- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of Monosaccharides from Arabidopsis Seed Mucilage and Whole Seeds Using HPAEC-PAD
Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3464 Views: 5780
Reviewed by: Yufang LuAgnieszka ZienkiewiczAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






![[<sup>14</sup>C] Glucose Cell Wall Incorporation Assay for the Estimation of Cellulose Biosynthesis](https://en-cdn.bio-protocol.org/imageup/arcimg/20150921035050714.jpg?t=1773968368)

Abstract
Arabidopsis seed coat epidermal cells deposit a significant quantity of mucilage, composed of the cell wall components pectin, hemicellulose, and cellulose, into the apoplast during development. When mature seeds are hydrated, mucilage extrudes to form a gelatinous capsule around the seed. Determining the monosaccharide composition of both extruded mucilage and whole seeds is an essential technique for characterizing seed coat developmental processes and mutants with altered mucilage composition. This protocol covers growth of plants to produce seeds suitable for analysis, extraction of extruded mucilage using water and sodium carbonate (used for mutants with impaired mucilage release), and extraction of alcohol insoluble residue (AIR) from whole seeds. The prepared polysaccharides are then hydrolyzed using sulfuric acid, which hydrolyses all polysaccharides including cellulose. Sensitive and reproducible quantification of the resulting monosaccharides is achieved using high-performance anion exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD).
Keywords: ArabidopsisBackground
In Arabidopsis, fertilization of the ovule initiates growth and differentiation of the ovule integuments into the seed coat (Beeckman et al., 2000; Western et al., 2000; Windsor et al., 2000). During differentiation, the outermost layer of integument cells begins to secrete large quantities of pectinaceous mucilage into the apoplastic space. This secretion is targeted to the point where the outer primary cell wall meets the radial cell wall, and leads to the formation of a donut-shaped pocket of mucilage surrounding a column of cytoplasm. After mucilage secretion is complete, the cytoplasmic column is replaced by a cellulosic secondary cell wall called the columella and the cells undergo programmed cell death prior to seed desiccation. When mature Arabidopsis seeds are hydrated, the mucilage expands and ruptures the primary cell wall. The extruded mucilage forms two distinct layers, a non-adherent layer that is easily detached, and an adherent layer that is strongly attached to the seed surface (reviewed in Šola et al., 2019a). Mucilage polysaccharides include pectin, hemicellulose, and cellulose. Ninety percent of the mucilage is pectin, of which 90% is unbranched Rhamnogalacturonan I (RG-I; reviewed in Voiniciuc et al., 2015).
The synthesis of seed coat mucilage is not necessary for plant viability under laboratory conditions (Western et al., 2001). In addition, seed mucilage is readily accessible in large quantities. This has allowed isolation of viable mutants and development of transgenic lines with altered seed coat mucilage. The analysis of these mutants and transgenic lines can be used to understand the biosynthesis, deposition, regulation and function of mucilage polysaccharides and serves as a model system for studying various aspects of cell wall biology (Arsovski et al., 2010; Haughn and Western, 2012; Western, 2012; North et al., 2014; Francoz et al., 2015; Voiniciuc et al., 2015; Griffiths and North, 2017; Francoz et al., 2018; Golz et al., 2018; Šola et al., 2019a).
An essential technique for analyzing mucilage is rapid, reliable, and simple quantification of monosaccharides. We have developed an HPAEC-PAD technique that has been employed successfully for over a decade (Dean et al., 2007; Huang et al., 2011; Mendu et al., 2011; Voiniciuc et al., 2013; Griffiths et al., 2014; Shi et al., 2019; Šola et al., 2019b). Our protocol for seed coat mucilage extraction includes modifications for mutants such as mucilage modified2 (mum2) that produce mucilage with altered properties that impair hydration and extrusion (Dean et al., 2007; Macquet et al., 2007). We also established a method for preparation of whole seed alcohol insoluble residue (AIR). As previous analysis in our lab has shown that much of the rhamnose (Rha) and galacturonic acid (GalA) in whole seed is derived from mucilage RG-I (Western et al., 2001), this analysis can be used to determine whether the total amount of mucilage synthesized is altered. The release of monosaccharides from mucilage polysaccharides and whole seed AIR is based on the secondary acid hydrolysis derived from the Klason procedure (Coleman et al., 2009). This protocol uses sulfuric acid instead of the trifluoracetic acid (TFA) used by other mucilage analysis protocols (e.g., Voiniciuc and Günl, 2016) to improve digestion of the cellulose in seed coat mucilage. Analysis of the resulting monosaccharides by HPAEC-PAD permits sensitive and reproducible detection of both neutral and acidic sugars. This protocol could also be adapted for use in other mucilage-producing species, including Plantago and flax (Western, 2012; Yang et al., 2012; Phan and Burton, 2018).
Materials and Reagents
- 35 mm Petri dishes (Thermo Fisher Scientific, catalog number: FB0875712)
- Serological pipettes, 50 ml
- 1 L autoclavable bottles for making and storing medium
- Plastic wrap
- Micropore tape (3M, catalog number: 1530-0)
- Plant pots, 12 cm
- Forceps
- Bamboo barbeque skewers, 30 cm
- Aluminum foil
- Small sieve OR WhatmanTM 3MM Chr Chromatography Paper (GE Healthcare, catalog number: 3030917) for removing chaff from seeds
- 2 ml boil-proof microcentrifuge tubes (VWR International, catalog number: 76332-080)
- 1.5 ml microcentrifuge tubes (VWR International, catalog number: 87003-294)
- 15 ml sterile polypropylene conical centrifuge tubes (e.g., Corning Life Sciences, catalog number: 431470)
- 50 ml sterile polypropylene conical centrifuge tubes (e.g., Corning Life Sciences, catalog number: 431472)
- Plastic rack for 1.5 ml and 2 ml tubes
- Plastic Pasteur pipettes, 5.8 ml (VWR International, catalog number: 612-4494)
- Plastic box to contain tubes on orbital shaker
- Tips for micropipettes
- Chemical resistant gloves (e.g., VWR International nitrile examination gloves, catalog number: 82026-426)
- Glass microscope slides with cavity (e.g., Ted Pella, catalog number: 260241)
- Face shield
- Safety glasses
- Ice bucket
- Bubble wrap
- Small box for vortexing samples (A tip box lid is ideal)
- 12 ml 16 x 100 mm KIMAX tubes with PTFE-faced rubber lined caps (VWR International, catalog number: SCERSP45066A-16100)
- Test tube brush for KIMAX tubes
- Porcelain pestles and mortars, 50 ml capacity (e.g., Coorstek, catalog number: 60310)
- 2 mm and 1 mm zirconia (zirconium dioxide) beads
- 2 ml screw-capped tubes (e.g., Precellys 2 ml Empty Reinforced Tubes with O-ring, catalog number: 9000538)
- UV syringe filters, 4 mm diameter, nylon, 0.45 μm pore size (Chromatographic Specialties, catalog number: CSUV04N451)
- 1 ml BD tuberculin syringes (VWR International, catalog number: 309659)
- Kimwipes (Kimtech, catalog number: 34120)
- 200 μl screw top microvials (Chromatographic Specialties, catalog number: C581311)
- Open top caps for HPLC vials (Chromatographic Specialties, catalog number: C223711M)
- 8 mm, 0.060” natural PTFE/silicone septa (Chromatographic Specialties, catalog number: C243M)
- Dry ice
- Ruthenium red, for microscopy (Sigma-Aldrich, catalog number: 00541)
