- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Lysosome Targeting RedGreen-assay: Selective Autophagy Sensing Assay for Mammalian Cells
(*contributed equally to this work) Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3455 Views: 5955
Reviewed by: Alka MehraMarco Di GioiaAlexandros C Kokotos
Original research article
The authors used this protocol in:

Mar 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The process of autophagy is an essential cellular mechanism, required to maintain general cell health through the removal of dysfunctional organelles, such as the ER, peroxisomes and mitochondria, as well as protein aggregates, and bacteria. Autophagy is an extremely dynamic process, and tools are constantly being developed to study the various steps of this process. This protocol details a method to study the end steps of autophagy-lysosomal fusion and the formation of the autolysosome. Many techniques have been used to study the various steps of the autophagy process. Here we describe the RedGreen-assay (RG-assay), an immunofluorescence-based technique used to visualize the targeting of substrates to the autolysosome in live cells. This technique takes advantage of the low lysosomal pH and over-expression of a tandem GFP-mCherry tagged protein targeted to an organelle of interest. While in the neutral cytosol or autophagosome, both GFP and RFP will fluoresce. However, within the autolysosome, the GFP signal is quenched due to the low pH environment and the RFP emission signal will predominate. This technique is readily quantifiable and amenable to high throughput experiments. Additionally, by tagging the GFP-RFP tandem fluorescent protein with organelle specific targeting sequences, it can be used to measure a wide range of substrates of autophagy.
Keywords: AutophagyBackground
Macroautophagy (herein referred to as autophagy) is an essential cellular process, required for the removal of large cellular material. It can be both general and selective, either removing bulk material or specific cytoplasmic material, respectively. The method described in this protocol can be used in the study of either process, but the focus here will be on selective autophagy. In brief, selective autophagy in mammalian cells begins with the tagging of damaged organelles or other cellular material, generally with the small protein ubiquitin. The collection of ubiquitinated proteins on the substrate allows the recruitment of autophagy receptors which interact with the ubiquitin protein and with the ATG8 family of autophagic factors on developing phagophores through their LC3 interacting region (LIR). This interaction results in the engulfment of the cargo by the phagophore, producing an autophagosome. Finally, the autophagosome fuses with the lysosome, producing an autolysosome and degrading the cargo within. Selective autophagy is responsible for degrading cargo such as peroxisomes (pexophagy), mitochondria (mitophagy), protein aggregates (aggrephagy) and bacteria (xenophagy). For a review, refer to Lahiri et al., 2019 and Morishita and Mizushima, 2019. As autophagy proceeds in many steps, different assays have been developed to study the various stages. Historically, the most popular assay to detect changes in autophagy induction is the activation of the ATG8 family of proteins, particularly LC3B. Another common assay for detecting targeting of substrate to the autolysosome is the co-localization between the cargo with either the autophagosome marker LC3, or with various lysosomal markers such as Lamp1. Co-localization studies are technically easy to perform, can be performed in a high throughout manner and can detect specific cargo within the lysosome easily through the use of fluorescent tagging. However, it is limited by the microscope resolution, and the cargo can be difficult to detect due to degradation. For a review describing these and other methods for detecting autophagy, refer to Klionsky et al., 2016.
The protocol described here, lysosome targeting Red-Green (RG) assay, can be used to study the end stages of autophagy, namely the localization of the cargo to the autolysosome. This assay takes advantage of the acidic pH of the lysosome (~4.6) and its ability to quench GFP (pKa ~6) fluorescence without affecting RFP (pKa = 4.5) fluorescence. Through the use of a tandem RFP-GFP fusion protein targeted to an organelle of choice, cargo that is cytosolic or within an autophagosome, and therefore exposed to neutral pH, will appear yellow when using pseudo colors green for GFP and red for RFP due to the fluorescence of both GFP and RFP. Upon lysosomal fusion, the labeled cargo will be exposed to an acidic pH, quenching GFP fluorescence and resulting in mainly RFP fluorescence (Figure 1). Again, this can be detected by microscopy and the ratio of cargo with RFP-only fluorescence can be compared to the total RFP fluorescence, to obtain a measure of autophagy. This method was first used to study aggrephagy (Pankiv et al., 2007). By tagging LC3 with a tandem GFP-mCherry, autophagic vesicles and autolysosomes were able to be readily visualized and distinguished. We have adopted the principle used with LC3 to visualize the targeting of peroxisomes to autolysosomes. This is done by tagging the peroxisome targeting transmembrane domain of PEX26 with the tandem fluorophore, allowing it to be localized to the cytosolic face of the peroxisomal membrane (Deosaran et al., 2013; Riccio et al., 2019). This allows for the visualization of pexophagy specifically. Others have also used a similar technique to study pexophagy, where the fluorescent proteins were targeted to the peroxisome matrix through the use of a matrix targeting sequence (Nazarko et al., 2014). Importantly, our method, targeting the fluorophore to the cytosolic face of the peroxisome membrane, allows for increased sensitivity of the tandem fluorophore to the acidic environment of the lysosome. Additionally, our quantification method differs, taking into account the relative GFP and mCherry fluorescence in order to determine the quantity of peroxisomes targeted to the lysosome. In addition to our use of the tandem fluorophore in pexophagy, we and others have used this method to quantify mitophagy (Allen et al., 2013; Kim et al., 2014; Wang et al., 2015).
