, catalog number 150350)
- Disposible sterile spreaders (e.g., VWR, catalog number: 612-5497) or autoclaved glass beads 2 mm (e.g., Supelco, catalog number: 1.04014)
- ZR-96 DNA Clean & Concentrator (Zymo Research, catalog number: D4024)
- Chemically competent Escherichia coli XL-1 Blue cells (Agilent, catalog number: 200249) or some other cell strain which is used for cloning (Mach1, DH5α, TOP10 etc.)
- Cloning primers
ColE1A: 5′-GGAGCGAACGACCTACACCGAACTGAGATACCTACAGCG-3′
ColE1B: 5′-CGCTGTAGGTATCTCAGTTCGGTGTAGGTCGTTCGCTCC-3′
Note: This pair of primers can be used for all plasmids containing the ColE1 origin of replication. - Plasmid to be used as a template in PCR
- Nicotinamide adenine dinucleotide, NAD+, 50 mM (New England Biolabs, catalog number: B9007L)
- Deoxynucleotide (dNTPs) solution mix, 10 mM each dNTP (New England Biolabs, catalog number: N0447S)
- T5 exonuclease, 10 U/μl (New England Biolabs, catalog number: M0363S)
- Phusion HF DNA polymerase, 2 U/μl (New England Biolabs, catalog number: M0530L)
- Taq DNA ligase, 40 U/μl (New England Biolabs, catalog number: M0208L)
- DpnI, 20 U/μl (New England Biolabs, catalog number: R0176L)
- Nuclease-free water (Cell Signaling Technology, catalog number: 12931S)
- Phusion High-Fidelity PCR Master Mix with GC Buffer (New England Biolabs, catalog number: M0532L) or some other appropriate DNA polymerase master mix (see Procedure B. PCR)
- PEG-8000 (Sigma-Aldrich, catalog number: P5413)
- Tris-HCl buffer, 1 M, pH 7.5 (Thermo Fischer Scientific, catalog number: 15567027)
- MgCl2, 1 M (Sigma-Aldrich, catalog number: M1028)
- DL-dithiothreitol (DTT), 1 M (Sigma-Aldrich, catalog number: 43816)
- SOC outgrowth medium (New England Biolabs, catalog number: B9020S)
- Antibiotic stock solution(s) (depending on an antibiotic resistance gene(s) carried by the plasmid of interest)
- Lysogeny broth (LB) broth with agar (Sigma-Aldrich, catalog number: L2897)
- Ultrapure deionized water with electrical resistivity of 18 MΩ cm at 25 °C
- 5x isothermal reaction (IT) buffer (see Recipes)
- 1.33x Gibson assembly mixture (see Recipes)
- LB-agar plates with the appropriate antibiotic(s)-Petri dishes (see Recipes)
Equipment
- Diverse pipettes including multi-channel pipettes and repeat pipettors
- Thermocycler that can accommodate 96-well PCR plates (e.g., EppendorfTM MastercyclerTM Nexus Thermal Cycler, catalog number: E6332000029)
- Centrifuge for deepwell plates (e.g., Eppendorf 5810 R with swing-bucket rotor A-4-62, catalog number: 5811000320)
- Laboratory incubator set at 37 °C
- Laboratory incubator/shaker set at 37 °C
Software
- AAscan (Sun et al., 2013; https://github.com/dmitryveprintsev/AAScan/)
- MutantChecker (Sun et al., 2013; https://github.com/dmitryveprintsev/AAScan/)
Procedure
- Primer design and ordering
- Download and open AAscan software (Sun et al., 2013) to design mutagenic primers for the site-directed scanning mutagenesis. A screenshot of the AAscan software interface is shown in Figure 2.
- Replace the default DNA sequence given in AAscan with a DNA sequence which is to be mutated, including the flanking regions (40-50 nucleotides) at the 5′ and 3′ ends (DNA seq. incl.40-50 nt flanking regions).
- Define the position of the first nucleotide in the first triplet codon (Nt position of 1st triplet). This relates the given DNA sequence with the amino-acid numbering. In the example given in Figure 2, the 50 nucleotides from the vector upstream of the protein coding sequence are included as a flanking region. Therefore, the nucleotide A from the ATG codon, which corresponds to amino-acid residue 1, is at the position 51. Whenever possible, we recommend using a standard numbering scheme for the protein of interest. This does not have to match the amino-acid numbering in the expressed protein construct (which might include a signal peptide, purification tags, protease cleavage sites, linkers or fusion protein partners).