- Sodium carbonate, anhydrous (Thermo Fisher Scientific, catalog number: S263)
- Sodium bicarbonate (Baking soda)
- meso-Erythritol (Sigma-Aldrich, catalog number: E7500)
- L-(-)-Fucose (Sigma-Aldrich, catalog number: F2252)
- L-(+)-Arabinose (Sigma-Aldrich, catalog number: A3256)
- L-Rhamnose (Sigma-Aldrich, catalog number: R3875)
- D-(+)-Galactose (Sigma-Aldrich, catalog number: G6404)
- D-(+)-Glucose (Sigma-Aldrich, catalog number: G8270)
- D-(+)-Mannose (Sigma-Aldrich, catalog number: M2069)
- D-(+)-Xylose (Sigma-Aldrich, catalog number: X3877)
- D-(+)-Galacturonic Acid (Sigma-Aldrich, catalog number: 48280)
- Sulfuric Acid, Certified ACS Plus (Thermo Fisher Scientific, catalog number: A300-212)
- 95% ethanol
- 70% ethanol (v/v) in ultrapure water
- HPLC-grade methanol (Sigma-Aldrich, catalog number: 34885)
- 80% HPLC-grade methanol (v/v) in ultrapure water
- HPLC-grade acetone (Sigma-Aldrich, catalog number: 439126)
- Sodium hydroxide solution (50% w/w Certified NaOH, Thermo Fisher Scientific, catalog number: SS254-4)
- Anhydrous sodium acetate (CH3COONa, Thermo Fisher Scientific, catalog number: S210-500)
- Ultrapure water (18.2 MΩ cm at 25 °C)
- Agar (Thermo Fisher Scientific, catalog number: BP1423)
- 1 M KOH for adjusting pH of medium
- Sunshine Mix #5 (Sungro) or growth medium of your choice (see Notes)
- AT minimal medium, with or without agar (see Recipes)
- 20 mM sodium carbonate solutions (see Recipes)
- Ruthenium red solution (see Recipes)
- 100 mM monosaccharide stock solution (see Recipes), stored at -20 °C
- 72% (w/w) sulfuric acid (see Recipes)
- Dilution series for monosaccharide standards (see Recipes)
- meso-Erythritol solution, 5 mg/ml (see Recipes), stored at -20 °C
- 1 M NaOH (HPAEC-PAD eluent) (see Recipes)
- 200 mM NaOH (HPAEC-PAD eluent) (see Recipes)
- 1 M sodium acetate (HPAEC-PAD eluent) (see Recipes)
Equipment
- Micropipettes for volumes ranging from 5 μl to 1 ml
- 5 ml, 10 ml, 100 ml, 1.5 L and 2 L volumetric flasks
- 100 ml beakers
- 3 x 2 L vacuum side arm flasks with stoppers and vacuum tubing for degassing HPAEC eluents
- pH meter
- Magnetic stirrer and stir bars
- Autoclave
- Growth chamber with light level and temperature controls (for plates, and growth of plants to maturity)
- Analytical balance
- Microcentrifuge
- Lab balance
- Vortex mixer with 3-inch platform (Scientific Industries, Vortex-Genie 2, catalog number: SI-0236)
- Orbital shaker (New Brunswick Scientific, Innova 2000 platform shaker)
- Dissecting or compound microscope with camera (e.g., Zeiss Axioscop2 with Leica DFC450C and Leica application suite 4.2)
- Fume hood
- Reacti-Vap evaporator (Thermo Fisher Scientific, catalog number: TS-18825) with Reacti-Therm heating module (Thermo Fisher Scientific, catalog number: TS-18822)
- Nitrogen (N2) gas (Praxair, NI M-T), connected to Reacti-Vap
- Ice machine
- Access to vacuum line (e.g., integral building vacuum) for degassing HPAEC-PAD solutions
- Ion chromatography (IC): Dionex DX600 system equipped as follows:
- AS50 autosampler
- GP50 gradient pump
- ED50 electrochemical detector
- CarboPac PA1 standard bore guard (4 x 50 mm) (Thermo Fisher Scientific, catalog number: 043096)
- CarboPac PA1 analytical column (4 x 250 mm) (Thermo Fisher Scientific, catalog number: 035391)
Software
- Chromeleon 6.8 chromatography data system software (Thermo Fisher Scientific, Dionex)
- Python 2.7 (https://www.python.org/download/releases/2.7/)
- Scientific Python (SciPy) package (https://www.scipy.org/install.html)
- Notepad ++ (Don Ho, https://notepad-plus-plus.org/)
- Microsoft Excel (Microsoft)
Procedure
- Growth of seeds for HPAEC analysis
Growing seeds correctly for HPAEC analysis is critical as mucilage release is impaired if seeds do not complete development. It is very important to use standard growth conditions, grow wild type and mutants together, and water until seed development is complete as described below. In addition, see the Notes for details on preparation of biological replicates.- Sow seeds of all genotypes onto Arabidopsis AT medium with agar in Petri dishes. Seeds do not need to be sterilized as this medium does not contain sugar.
- Seal plates with micropore tape.
- Place in growth chamber 20 °C under continuous light (100-150 μmol/(s m2) photosynthetically active radiation, PAR).
- Transplant the seedlings after 7-10 days.
- Prepare one 12 cm pot of soil mix for each genotype (see Notes).
- Add 30 ml of AT medium without agar to the top of the soil.
- Use forceps to transplant 8 seedlings from each genotype into one pot of soil.
- Cover with plastic wrap to maintain humidity.
- Return to the growth chamber.
- Remove plastic wrap after 3-4 days.
- Once the plants have bolted (3-4 weeks), gather the inflorescence stems of all eight plants in each pot and tie them to a bamboo skewer stake using aluminum foil.
- Water plants until the last siliques are yellow and beginning to dry out.
- Harvest seeds, and pool seeds of the same genotype together. Be sure to remove all chaff from the seeds. This can be done using a small sieve (being careful to clean it between different seed batches) or by collecting seeds on rough paper (e.g., Whatman 3MM paper) then tilting and gently shaking the paper so that healthy seeds roll onto a second piece of paper leaving the chaff and undeveloped seeds behind. Seeds can be transferred several times between the two pieces of paper until the seeds are free of chaff.
- Preparation of Arabidopsis seed extruded mucilage. Wear chemical-resistant gloves and a lab coat
- Determine optimal mucilage release for mature seeds hydrated in aqueous solution.
- Place approximately 50 seeds per genotype (including wild type) into separate 2 ml tubes.
- Add 1 ml of ultrapure water, and vortex on maximum speed for 5 s to wet seeds.
- Place tubes in a box large enough for the tubes to roll around freely and place on an orbital shaker at 130 rpm.
- After 1 h shaking, remove the tubes, place upright in a tube rack and allow the seeds to settle for 1-2 min.
- Using a plastic Pasteur pipette, remove and discard as much water as possible without disturbing the seeds.
- Add 1 ml of 0.01% ruthenium red solution and return the tubes to the box on the shaker at 130 rpm.
- After 1 h, remove the tubes to the rack, allow the seeds to settle for 1-2 min, then remove and discard the ruthenium red as before.
Note: Do not let the seeds dry out as once they are stained, the mucilage will not rehydrate to form a halo around the seed. - Add 0.5 ml of ultrapure water and transfer the seeds to cavity slide (no need for cover slips).
- View the seeds using the compound microscope at low magnification (5x) and determine if mucilage is efficiently released using water by comparing halo size between wild-type controls and genotypes of interest [Figure 1, compare wild type (A) with mum2 (B)].
- If mucilage is not released efficiently using water, repeat the entire process using sodium carbonate solution instead of water. Many genotypes e.g., mum2, will release more mucilage in sodium carbonate than in water [Figure 1, compare mum2 in water (B) with mum2 in sodium carbonate (D). Wild type treated with sodium carbonate (C) is included for comparison]. It is important to determine the minimum concentration of sodium carbonate that will allow efficient mucilage release, as sodium ions interfere with HPAEC-PAD detection of monosaccharides. Mucilage is released from mum2 using 20 mM sodium carbonate. It may also be useful to compare the composition of mucilage released using water with mucilage released with sodium carbonate.