We have shown that the use of the tandem fluorophore is a reproducible method for observing and quantifying selective autophagy. As displayed above, the tandem mCherry-GFP lysosome targeting assay is extremely flexible, and can be used to measure various forms of selective autophagy, as well as general autophagy. Through the pH sensitivity of the fluorophores, it is a reliable method to detect lysosomal targeting. Additionally, because GFP and mCherry are expressed at equal levels, comparing the fluorescent intensities of mCherry to GFP allows for a reproducible method to quantify pexophagy.
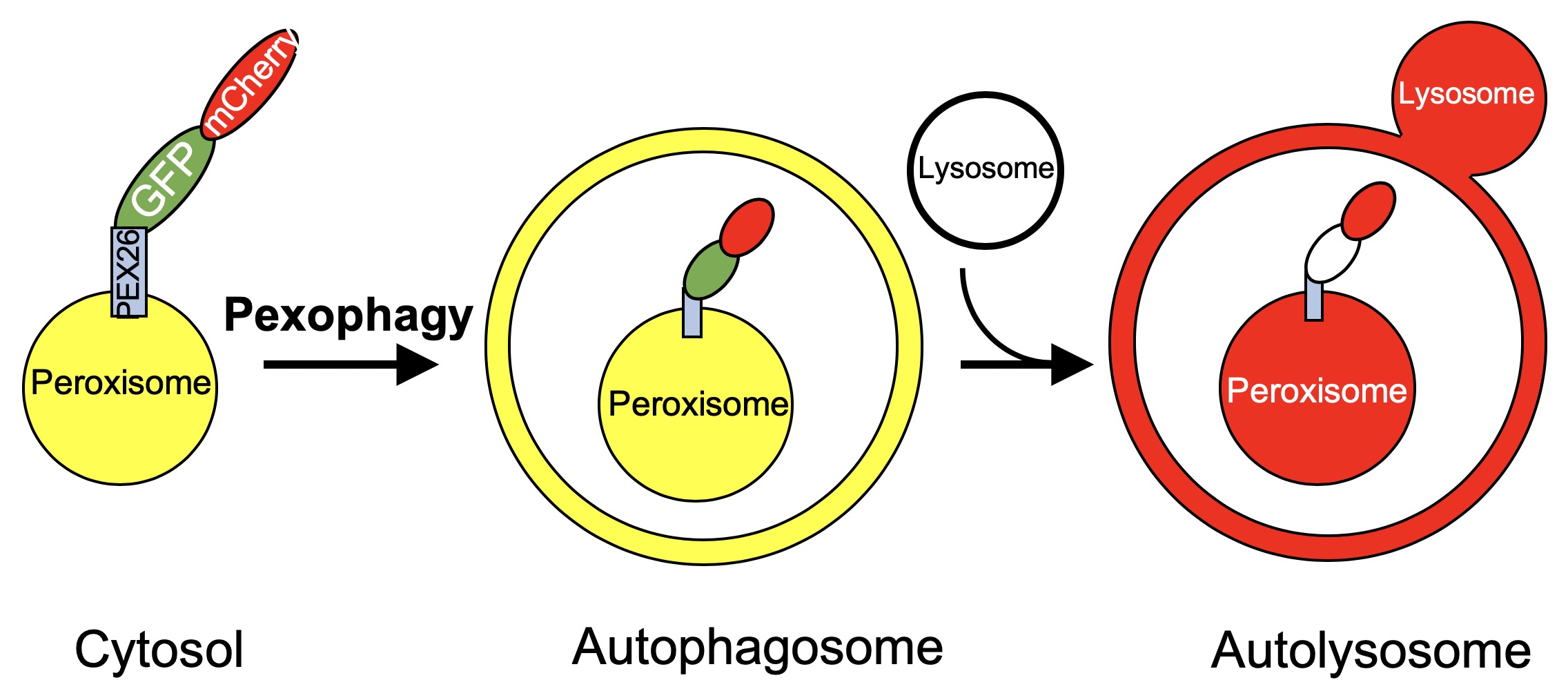
Figure 1. Schematic of the lysosome targeting RG assay. Peroxisomes expressing the PEX26TM-RG protein will appear yellow within the cytosol or the autophagosome due to fluorescence of both GFP and mCherry. When targeted to the lysosome, GFP fluorescence will be quenched and the peroxisome will appear red.