- Define up to two codons coding for an amino acid (Xxx) by which you want to replace other amino acids in the scanning mutagenesis, e.g., ‘GCG’ and ‘GCA’ for Ala (mut codon 1 and 2). If only one particular codon is to be used, include it into both mut codon 1 and 2 text fields. We recommend choosing the codons based on their frequency in the expression host and the GC content of the gene of interest. Select those codons which are most frequent in the expression host and, if possible, avoid those with high or low GC content if your gene of interest has a very high (> 60%) or very low (< 40%) GC content, respectively. For several constructs with GC content in a range 50-55 % which were to be expressed in human cell culture (HEK 293S), we used the most frequent codons for this host: GCC/GCT for Ala, GGC/GGA for Gly, and GTG/GTC for Val. On the other hand, if the gene of interest has a high GC content, it might be better to use codons with lower GC content, i.e., GCT/GCA for Ala, GGA/GGT for Gly and GTT/GTA for Val. Such codon choice minimizes the number of designed mutagenic primers with relatively high annealing temperatures.
- If a particular amino acid in the scanning mutagenesis region is already Ala, replace it with another amino acid Yyy by defining up to two corresponding codons, e.g., in alanine scanning mutagenesis replacing an existing alanine with a glycine (if Ala, use Gly). In choosing the codons, use the recommendations given in the previous step.
- Enter an amino acid mutagenesis range (text fields From and To) by using the amino-acid numbering scheme defined in Step A3.
- Define the primer design parameters. Based on our experience (Sun et al., 2013; Heydenreich et al., 2017), we recommend using the following parameters: minimal primer length (min length)–18 nucleotides (nt); maximal primer length (max length)–60 nt; minimal GC clamp (MinGCclamp)–2; optimized GC clamp (OptimisedGCclamp); minimal primer melting temperature (MinTm)–60 °C; maximal primer melting temperature (MaxTm)–70 °C; maximal difference between melting temperatures of forward and reverse primer (MaxDeltaTm)–5 °C; minimal distance from the mutation codon to the 3′ end of the primer (MinAnnealLen)–15 nt; length of the overlap sequence between the ends of the resulting PCR fragment (MinOverlap and MaxOverlap)–between 15 and 27 nt; the number of primer pairs designed for the mutation of every specified amino acid position (MaxSuggestions)–1.
- Define the output format. Use separate result columns for forward and reverse primers (Separate F and R). User can choose among the more (Long1 and Long2) or less (Short and mFASTA) detailed outputs.
- Click or touch Batch button. This gives two separate lists of the designed forward (‘n_F’) and reverse (‘n_R’) primers including their names and sequences (and other primer properties if output format Long1 or Long2 was chosen). ‘n’ in a primer name corresponds to an amino acid number which is to be mutated by using that primer.
- Order the designed primers from a company that provides synthesis of DNA oligonucleotides. Such company usually makes available an order sheet template file into which the AAscan output (obtained in Step A10) can easily be copied. When ordering, use a 96-well plate format with forward and reverse primers separated into different 96-well plates, but in such a way that a position of each forward primer corresponds to exactly the same position of the related reverse primer in the other 96-well plate (e.g., primers 103_F and 103_R at the position A6 in each 96-well plate). Order desalted DNA oligonucleotides already dissolved at concentration 100 μM either in nuclease-free water or in some standard buffer provided by the company (usually 10 mM Tris-HCl, 0.1 mM EDTA, pH 7.5); see Note 1.
- Purchase a pair of the desalted reverse-complementary primers which anneal approximately on the opposite side of the plasmid. It is advisable to choose such primers that bind either to the origin of replication or the antibiotic resistance gene. In this case, only the properly assembled plasmid DNA will allow growing the transformed bacteria on a plate with selection antibiotic. We use origin-of-replication ColE1 primers (see Materials and Reagents).