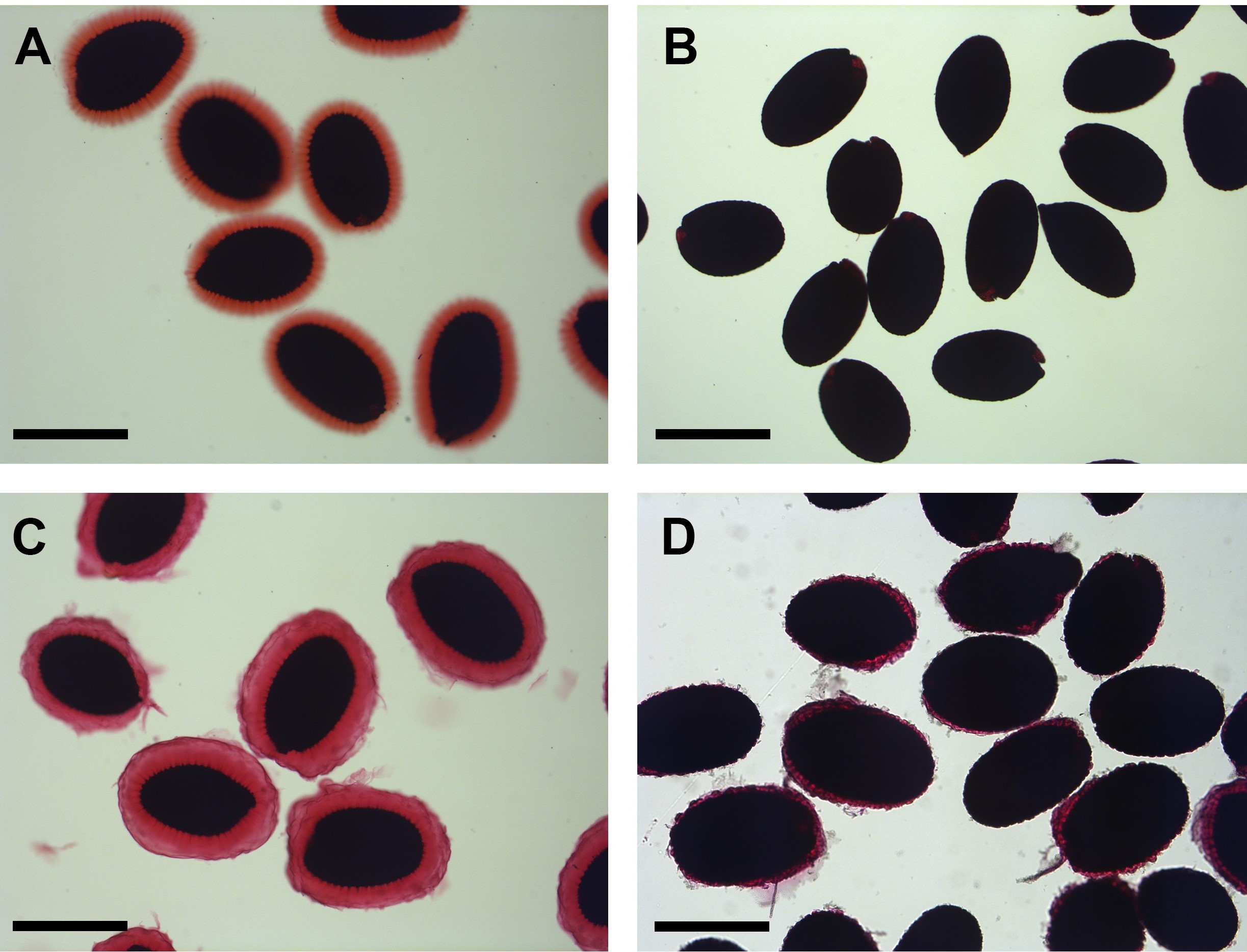
Figure 1. Mucilage release after treatment with water in wild type (A) and mum2 (B), and after treatment with 20 mM sodium carbonate in wild type (C) and mum2 (D). Scale bars = 500 μm.
Note: Non-adherent mucilage has been removed during the staining procedure. - Once optimal release of mucilage has been determined, proceed to either Step B2. (Preparation of extruded mucilage in water) or B3 (preparation of extruded mucilage in sodium carbonate) below.
- Preparation of extruded mucilage in water. Use seeds grown as described in A, and record the batch.
- Prepare 4 replicates for each genotype by weighing out approximately 20 mg of seed per replicate. Record the exact weight of each replicate.
- Place each replicate into a 2 ml tube and add 1.4 ml of ultrapure water. Make sure all seeds are suspended in the solution rather than floating on the surface by vortexing for 5 s.
- Add 5 μl of 5 mg/ml meso-Erythritol internal standard.
- Prepare the samples for vortexing by taping a plastic box onto a vortex mixer 3-inch platform and fill it with tubes laid on their sides, with all tubes in the same orientation. Fill the box with bubble wrap and tape it in place securely to ensure the tubes do not move. See Notes for further details on mucilage removal.
- Vortex for 2 h on the “vortex #3” setting of the vortex mixer.
- While the samples are vortexing, prepare the standards (see Notes for important technical considerations). New standards must be prepared, processed, and run with each set of samples.
- Dilute the 100 mM monosaccharide stock to 2 mM and then make a serial dilution to give 1 mM, 0.5 mM, 0.25 mM, 0.125 mM, 0.0625 mM solutions, to give a total of 6 sugar standards (see Recipes).
- Transfer 0.5 ml of each sugar standard to a KIMAX tube.
- Add 5 μl 5 mg/ml meso-Erythritol to each standard.
- Remove the samples from the vortex mixer, place in a tube rack and allow seeds to settle for 1-2 min. Do not centrifuge as mucilage will pellet.
- Remove 1 ml of the mucilage-containing supernatant to a KIMAX tube. See Notes for proper cleaning of tubes.
- After mucilage removal, stain the seeds with ruthenium red, as described in Steps B1e-B1i, and photograph to record how much mucilage was removed.
- Using the Reacti-Vap, dry the samples and the standards (in batches) under a stream of N2 gas at 60 °C. Once dry, the tubes can be capped and stored at -20 °C if required.
- In the fume hood, wearing safety glasses, add 17.4 μl 72% sulfuric acid to each sample and standard, and cap the tubes.
- Incubate samples and standards on ice for 2 h, mixing every 30 min by vortexing on maximum for 5-10 s.
- In the fume hood, add 482.6 μl of ultrapure water to give a final concentration of 4% sulfuric acid.
- Tighten the caps and autoclave at 121 °C (15 psi/1 Bar/1 ATM) for 1 h.
- Remove from autoclave and allow to cool.
- In the fume hood, transfer samples into clean 1.5 ml tubes, and centrifuge at 16,000 x g for 1 min to pellet debris.
- Filter the samples and standards into clean 1.5 ml tubes using 1 ml syringes and 0.45 μm nylon syringe filters. Wrap the junction between the syringe and the filter with a Kimwipe in case it “pops”.
Note: If the sample is very difficult to filter then the filter is blocked and should be replaced. - Load 200 μl of each sample into HPLC vials with caps and septa. Samples and standards can be stored at -20 °C in HPLC vials or 1.5 ml tubes to prevent growth of microbes.
- Preparation of extruded mucilage in sodium carbonate. The protocol described here is for extrusion of mucilage in 20 mM sodium carbonate. If a different concentration is used, the amount of 72% sulfuric added should be to be adjusted, as some carbonate will react with sulfuric acid and alter the final concentration. See Notes for more details. Use seeds grown as described in A, and record the batch.
- Prepare 4 replicates for each genotype by weighing out approximately 20 mg of seed per replicate. Record the exact weight of each replicate.
- Place each replicate into a 2 ml tube and add 1.4 ml 20 mM sodium carbonate. Make sure all seeds are suspended in the solution rather than floating on the surface by vortexing for 5 s.
- Add 5 μl of 5 mg/ml meso-Erythritol internal standard.
- Prepare the samples for vortexing by taping a plastic box onto a vortex mixer 3-inch platform and fill it with tubes laid on their sides, with all tubes in the same orientation. Fill the box with bubble wrap and tape it in place securely to ensure the tubes do not move. See Notes for further details on mucilage removal.
- Vortex for 2 h on the “vortex #3” setting of the vortex mixer
- While the samples are vortexing, prepare the standards (see Notes for important technical considerations). New standards must be prepared, processed, and run with each set of samples.
- Dilute the 100 mM monosaccharide stock to 2 mM and then make a serial dilution to give 1 mM, 0.5 mM, 0.25 mM, 0.125 mM, 0.0625 mM solutions, to give a total of 6 sugar standards (see Recipes).
- Transfer 0.5 ml of each sugar standard to a KIMAX tube.
- Add 5 μl 5 mg/ml meso-Erythritol and 1 ml 20 mM sodium carbonate. If a different concentration of sodium carbonate is used for mucilage extraction, the amount added here should be adjusted to be the same as in the 1 ml of sample removed in Step B3h below.
- Remove the samples from the vortex mixer, place in a tube rack and allow seeds to settle for 1-2 min. Do not centrifuge as mucilage will pellet.
- Remove 1 ml of the mucilage-containing supernatant to a KIMAX tube. See Notes for proper cleaning of tubes.
- After mucilage removal, stain the seeds with ruthenium red, as described in Steps B1e-B1i to record how much mucilage was removed.
- Using the Reacti-Vap, dry the samples and the standards (in batches) under a stream of N2 gas at 60 °C. Once dry, the tubes can be capped and stored at -20 °C if required.
- In the fume hood, wearing safety glasses, add 19.07 μl of 72% sulfuric acid to each sample and standard, and cap the tubes.
- Incubate samples and standards on ice for 2 h, mixing every 30 min by vortexing on maximum for 5-10 s.
- In the fume hood, add 480.9 μl of ultrapure water to give a final concentration of 4% sulfuric acid.
- Tighten the caps and autoclave at 121 °C (15 psi/1 Bar/1 ATM) for 1 h.
- Remove from autoclave and allow to cool.
- In the fume hood, transfer samples into clean 1.5 ml tubes, and centrifuge at 16,000 x g for 1 min to pellet debris.
- Filter the samples and standards into clean 1.5 ml tubes using 1 ml syringes and 0.45 μm nylon syringe filters. Wrap the junction between the syringe and the filter with a Kimwipe in case it “pops”.
Note: If the sample is very difficult to filter then the filter is blocked and should be replaced. - Load 200 μl of each sample into HPLC vials with caps and septa. Samples and standards can be stored at -20 °C in HPLC vials or 1.5 ml tubes to prevent growth of microbes.