Materials and Reagents
- Stericup Quick Release Millipore Express PLUS 0.22 μm PES filter, 500 ml (Millipore Sigma, catalog number: S2GPU05RE)
- T25 flask (Corning Life Sciences, catalog number: C353109)
- 4-well NuncTM Lab-TekTM Chambered Coverglass (Thermo Fisher Scientific, catalog number: 155383)
- Lipofectamine 2000 (Life Technologies, catalog number: 11668-019)
- E-64 (Enzo Life Sciences, catalog number: BML-PI107-0001)
- Leupeptin (Bioshop, catalog number: LEU001)
- HeLa human cervical cancer cell line (ATCC CCL-2)
- GFP-mCherry (RG)-PEX26TM plasmid
- Dulbecco’s Modified Eagle’s Medium (Hyclone, catalog number: SH3008101)
- L-Glutamine (Hyclone, catalog number: SH30034.01)
- Fetal Bovine Serum (Life Technologies, catalog number: A12617)
- OptiMEM Reduced Serum Medium (Thermo Fisher, catalog number: 31985062)
- Trypsin-EDTA (0.05%), phenol red (Thermo Fisher, catalog number: 25300062)
- CO2 independent medium (Life Technologies, catalog number: 18045)
- Hank’s Balanced Salt Solution (HBSS) (Thermo Fisher, catalog number: 14025092)
Equipment
- Zeiss LSM710 equipped with a 63x 1.4 NA oil immersion objective and the appropriate lasers (488 nm on a tuneable Argon 458/477/488/514 nm at 30 mM with a 493-565 nm bandpass filter and 561 nm DPSS laser with a 600-700 nm bandpass)
- Forma Steri-Cycle CO2 Incubator (Thermo Fisher)
Software
- Volocity (Perkin Elmer, version 6.3)
- Zeiss Zen (Zeiss, https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html)
- Microsoft® Office Excel (Microsoft Office 365)
Procedure
- Gene cloning
GFP-mCherry-PEX26TM was constructed in several steps as described in Deosaran et al., 2013. First, the PEX26 ORF from pSPORT-PEX26 (Invitrogen) was cloned into the EcoRI and SalI sites of pmEGFP-C1, to insert the entire ORF of PEX26 at the C-terminus of GFP. Next, the entire PEX26 but its C-terminus tail-anchor domain (TM) was removed by PCR using two primers containing a BglII site. Finally, primers containing a BglII or BamHI sites were used to PCR amplified mCherry. Following digestion with BglII and BamHI, the PCR product was inserted into the BglII site on the pmEGFP-PEX26TM vector to generate the GFP-mCherry-PEX26TM (RG-PEX26TM) vector. Refer to Figure 2 for a schematic.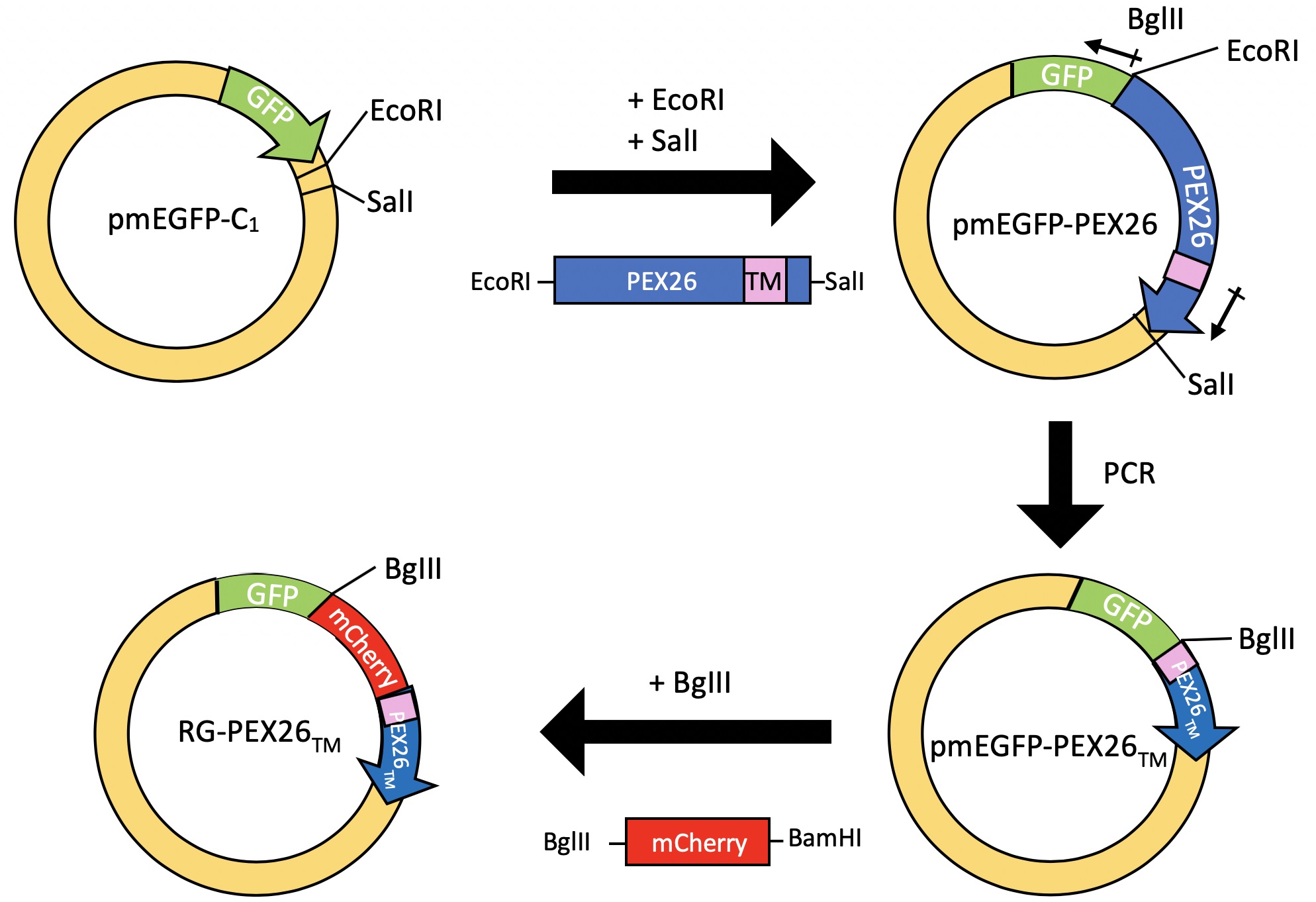