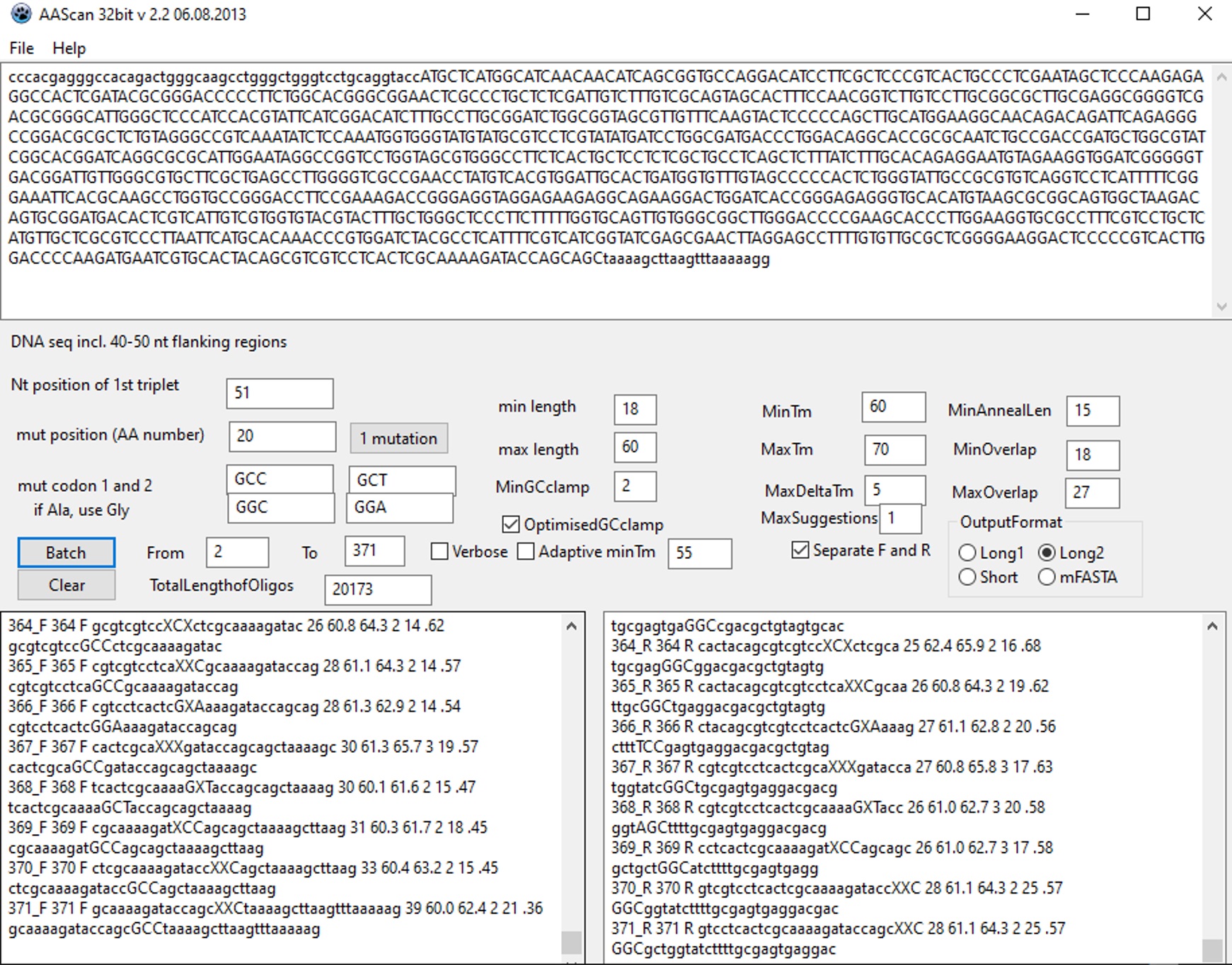
Figure 2. AAscan software interface. In the example shown, the coding sequence is written in capital letters and the flanking regions are given in small letters (DNA seq incl. 40-50 nt flanking regions). The first nucleotide in the first triplet codon (ATG) is at the position 51 of the specified DNA sequence (Nt position of 1st triplet). The mutagenic primers designed for amino-acid residues 2-371 are separated into two output windows: the forward primers are listed in the left one and the reverse primers in the right one.
- PCR
Each mutation is generated in two separate PCRs (denoted A and B) resulting in two PCR fragments that are combined in subsequent Gibson assembly. Before performing PCR for all of the target mutations, we recommend testing PCR conditions on a small number (20-30) of randomly chosen mutations and modifying the protocol accordingly, as detailed in the original article by Heydenreich et al. (2017). We recommend testing different polymerases and buffer condition as well as DMSO addition. To identify efficient PCR conditions, we analyze each PCR product from the test mutation set in agarose gel electrophoresis. Nevertheless, we do not include this or any other control in the large-scale mutagenesis.- Before doing the experiment, check the orientation of the gene of interest in its plasmid. This is important to determine how to combine in PCR mutagenic primers with the origin-of-replication ColE1 primers. A mutagenic primer and the appropriate ColE1 primer should anneal to complementary DNA strands; e.g., if the forward mutagenic primers are combined with ColE1B, the reverse mutagenic primers should be combined with ColE1A (and vice versa).
- Dilute the plasmid DNA template with nuclease-free water to concentration 1 ng/μl.
- Dilute the primers ColE1A and ColE1B with nuclease-free water to concentration 6 μM.
- Dilute the mutagenic primers in a 96-well plate with nuclease-free water to 6 μM.
- Two sets of PCRs are necessary. For each set of x mutagenesis PCR reactions, prepare a PCR master mix as defined in Table 1.