- Determine optimal mucilage release for mature seeds hydrated in aqueous solution.
- Preparation of Alcohol Insoluble Residues (AIR) from mature whole seeds. Use seeds grown as described in A, and record the batch
- Prepare 4 replicates for each genotype by weighing out approximately 20 mg of seed per replicate. Record the exact weight of each replicate.
- Homogenize seeds with one of the following techniques:
- Place each replicate into a 1.5 ml tube and freeze the seeds in 1.5 ml tubes in dry ice. Cool pestles and mortars with dry ice, and grind the frozen seeds to a fine powder, then transfer the frozen powder back to a 1.5 ml tube and store on dry ice. This procedure gives very good results but is time-consuming.
- Place each replicate into a 2 ml screw-capped tube with three to five 2 mm zirconia beads, freeze on dry ice and disrupt in a tissue homogenizer at 6,500 rpm for 20 s. It is important to choose tubes that will not break in the tissue homogenizer.
- Add 1 ml 70% (v/v) ethanol to the tubes containing the frozen, ground seeds, and resuspend the tissue fully by vortexing. If grinding was done with a pestle and mortar, add three to five 2 mm zirconia beads to the tube to help to resuspend the tissue.
- Incubate at 65 °C (use the heat block in the Reacti-Vap) for 10 min.
- Vortex the samples in a box attached to the vortex mixer (as described in Step B2d) for 10-15 min to solubilize metabolic sugars.
- Centrifuge at 16,000 x g for 15 s. Do not spin for longer or the pellet will become hard to resuspend.
- Remove the supernatant, and replace with 1 ml fresh 70% (v/v) EtOH in ultrapure water.
- Vortex for 10-15 min and centrifuge again, as described above.
- Remove the supernatant, and replace with 1 ml 80% (v/v) HPLC-grade MeOH in ultrapure water.
- Vortex for 10-15 min and centrifuge again, as described above.
- Remove the supernatant, and replace with 1 ml 100% HPLC-grade MeOH.
- Vortex for 10-15 min and centrifuge again, as described above.
- Remove the supernatant, and replace with 1 ml fresh 100% HPLC-grade MeOH.
- Vortex for 10-15 min and centrifuge again, as described above.
- Remove the supernatant, and replace with 1 ml 100% HPLC-grade acetone.
- Vortex for 10-15 min and centrifuge again, as described above.
- Remove the supernatant, and replace with 1 ml fresh 100% HPLC-grade acetone.
- Vortex for 10-15 min and centrifuge again, as described above. Remove most of the acetone, leaving a small amount which can be used to resuspend the pellet by vortexing briefly.
- Using a micropipette, transfer the resuspended pellet (in acetone) to a clean 2 ml tube, leaving the beads behind.
- Dry the AIR by leaving the tubes open in the fume hood. Acetone will evaporate quickly, leaving a light brown AIR pellet.
- Weigh the resulting AIR (there should be over 3 mg of AIR) and record the exact weight, and then transfer the AIR to a KIMAX tube.
- Add 20-30 1 mm zirconia beads to each tube, cap the tubes, and vortex briefly to break the pellet into powder. The beads will also help with mixing samples in sulfuric acid.
- Add 500 μl of 100% HPLC-grade acetone and 20 μl of 5 mg/ml meso-Erythritol to each sample and dry under a stream of N2 gas at 60 °C using the Reacti-Vap. Acetone helps to evaporate the water, leaving a less clumped pellet than evaporating 20 μl of water alone.
- Prepare the standards (see Notes for important technical considerations). New standards must be prepared, processed, and run with each set of samples.
- Dilute the 100 mM monosaccharide stock to 2 mM and then make a serial dilution to give 1 mM, 0.5 mM, 0.25 mM, 0.125 mM, 0.0625 mM solutions, to give a total of 6 sugar standards (see Recipes).
- Transfer 0.5 ml of each sugar standard to a KIMAX tube.
- Add 5 μl of 5 mg/ml meso-Erythritol to each tube.
- Using the Reacti-Vap, dry standards under a stream of N2 gas at 60 °C using the Reacti-Vap.
- Once dry, the tubes containing standards and samples can be capped and stored at -20 °C if required.
- In the fume hood, wearing safety glasses, add 70 μl of 72% sulfuric acid to each sample, and 17.4 μl of 72% sulfuric acid to each standard, and cap the tubes.
- Incubate samples and standards on ice for 2 h, mixing every 30 min by vortexing for 5-10 s.
- In the fume hood, add 1.93 ml of ultrapure water to the samples, and 482.6 μl to the standards, to give a final concentration of 4% sulfuric acid.
- Tighten the caps and autoclave at 121 °C (15 psi/1 Bar/1 ATM) for 1 h.
- Remove from autoclave and allow to cool.
- In the fume hood, transfer samples into clean 1.5 ml tubes, and centrifuge at 16,000 x g for 1 min to pellet debris.
- Filter the samples and standards into clean 1.5 ml tubes using 1 ml syringes and 0.45 μm nylon syringe filters. Wrap the junction between the syringe and the filter with a Kimwipe in case it “pops”.
Note: If the sample is very difficult to filter then the filter is blocked and should be replaced. - Load 200 μl of each sample into HPLC vials with caps and septa. Samples and standards can be stored at -20 °C in HPLC vials or 1.5 ml tubes to prevent growth of microbes.
- Separation and quantification of monosaccharides using high performance anion-exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD) using a CarboPac PA1 Guard and Analytical column at 25 °C
- Separation of acid sugars.
- Use a constant eluant flow rate of 0.4 ml/min.
- Equilibrate the column with 10% 1 M NaOH, 1% 1 M sodium acetate, and 89% ultrapure water for 10 min.
- Independently inject 10 μl of each sample and standard.
- Separate acid sugars using a 30-minute linear gradient reaching 40% 1 M sodium acetate, and keeping the 1 M NaOH constant (Figure 2).
- Clean the column with a linear gradient of 1 M NaOH that reaches 30% over 10 min, while decreasing 1 M sodium acetate to 1% (Figure 2).
- Re-equilibrate the column for 10 min with the eluents at their starting concentrations for 10 min (10% 1 M NaOH, 1% 1 M sodium acetate, and 89% ultrapure water) before injection of the next sample (Figure 2).
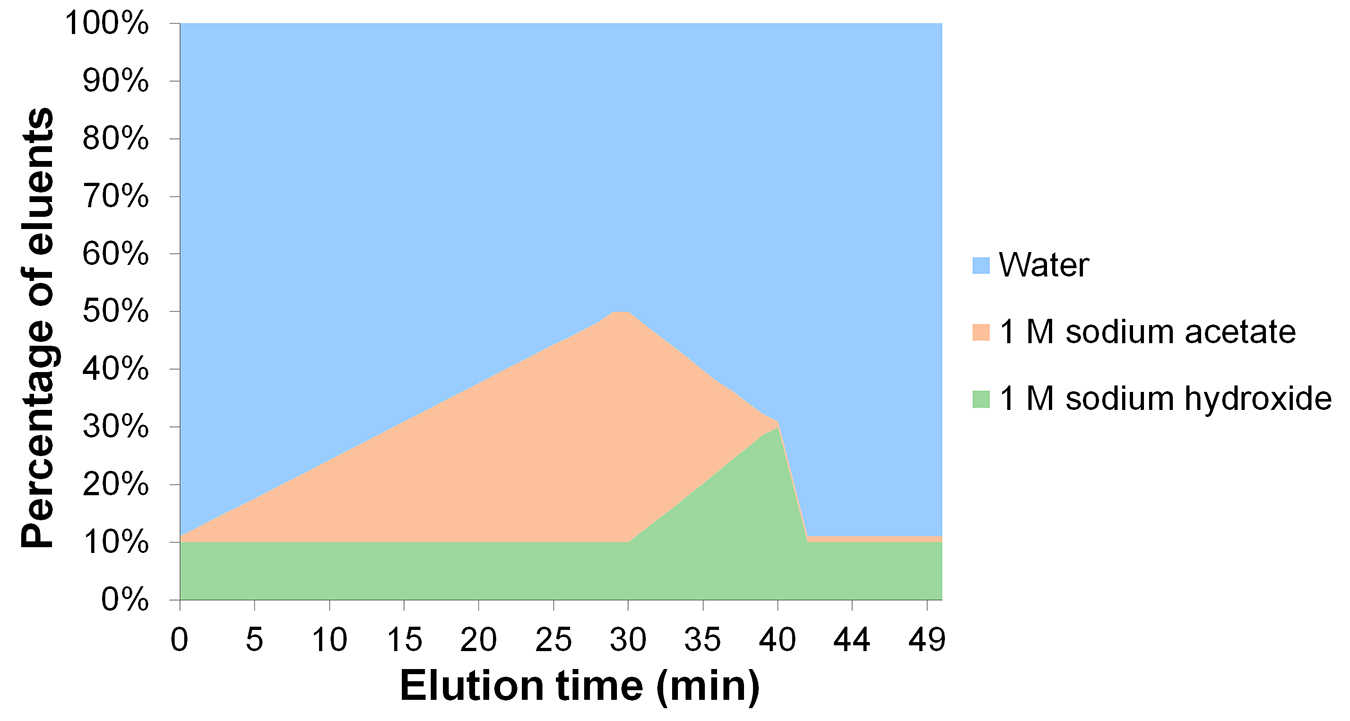
Figure 2. Eluent gradient for the separation of acid sugars
- Separation of neutral sugars.