Table 1. PCR master mixes. Each mixture is enough for x mutagenesis PCRs in 20 μl reaction volume.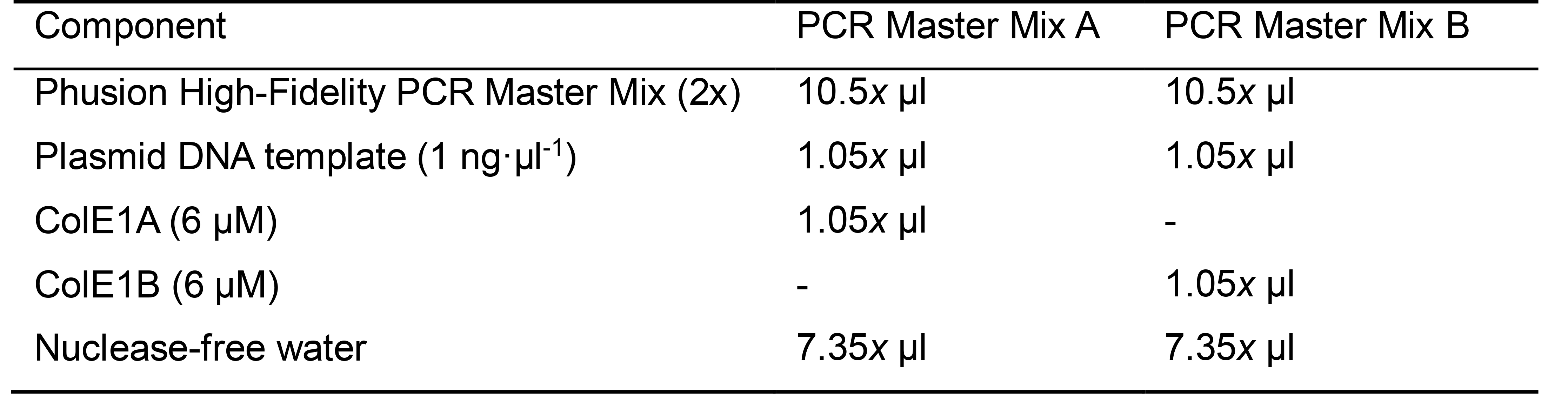
- To prepare PCR mixtures A, pipette using a multichannel pipette 19 μl PCR Master Mix A into each used well of a 96-well 200-μl PCR plate on ice and add to it 1 μl mutagenic primer (6 μM) from the appropriate primer set (forward or reverse, as discussed in Step B1). Close the 96-well PCR plate with a PCR seal.
- Prepare a PCR program as in Table 2 (see Note 2).
Table 2. PCR program for DNA multiplication using Phusion polymerase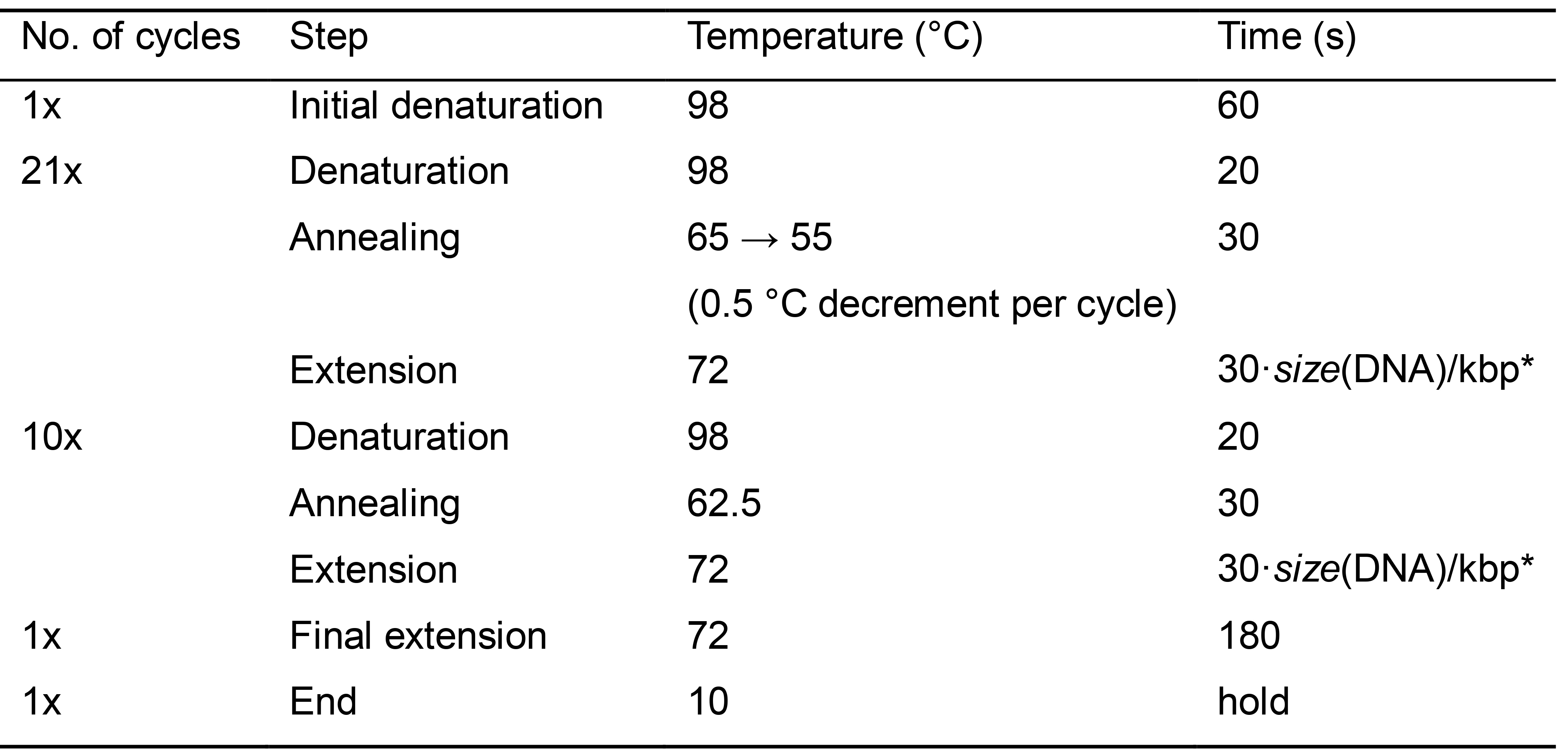
*Size of the longest DNA fragment in kilobase pairs (kbp) to be amplified in PCR; the extension time is 30 s per 1 kbp. - Preheat a thermocycler: a block to 98 °C (denaturation temperature) and a lid to 103 °C (see Note 3).
- Transfer the sealed 96-well PCR plate with the reaction mixtures directly from ice to the preheated thermocycler, close the lid and start the PCR program defined in Step B7.
- Keep the 96-well PCR plate with completed PCR on ice until further processing.
- To prepare PCR mixtures B, repeat the Steps B6-B9 by using PCR Master Mix B and the corresponding mutagenic primers (the second mutagenic primer set, as discussed in Step B1).
- DNA template digestion and reaction clean-up
- After completion of both PCRs A and B, transfer 20 μl PCR mixture A to the corresponding 20 μl PCR mixture B in a 96-well PCR plate using a multichannel pipette. In this step, we combine two fragment PCR products possessing the same mutation.
- Add 0.5 μl DpnI (20 U·μl-1) to each mixture of fragments, seal the 96-well plates and incubate them at 37 °C to digest methylated DNA template. DpnI digestion for 1 h is already enough, but the mixtures can be incubated for up to 18 h if this is more convenient in terms of time management.
- In order to purify the PCR fragments from the reaction mixture after the DpnI digestion, use ZR-96 DNA Clean & Concentrator-5 KitTM (Zymo Research) according to the manufacturer’s protocol, but use 10 μl elution buffer in the last step. This small volume should result in higher DNA concentrations for the subsequent Gibson assembly.
- Gibson assembly
- In a 96-well PCR plate on ice, mix 1 μl each mixture of cleaned PCR fragments with 3 μl 1.33x Gibson assembly mix (Recipe 2) by pipetting gently up and down ten times using a multichannel pipette set at volume 3 µl to avoid foaming (see Note 4).
- Transfer the sealed 96-well plate with Gibson assembly reaction mixtures into a preheated thermocycler (a block at 50 °C and a lid at 53 °C) and incubate it for 10 min at 50 °C followed by 1 h at 37 °C and a hold at 10 °C. Alternatively and especially for longer nucleotide overlaps within a pair of mutagenic primers (e.g., 21 or more nt; see Step A8), the Gibson assembly reaction can be conducted for 1 h at 50 °C followed by a hold at 10 °C. Which Gibson assembly conditions are better suited can be checked first on a small number (20-30) of randomly chosen mutations, as detailed by Heydenreich et al., 2017 (see Note 5).
- Cell transformation
- Thaw chemically competent Escherichia coli XL-1 Blue cell suspension on ice (see Note 6).
- Onto a 96-well PCR plate on ice, distribute 20 μl cell suspension per each well and mutation using a repeat pipette.
- Add 2 μl Gibson assembly mixture to each 20-μl cell aliquot, seal the plate with a PCR plate seal and incubate on ice for 10-20 min.
- Transfer the 96-well plate with cells-DNA mixtures into a preheated thermocycler (a block at 42 °C and a lid at 42 °C) and incubate it for 60 s at 42 °C.
- Immediately put the cells-DNA mixtures on ice and incubate for 2-5 min. If the plasmid carries the ampicillin resistance gene, you can proceed immediately to Step E8 (although Steps E6 and E7 increase cloning efficiency in this case as well).
- Using a multichannel pipette, transfer the transformed cells to a 96-deep-well plate filled with 300 μl SOC media (without any antibiotic), close the plate with a gas-permeable seal and incubate for 1 h at 37 °C while shaking at 160 rpm.