- Use a constant eluant flow of 1 ml/min.
- Equilibrate the column with 100% ultrapure water for 10 min, and start the post column pump which supplies 200 mM NaOH to the detector for post-column separation of monomeric neutral sugars.
- Inject 10 μl of each sample.
- Elute the neutral sugars with 100% ultrapure water for 35 min with post column detection.
- Clean the column for 10 min with 20% 1 M NaOH.
- Re-equilibrate the column with a 10-min rinse of 100% ultrapure water before next injection.
- Peak integration and data export from Chromeleon.
- View the files produced in Chromeleon (see examples in Figures 3 and 4).
- Manually integrate chromatographs, keeping the limits of the peaks in the samples at similar retention times as the limits of the peaks in the standards. Baselines for the meso-Erythritol internal standard and peak of interest (GalA) should have a flat baseline; adjust baseline with the vertical/horizontal tool from the Chromeleon tool ribbon. Save changes to peak integrations.
- Export values for area under the curve from the integration tab in the Chromeleon software to Excel.
- Before running further analysis, make sure that the standards show linear response, i.e., for any given monosaccharide, the peak area for the 1 mM standard should be exactly half of the 2 mM standard, and so on. This can be tested by plotting peak area vs. concentration of the standards in Excel and performing linear regression. The intercept should be close to 0 and R2 close to 1 to ensure accurate quantification.
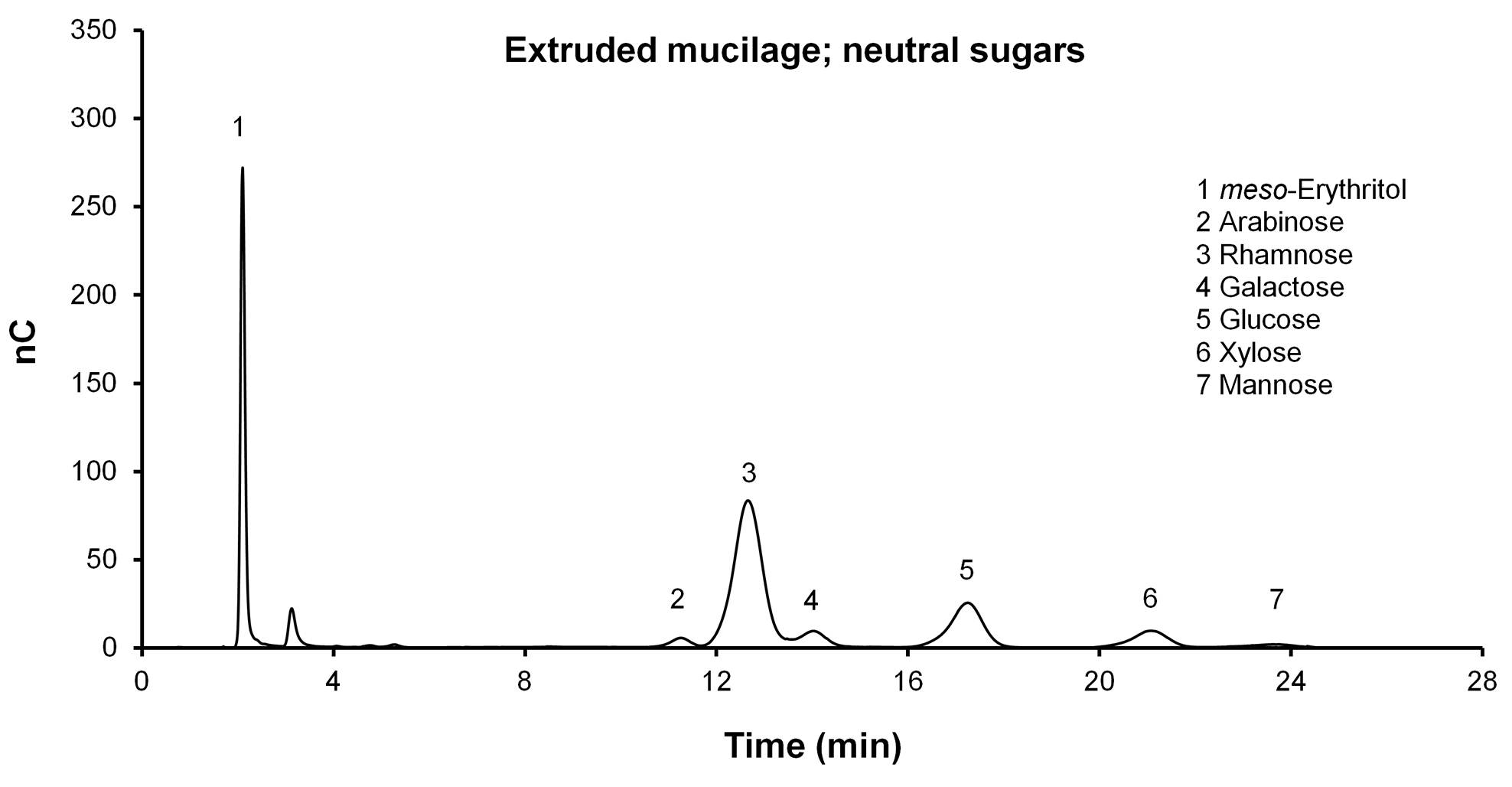
Figure 3. Separation of neutral sugars (Ara, Rha, Gal, Glu, Xyl, Man) with the CarboPac PA1 analytical column. Fuc is not detectable in this sample but has a retention time of approximately 5.5 min.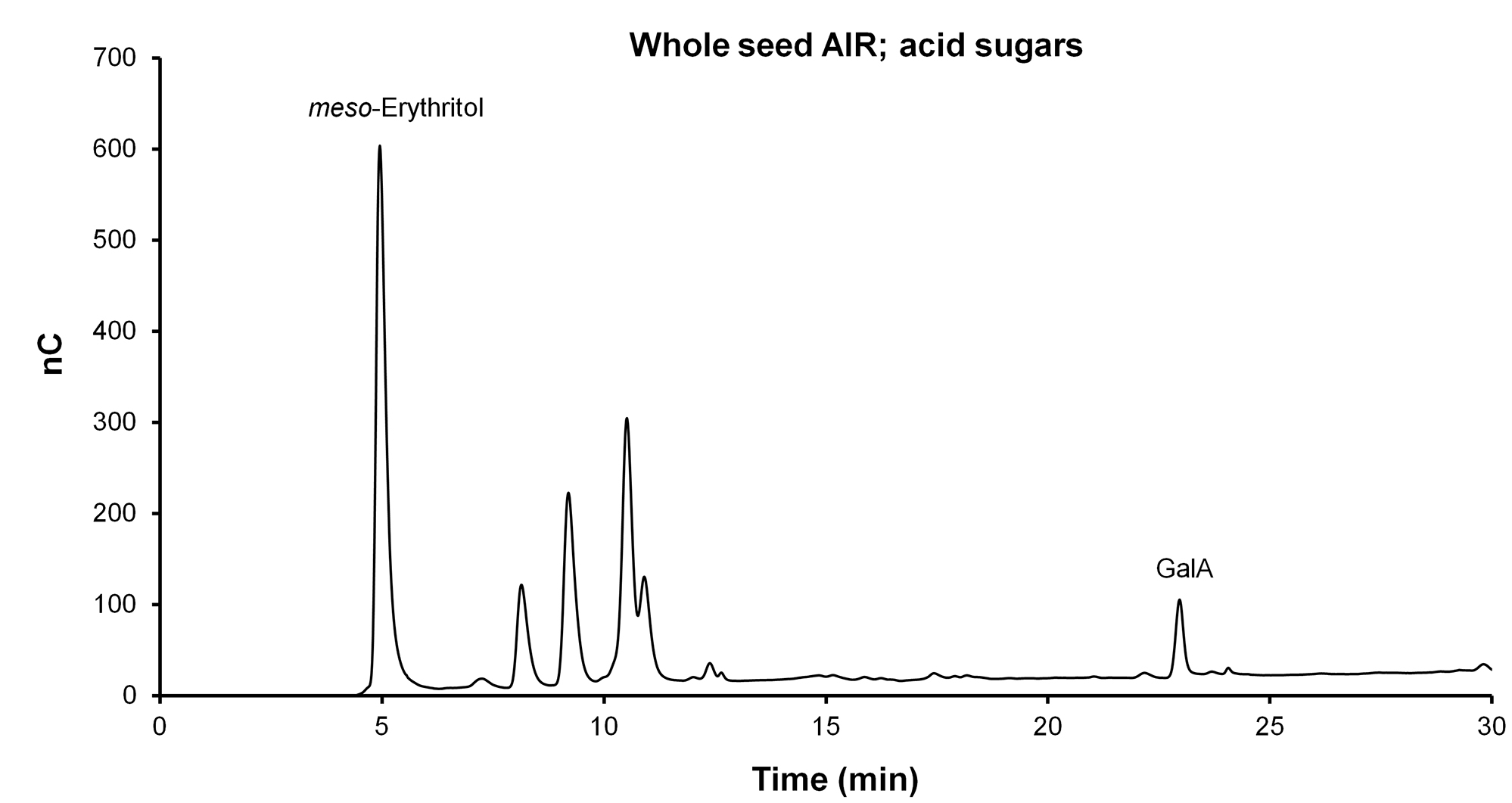
Figure 4. Separation of acid sugars (GalA) with the CarboPac PA1 analytical column
- Separation of acid sugars.