- Spin down the cells (3,000 x g, 5 min) and carefully remove 220 μl supernatant using a multichannel pipette.
- Resuspend a cell pellet in the remaining supernatant (ca. 100 μl) by pipetting up and down using a multichannel pipette set at 80 µl (to avoid foaming) until all cell pellets are resuspended without visible clumps during pipetting. Plate the cell suspension onto an LB-agar plate with the appropriate antibiotic. To save some storage space and material, you can plate four different mutants per LB-agar plate (one mutant onto each plate quadrant) or 24 mutants on a 24-well LB-agar plate (one mutant per well) by using an expanding multi-channel pipette.
- Allow the plates to dry for ca. 10 min and then incubate them upside-down at 37 °C overnight. Next day, you should find bacterial colonies on the LB-agar plates.
- Plasmid DNA preparation and sequencing
For high-throughput projects, sequencing companies offer plasmid preparation and sequencing directly from a bacterial colony on 96-well plates without extra costs in comparison to sending already extracted plasmid samples. We strongly recommend this option.- Label a position of a selected colony on an LB-agar plate, pick it by a sterile pipette tip or a toothpick and transfer it to LB-agar with the appropriate antibiotic in a 96-well plate. Such plates are usually offered by a sequencing company; if not, you can easily prepare them by your own (Recipe 3).
- Order the sequencing with the appropriate sequencing primer (forward or reverse). For reliable results, the sequencing primer should anneal at least 30-40 nucleotides upstream from the expected mutation. Depending on a sequencing service, one can usually expect reliable sequence read lengths of at least ca. 600 nucleotides (which corresponds to ca. 200 amino acids). For longer sequences, one should use additional sequencing primer(s) to cover the whole region of interest. Send the 96-well LB-agar plates with one colony per mutant to the sequencing company and wait for the sequencing results. Sending one mutant per sequencing round reduces sequencing costs.
- Analyze the sequencing results in MutantChecker software (Sun et al., 2013), as detailed in the Data analysis section.
- If the mutation is not confirmed in a sequencing round, send a new colony in the next sequencing round (see Note 8).
- You can obtain plasmid solutions in a 96-well plate from the sequencing company. Alternatively or in addition, you can prepare plasmids by yourself from the colonies labeled on LB-agar plates which were positive in sequencing.
Data analysis
Analysis of sequencing data
- Save the sequencing data as separate .fasta, .seq or .txt files in a folder on the computer.
- Open MutantChecker (Figure 3) and paste the reference non-mutated DNA sequence into the indicated field (Reference sequence). The sequence should start with the first codon as defined in Procedure, Step A3. Analyze mutant DNAs sequenced with forward primers separately from those sequenced with reverse primers. For more detailed information see Sun et al. (2013).
- For DNA sequenced with forward primers, change the default values provided in Report mut from # and To in the respective fields, using amino acid numbers. Generally, a forward sequence should cover about 200 to 250 amino acids.
- Once you have checked the program settings, click on “File” → “Load File List”. Select one or multiple files to be checked. We recommend checking files one by one initially. If several files are analyzed at once, an alignment will only be displayed for the last file selected.
- The largest window will now display the alignment of the reference sequence and the sequencing result (Figure 3). The graph on the right indicates how many possible alignments have been found. Ideally, you should see a single sharp peak. The window on the lower right displays sequence names followed by any mutation found within the limits entered in step 3 (see Note 7).
- For DNA sequenced with reverse primers, indicate the range of amino acids where a mutation should be reported using the fields RC From and RC To. RC stands for reverse complement. Tick the ReverseComplement box when checking DNA sequenced with reverse primers.
- Repeat steps 4-5 to analyze the remaining sequencing data.
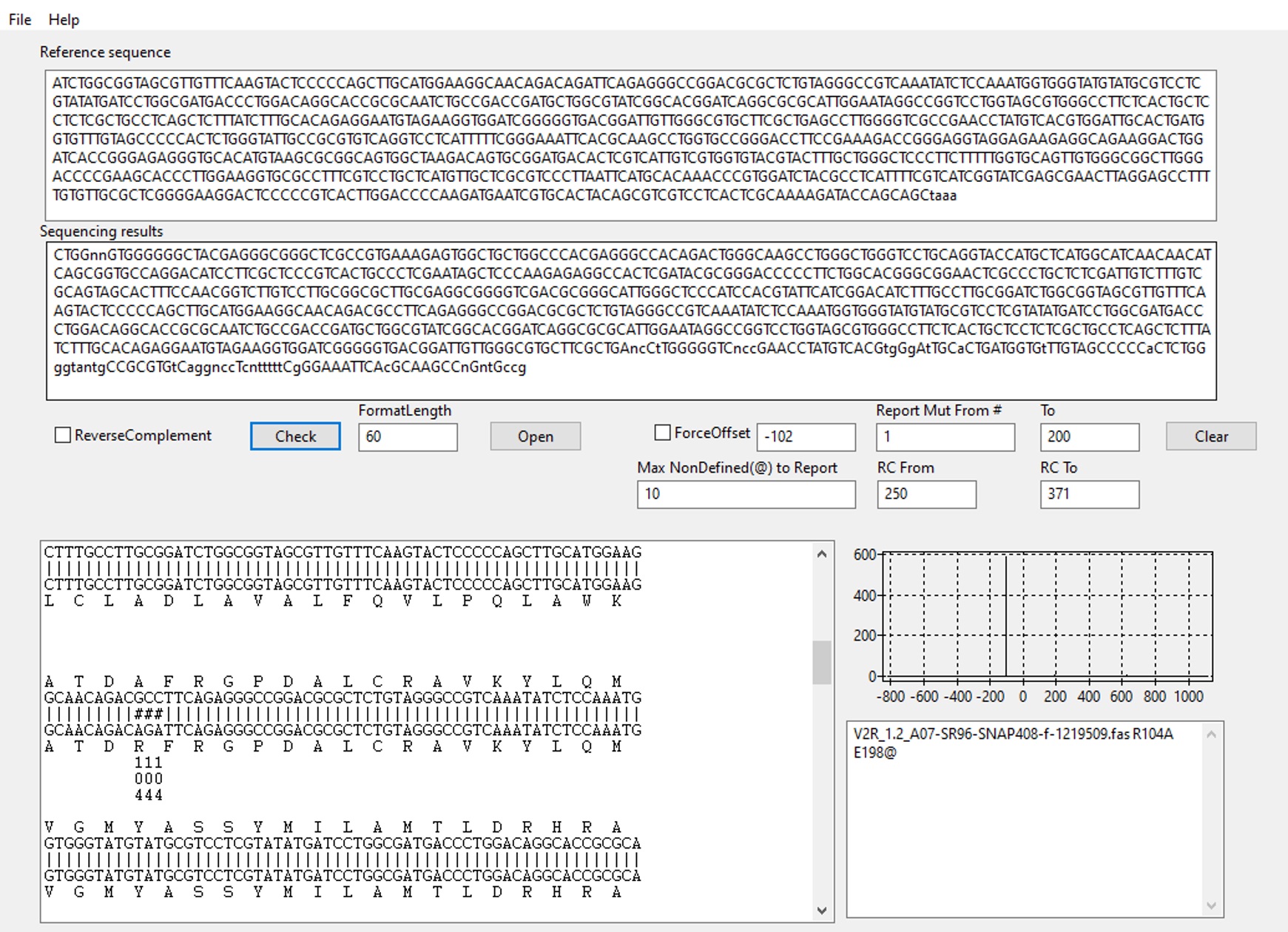
Figure 3. MutantChecker software interface. Sequencing data analysis for mutation R104A is shown as an example. Graph on the right side depicts the alignment score function vs. offset between the sequencing result and the reference sequence (Sun et al., 2013). In this case, there is a single peak which indicates that one possible alignment was found. Multiple peaks would have implied several alternative alignments due to repeated sequences (usually an indication of a cloning artifact). Mutations identified in the specified amino acid range (1-200) are listed in the lower right output window. ‘E198@’ denotes that sequencing trace starts to be unclear (“n” in the sequencing result) from amino acid E198.
Notes
- No primer modification (like phosphorylation) is necessary for Gibson assembly and the minimum synthesis scale of DNA oligonucleotides is usually more than sufficient. Store the frozen primers at -20 °C for up to 2 years. Several freeze-thaw cycles have no considerable influence on the stability of DNA oligonucleotides.
- Different PCR protocols might be needed for DNA polymerases other than Phusion.
- This step minimizes mis-priming during initial denaturation. It may not be necessary if using hot-start polymerases.
- Instead of the homemade Gibson assembly mix, one can use a commercial Gibson assembly master mix or a similar DNA assembly product.
- More recently, we successfully used incubation for 1 h at 45 °C for which it was shown to increase the efficiency of Gibson assembly (Gibson et al., 2010). This may be especially beneficial if the primers were designed with relatively short overlap to improve their specificity (e.g., 15 bp).
- The cells should be prepared by the Inoue method (Sambrook and Russell, 2006) and have a transformation efficiency of at least 107 colonies per 1 μg pBR322 (or a similar plasmid). Alternatively, one can use some other E. coli strain that is customary for routine cloning, such as Mach1, DH5α or TOP10.