Data analysis
- Install Python and SciPy, if not already installed on your workstation e.g., as follows:
- Install Python 2.7.16 (https://www.python.org/downloads/release/python-2716/) and make a note of the directory where Python is installed.
- Open the Command Prompt and type in “cd” (without the quotation marks).
- Open File Explorer and navigate to the Python27 folder.
- Drag and drop the Python27 folder into the Command Prompt and press Enter. The Command Prompt should look as shown in Figure 5.
Figure 5. Command prompt after setting up Python27 - Go to scipy.org/install.html, and copy the code highlighted in dark grey shown in Figure 6:
Figure 6. Code to install SciPy - Paste the code into the Command Prompt and press Enter.
- The SciPy package will be installed.
- Customize the MonosaccharideComposition.py Python code provided as part of this protocol. The code must be changed between mucilage and AIR data processing, as the final sample volumes are different
- Use Notepad++ to open the “MonosaccharideComposition.py” Python file (see Supplemental 1).
- Edit the final sample volumes. Mucilage samples are prepared in a final volume of 0.5 ml, and AIR samples are prepared in a final volume of 2 ml. To adjust different final volumes that are employed during sample preparation, change line 162 of the code: “finalCor = finalCorrections (calibrated, corrected, 6, 0.5)”, where the last number, shown here in red and bold, represents volume in ml.
- Edit the number of standards used. Go to line number 161 and 162 and find the code that reads “calibrated = calibration (corrected, 6)” and “finalCor = finalCorrections (calibrated, corrected, 6, 0.5)”. Replace the number 6 (Colored red) in the code with the number of standards used, if different.
- Save the customized Python file.
- Prepare HPAEC data for analysis
- Paste integrated peak areas obtained from Chromeleon into the template Excel spreadsheet. Prepare separate spreadsheets for mucilage and whole seed AIR samples as the final sample volumes are different.
- Acid and neutral monosaccharides are analyzed separately, and therefore need to be processed as separate files; the template Excel spreadsheet has separate sheets for inputting acid and neutral sugar data (see Supplemental 2).
- Make sure to use monosaccharide abbreviations: Ara for arabinose, Xyl for xylose, Fuc for fucose, Rha for rhamnose, GalA for galacturonic acid, Gal for galactose, Man for mannose, and Glc for glucose, as they will be used to determine correction coefficients for hydrolysis.
- Enter the mass, in mg, of either seeds used for mucilage extraction, or of AIR used for whole seeds.
- Do not include spaces in sample and standard names.
- Add “mM” after standard concentrations. This is required so that standards can be distinguished from samples during processing.
- For each genotype, mark replicates by adding “#” followed by replicate number after the genotype name. This must be included so that replicates are grouped together.
- Copy the data from Excel, paste it into Notepad++, and save as a .txt file. Make sure there is no extra row at the end of the .txt file underneath the data or it will not run properly.
- Paste integrated peak areas obtained from Chromeleon into the template Excel spreadsheet. Prepare separate spreadsheets for mucilage and whole seed AIR samples as the final sample volumes are different.
- Run the customized Monosaccharide Composition.py Python code saved in B
- Drag and drop the Monosaccharide Composition.py file into the Command Prompt, and press Enter.
- A popup window will open: navigate to and select the .txt file of integrated peak areas.
- A popup window will open: choose a file name and a location for the processed data, and press Enter. The processed data will be saved.
- Open the processed tab-delimited data file with Excel. The data for each sample and sugar can be cut and pasted into Excel for processing. Calculate average nmol/mg and standard deviation for each sugar for each genotype, and plot graphs.
Notes
- Arabidopsis prefers a light peaty soil and previously we have used 1:1:1 peat: perlite: vermiculite fertilized with AT medium, but have found that prepared peat-rich medium such as Sunshine Mix #5 (Sungro) works well and is more convenient. As Sunshine Mix #5 does not contain fertilizer, we water once before planting with liquid AT minimal medium (see Recipes) as described in Procedure A. We recommend that plants be grown according to your usual procedure. It is critical to water plants until the last siliques are turning yellow as described in Procedure A. It is also very important to grow several independent sets of seeds, including wild type, at different times to capture any variation caused by slight differences in growth conditions. We recommend growing 3 independent replicates of seeds as described in Procedure A, and performing monosaccharide analysis independently on each one. Please note that within one HPAEC experiment (as outlined in Procedure B), the 4 replicates prepared are biological and not technical replicates. In our hands, there is very little variation among replicates from the same seed batch that are prepared and analyzed together.
- Mucilage removal from seeds can be increased by splitting the 20 mg of seeds used for extraction into 4 x 5 mg aliquots (exact weights recorded), and extracting each 5 mg aliquot with 0.35 ml water/sodium carbonate and 1.25 μl of 5 mg/ml meso-Erythritol. After vortexing as described, recombine the 4 aliquots, then take 1 ml and continue with the standard protocol.
- Proper cleaning of the KIMAX tubes used for acid hydrolysis is important to prevent contamination. Wash the tubes with HPLC-grade methanol to remove any soluble sugars and dry in the fume hood. After sample processing, first prepare a solution of sodium bicarbonate (baking soda) in water. Wearing safety glasses, lab coat, and chemical resistant gloves, fill the KIMAX tubes containing debris and residual sulfuric acid with the baking soda solution. Leave to soak for at least 2 h, then scrub the debris from the tubes using a test tube brush and wash thoroughly with tap water. Finally, either wash in a dishwasher or rinse thoroughly with ultrapure water before air drying.
- Careful preparation of standard curves to convert peak area to amount of monosaccharide is essential, as is accurate addition of the meso-Erythritol internal standard. Test micropipette accuracy using an analytic balance (1 ml of ultrapure water weighs 1 g). The sample values detected by the PAD detector need to be within the calibration range of the external standards. If the samples have higher reads than the highest concentration standard (2 mM), repeat the procedure with lower amounts of tissue or higher standard concentrations.
- If a different concentration of sodium carbonate is used for mucilage extrusion, then the amount of 72% sulfuric acid added has to be adjusted as the sodium carbonate will react with the acid, decreasing the concentration. The reaction between sodium carbonate and sulfuric acid is as follows:
H2SO4 + Na2CO3 → Na2SO4 + CO2 + H2O
1 mole of sulfuric acid reacts with 1 mole of sodium carbonate. If 20 mM sodium carbonate is used, then 0.02 mmoles of sodium carbonate is present in the 1 ml mucilage sample, and 0.02 mmoles of sulfuric acid is required for neutralization. The molarity of 72% sulfuric acid is 12 M (specific gravity 1.622 g/cm3), hence 1.67 μl of 72% sulfuric acid is required for neutralization. Therefore, the total volume of sulfuric acid required is 1.67 μl + 17.4 μl = 19.07 μl.
Recipes
- AT minimal medium
We use an AT minimal medium which was developed in our lab (Haughn and Somerville, 1986), but the Arabidopsis growth medium of your choice may be substituted. For AT minimal medium, begin by making the stock solutions and autoclaving them in 1 L bottles (see Table 1). To prepare the medium, add the correct amount of each nutrient stock solution to 800 ml of ultrapure water (see Table 1). Add stocks to water to prevent precipitation of insoluble calcium phosphate complexes. Make volume up to 1 L.
Table 1. AT minimal medium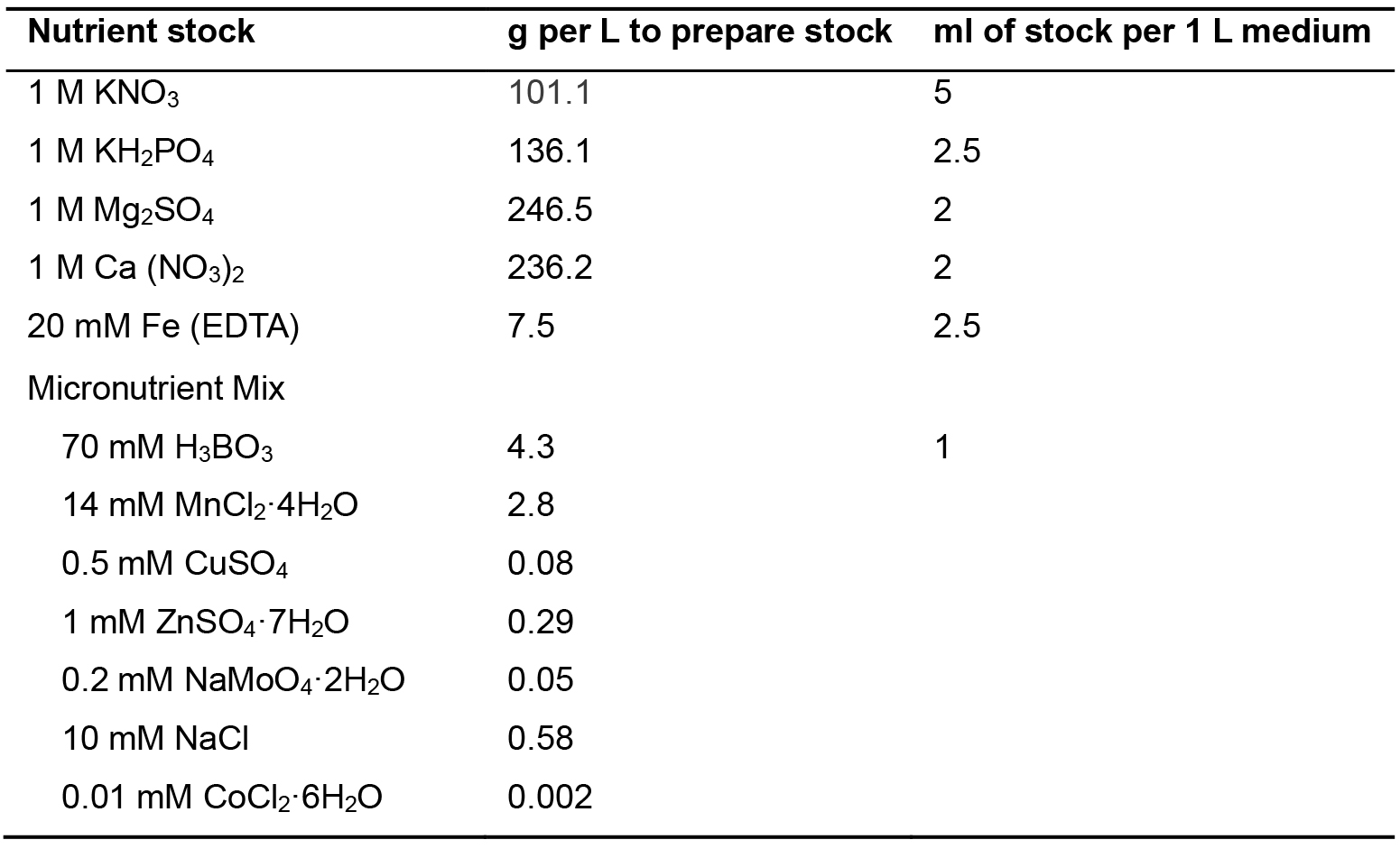
Note: We water our soil mix with AT minimal medium without agar, but this may not be necessary depending on your choice of soil mix (see Notes). AT minimal medium that will be used for watering does not need to be autoclaved, but should be stored in dark to prevent growth of algae.
For plates, adjust the pH of the medium to 5.8 with KOH, add 7 g/L of agar and autoclave. After autoclaving, medium can be stored in bottles at room temperature, and plates stored wrapped in plastic wrap at 4 °C. - Ruthenium red solution
Stock solution of 0.1% (w/v) ruthenium red
Dissolving 50 mg of ruthenium red into 50 ml of ultrapure water
Store in a 50 ml conical tube at 4 °C
Working solution of 0.01% (w/v) ruthenium red
Dilute 5 ml into 50 ml of ultrapure water in a 50 ml conical tube and store at 4 °C
Note: These solutions are good as long as the color is not purple or brown. Once discolored, they should be discarded and fresh solutions prepared. - Sodium carbonate solution(20 mM)
- Dissolve 212 mg of anhydrous sodium carbonate in 80 ml ultrapure water
- Make up to 100 ml with ultrapure water using a volumetric flask
- Store at room temperature
- Sulfuric acid, 72% (w/w), specific gravity 1.634 g/cm3
Note: Prepare in the fume hood with face shield and chemical-resistant gloves, while wearing a lab coat.
Slowly add 665 ml of concentrated sulfuric acid (specific gravity 1.84 g/cm3, 96% solution) to 333 ml ultrapure water while mixing slowly and continuously
Formula for calculations:
Volume1 × Specific Gravity1 × % Solution1=Volume2 × Specific Gravity2 × % Solution2 - Monosaccharide stock solution, 100 mM
- To prepare a 100 mM monosaccharide stock solution containing 100 mM each of L-(-)-Fucose, L-(+)-Arabinose, L-Rhamnose, D-Galactose, D-Glucose, D-(+)-Mannose, D-(+)-Xylose, D-(+)-Galacturonic acid, weigh the appropriate amount of each sugar in Table 2 below
- Dissolve in ultrapure water and make up to 5 ml in a volumetric flask
- Store as 1 ml aliquots in sterile 1.5 ml tubes at -20 °C
Table 2. Preparation of monosaccharide stock solution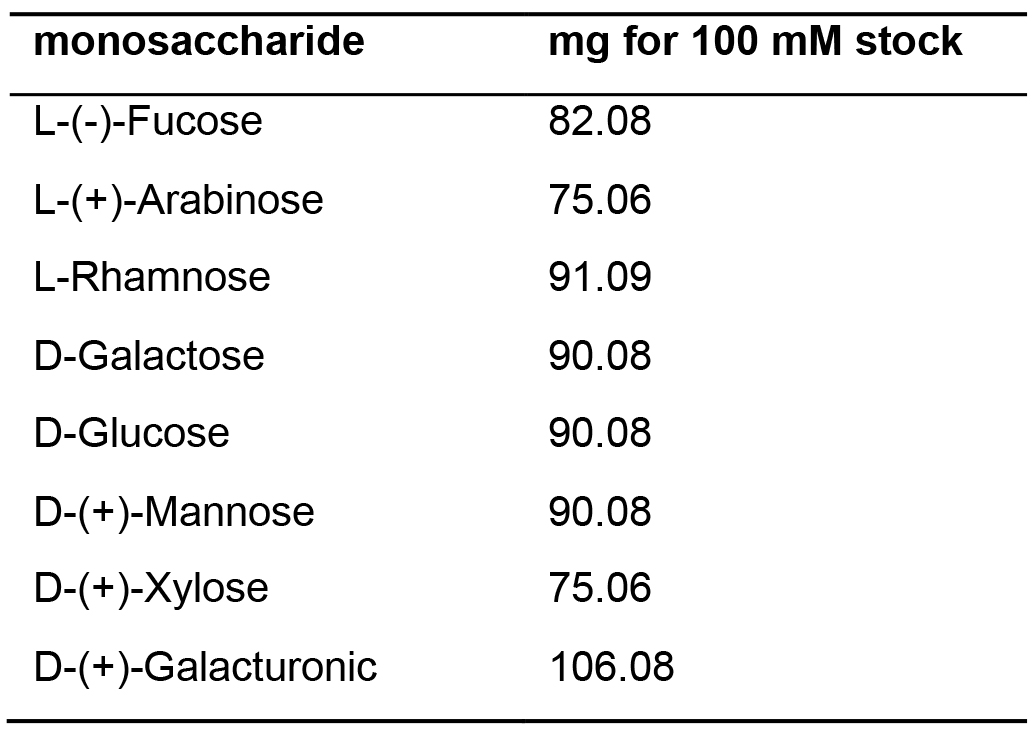
- Dilution series for monosaccharide standards
Prepare a dilution series as shown in Table 3. Use ultrapure water for the dilutions.