- Depending on the company which sequenced the DNA, the annotation of the traces may vary. Some companies decide not to display an “N” when the trace is unclear, but instead opt for the highest peak. In both cases, a mismatch will be reported, but it may be perceived as a real mutation when the trace does not contain Ns. We recommend that you check the trace in cases like that even if the reported mutation occurs at the very beginning or end of a sequencing result. Especially towards the end of a sequence, a repeat of four or more bases of the same type (all Gs, all Cs, all As or all Ts) may lead to difficult-to-interpret traces, which can lead to n bases being read as n-1 or n+1, resulting in a frameshift. This is usually a sequencing problem rather than a true frameshift. These cases, or sequences trailed by many Ns may lead to additional peaks close to the main peak.
- For troubleshooting, please refer to the original publication (Heydenreich et al., 2017).
Recipes
- 5x Isothermal reaction (IT) buffer (6 ml)
1.5 g PEG 8000
3 ml Tris-HCl (1 M, pH 7.5)
300 μl MgCl2 (1 M)
300 μl DTT (1 M)
600 μl NAD+ (50 mM)
600 μl dNTPs solution mix (10 mM each dNTP)
Prepare 500-μl aliquots of 5x IT buffer and store them at -80 °C - 1.33x Gibson assembly mixture (1.8 ml)
480 μl 5x IT buffer
0.96 μl T5 exonuclease (10 U/μl)
30 µl Phusion HF DNA polymerase (2 U/μl)
240 µl Taq DNA ligase (40 U/μl)
1,049 μl nuclease-free water
Make 50-μl aliquots and store at -20 °C (or -80 °C for long-term storage)
Note: Gibson assembly mixture can be stored for more than a year at -20 °C and the enzymes are still active after more than 10 freeze-thaw cycles (Gibson et al., 2009). - LB-agar plates with antibiotic
- Add 35 g LB broth with agar in 1 L ultrapure deionized water
- Autoclave at 121 °C for 15 min to dissolve the powder and sterilize LB-agar
- Let it cool down to hand-hot temperature (ca. 45 °C), add stock solution of the appropriate antibiotic to achieve the target antibiotic concentration and swirl vigorously to mix
- Pour ca. 15 ml melted LB-agar with antibiotic into each 10-cm-diameter Petri dish and let it solidify
- For 96-well LB-agar plates, use a multi-channel pipette to transfer 150 μl LB-agar with antibiotic per well into a 96-well plate with flat bottom and let the agar solidify
- The plates should be stored upside-down in sealed plastic bags at 4 °C until use
Acknowledgments
This protocol was derived from our previous publication (Heydenreich et al., 2017). This work was supported by Swiss National Science Foundation Doc.Mobility fellowship 165219 to FMH, Erasmus, EMBO Short Term Fellowship ASTF 420–2016 and Swiss National Science Foundation Doctoral Mobility Grant 31003A_159748/2 travel fellowships, and NanoMem ITN network funding to TM, NRW Strategy Project BioSC BOOST FUND “RIPE” to the Institute of Biochemical Plant Physiology at the Heinrich Heine University, Marie Curie (274497) and UBS Promedica Foundation post-doctoral fellowships to DM, and Swiss National Science Foundation grants 141898, 159748 to DBV and COST Action CM1207 GLISTEN.
Competing interests
The authors declare no competing financial or non-financial interests.
References
- Edelheit, O., Hanukoglu, A. and Hanukoglu, I. (2009). Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol 9: 61.
- Gibson, D. G., Smith, H. O., Hutchison, C. A., 3rd, Venter, J. C. and Merryman, C. (2010). Chemical synthesis of the mouse mitochondrial genome. Nat Methods 7(11): 901-903.
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6(5): 343-345.
- Heydenreich, F. M., Miljuš, T., Jaussi, R., Benoit, R., Milić, D. and Veprintsev, D. B. (2017). High-throughput mutagenesis using a two-fragment PCR approach. Sci Rep 7(1): 6787.
- Liu, H. and Naismith, J. H. (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8: 91.
- Ortega, C., Abreu, C., Oppezzo, P. and Correa, A. (2019). Overview of high-throughput cloning methods for the post-genomic era. Methods Mol Biol 2025: 3-32.
- Sambrook, J. and Russell, D. W. (2006). The inoue method for preparation and transformation of competent E. coli: "ultra-competent" cells. CSH Protoc 2006(1).
- Sun, D., Ostermaier, M. K., Heydenreich, F. M., Mayer, D., Jaussi, R., Standfuss, J. and Veprintsev, D. B. (2013). AAscan, PCRdesign and MutantChecker: a suite of programs for primer design and sequence analysis for high-throughput scanning mutagenesis. PLoS One 8(10): e78878.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Heydenreich, F. M., Miljuš, T., Milić, D. and Veprintsev, D. B. (2020). High-throughput Site-directed Scanning Mutagenesis Using a Two-fragment PCR Approach. Bio-protocol 10(1): e3484. DOI: 10.21769/BioProtoc.3484.
Category
Molecular Biology > DNA > DNA cloning
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.