Table 3. Preparation of monosaccharide standard dilution series
- meso-Erythritol solution, 5 mg/ml
- To prepare a 5 mg/ml stock solution, weigh out 50 mg of meso-Erythritol. Dissolve in ultrapure water and make up to 10 ml in a volumetric flask
- Transfer to a sterile 15 ml conical tube
- Store at -20 °C
- 1 M NaOH (HPAEC-PAD eluent)
- Weigh out 120 g of 50% (w/w) NaOH into a clean beaker using a plastic 50 ml serological pipette and transfer into a 1.5 L volumetric flask
- Using ultrapure water, rinse the beaker into the volumetric flask and make up to volume
- Transfer to a vacuum flask with a magnetic stir bar and degas by vacuum for at least 2 h
- 200 mM NaOH (HPAEC-PAD eluent)
- Weigh out 32 g of 50% (w/w) NaOH into a clean beaker using a plastic 50 ml serological pipette and transfer into a 2 L volumetric flask
- Using ultrapure water, rinse the beaker into the volumetric flask and make up to volume
- Transfer to a vacuum flask with a magnetic stir bar and degas by vacuum for at least 2 h
- 1 M sodium acetate (HPAEC-PAD eluent)
- Weigh out 82.03 g of anhydrous sodium acetate
- Dissolve in ultrapure water and make up to 1 L in a volumetric flask
- Transfer to a vacuum flask with a magnetic stir bar and degas by vacuum for at least 2 h
Acknowledgments
This protocol was derived from the original protocol in Dean et al. (2007) with minor modifications in Šola et al. (2019b). This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants to G.W.H. and S.D.M.
Competing interests
The authors have no financial or non-financial competing interests.
References
- Arsovski, A. A., Haughn, G. W. and Western, T. L. (2010). Seed coat mucilage cells of Arabidopsis thaliana as a model for plant cell wall research. Plant Signal Behav 5(7): 796-801.
- Beeckman, T., Rycke, R. De., Viane, R. and Inzé, D. (2000). Histological study of seed coat development in Arabidopsis thaliana. J Plant Res 113: 139-148.
- Coleman, H. D., Yan, J. and Mansfield, S. D. (2009). Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc Nati Acad Sci U S A 106(31): 13118-13123.
- Dean, G. H., Zheng, H., Tewari, J., Huang, J., Young, D. S., Hwang, Y. T., Western, T. L., Carpita, N. C., McCann, M. C., Mansfield, S. D. and Haughn, G. W. (2007). The Arabidopsis MUM2 gene encodes a β-galactosidase required for the production of seed coat mucilage with correct hydration properties. Plant Cell 19(12): 4007-4021.
- Francoz, E., Lepiniec, L. and North, H. M. (2018). Seed coats as an alternative molecular factory: thinking outside the box. Plant Reprod 31: 327-342.
- Francoz, E., Ranocha, P., Burlat, V. and Dunand, C. (2015). Arabidopsis seed mucilage secretory cells: Regulation and dynamics. Trends Plant Sci 20: 515-524.
- Golz, J. F., Allen, P. J., Li, S. F., Parish, R. W., Jayawardana, N. U., Bacic, A. and Doblin, M. S. (2018). Insights into the role of transcription factors controlling mucilage production in the Arabidopsis seed coat. Plant Sci 272: 179-192.
- Griffiths, J. S. and North, H. M. (2017). Sticking to cellulose: exploiting Arabidopsis seed coat mucilage to understand cellulose biosynthesis and cell wall polysaccharide interactions. New Phytol 214(3): 959-966.
- Griffiths, J. S., Tsai, A. Y., Xue, H., Voiniciuc, C., Šola, K., Seifert, G. J., Mansfield, S. D. and Haughn, G. W. (2014). SALT-OVERLY SENSITIVE5 mediates Arabidopsis seed coat mucilage adherence and organization through pectins. Plant Physiol 165(3): 991-1004.
- Haughn, G. W. and Somerville, C. (1986). Sulfonylurea-resistant mutants of Arabidopsis thaliana. Mol Gen Genet 204: 430-434.
- Haughn, G. W. and Western, T. L. (2012). Arabidopsis seed coat mucilage is a specialized cell wall that can be used as a model for genetic analysis of plant cell wall structure and function. Front Plant Sci 3: 64.
- Huang, J., DeBowles, D., Esfandiari, E., Dean, G., Carpita, N. C. and Haughn, G. W. (2011). The Arabidopsis transcription factor LUH/MUM1 is required for extrusion of seed coat mucilage. Plant Physiol 156(2): 491-502.
- Macquet, A., Ralet, M. C., Loudet, O., Kronenberger, J., Mouille, G., Marion-Poll, A. and North, H. M. (2007). A naturally occurring mutation in an Arabidopsis accession affects a β-D-galactosidase that increases the hydrophilic potential of rhamnogalacturonan I in seed mucilage. Plant Cell 19(12): 3990-4006.
- Mendu, V., Griffiths, J. S., Persson, S., Stork, J., Downie, A. B., Voiniciuc, C., Haughn, G. W. and DeBolt, S. (2011). Subfunctionalization of cellulose synthases in seed coat epidermal cells mediates secondary radial wall synthesis and mucilage attachment. Plant Physiol 157(1): 441-453.
- North, H. M., Berger, A., Saez-Aguayo, S., and Ralet, M. C. (2014). Understanding polysaccharide production and properties using seed coat mutants: future perspectives for the exploitation of natural variants. Ann Bot 114(6): 1251-1263.
- Phan, J. L. and Burton, R. A. (2018). New insights into the composition and structure of seed mucilage. Annual Plant Reviews 1: 1-41.
- Shi, L., Dean, G. H., Zheng, H., Meents, M. J., Haslam, T. M., Haughn, G. W. and Kunst, L. (2019). ECERIFERUM11/C-TERMINAL DOMAIN PHOSPHATASE-LIKE2 Affects secretory trafficking. Plant physiol 181: 901-915.
- Šola, K., Dean, G. H. and Haughn, G. W. (2019a). Arabidopsis seed mucilage, a specialized extracellular matrix that demonstrates the structure-function versatility of cell wall polysaccharides. Annual Plant Reviews 2(4).
- Šola, K., Gilchrist, E. J., Ropartz, D., Wang, L., Feussner, I., Mansfield, S. D., Ralet, M. C. and Haughn, G. W. (2019b). RUBY, a putative galactose oxidase, influences pectin properties and promotes cell-to-cell adhesion in the seed coat epidermis of Arabidopsis thaliana. Plant Cell 31(4): 809-831.
- Voiniciuc, C., Dean, G. H., Griffiths, J. S., Kirchsteiger, K., Hwang, Y. T., Gillett, A., Dow, G., Western, T. L., Estelle, M. and Haughn, G. W. (2013). FLYING SAUCER1 is a transmembrane RING E3 ubiquitin ligase that regulates the degree of pectin methylesterification in Arabidopsis seed Mucilage. Plant Cell 25: 944-959.
- Voiniciuc, C. and Günl, M. (2016). Analysis of monosaccharides in total mucilage extractable from Arabidopsis seeds. Bio-protocol 6: e1801.
- Voiniciuc, C., Yang, B., Schmidt, M. H., Günl, M. and Usadel, B. (2015). Starting to gel: how Arabidopsis seed coat epidermal cells produce specialized secondary cell walls. Int J Mol Sci 16: 3452-3473.
- Western, T. L. (2012). The sticky tale of seed coat mucilages: production, genetics, and role in seed germination and dispersal. Seed Sci Res 22: 1-25.
- Western, T. L., Burn, J., Tan, W. L.,. Skinner, D. J., Martin-McCaffrey, L., Moffatt, B. A. and Haughn, G. W. (2001). Isolation and characterization of mutants defective in seed coat mucilage secretory cell development in Arabidopsis. Plant Physiol 127: 998-1011.
- Western, T. L., Skinner, D. J. and Haughn, G. W. (2000). Differentiation of mucilage secretory cells of the Arabidopsis seed coat. Plant Physiol 122: 345-356.
- Windsor, J. B., Symonds, V. V., Mendenhall, J. and Lloyd, A. M. (2000). Arabidopsis seed coat development: morphological differentiation of the outer integument. Plant J 22: 483-493.
- Yang, X., Baskin, J. M., Baskin, C. C. and Huang, Z. (2012). More than just a coating: Ecological importance, taxonomic occurrence and phylogenetic relationships of seed coat mucilage. Perspect Plant Ecol 14(6): 434-442.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dean, G. H., Sola, K., Unda, F., Mansfield, S. D. and Haughn, G. W. (2019). Analysis of Monosaccharides from Arabidopsis Seed Mucilage and Whole Seeds Using HPAEC-PAD. Bio-protocol 9(24): e3464. DOI: 10.21769/BioProtoc.3464.
Category
Plant Science > Plant cell biology > Cell wall
Plant Science > Plant physiology > Tissue analysis > Cell wall
Biochemistry > Carbohydrate
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.