Figure 2. Schematic diagram of the construction of the RG-PEX26TM construct. Restriction enzymes used are indicated. Small arrows indicate the location of primer used. - Cell seeding
Here we will describe work done on HeLa cells which were grown in Dulbecco's Modified Eagle Media (DMEM) supplemented with 10% FBS and 5% CO2. Media was filter sterilized with FBS. However, this protocol can be adapted to most transfectable cell lines. We have tested this protocol in a number of transfectable cells such as MEFs, Cos7, U2OS, Hek293 and MDCKs.- To seed cells, first warm sterile media and PBS to 37 °C. Thaw trypsin and warm to room temperature.
- Remove cells from the incubator and aspirate growth media. Wash with PBS (about 5 ml) and aspirate. Add trypsin to the flask at an appropriate volume to ensure cells will not dry (i.e., 1 ml trypsin in a T25 flask).
- Incubate cells in trypsin at 37 °C for 3-5 min.
- Once cells are no longer adhered to the flask, add media to dilute and inactivate the trypsin (about 5 ml). Collect media containing cells in a 15 ml tube and centrifuge at 1,000 x g for 5 min to pellet cells.
- Following centrifugation, aspirate media and resuspend the cells in fresh media.
- Count the cells using a hemocytometer or other cell counting device.
- Seed cells in a 4-well Lab-Tek chamber (or equivalent coverslip system that allows for live cell imaging) at a density of 25,000 cells/well. Total volume within the well should be 0.5 ml. For other cell types or longer procedures, seed cells at an appropriate density for transfection.
- Allow cells to adhere and rest for 24 h before transfection in the 37 °C incubator.
- Cell transfection
- On Day 2, transfect with the RG-PEX26TM construct (and any other construct required for the experiment). Use 1-2 μg of DNA per well and Lipofectamine2000 transfection reagent according to the manufacturer’s instructions. In brief, combine 0.5 μl of Lipofectamine2000 with 50 μl of Opti-mem medium in a 1.5 ml tube. Allow it to sit for 5 min. In another 1.5 ml tube, combine 50 μl of Opti-mem medium and DNA. Combine the lipofectamine and DNA containing mixtures and let sit for 20 min. Do this for each transfected well. Add complete mixture (100 μl) to each well.
- Let cells grow for 24-36 h.
- Change media to DMEM + FBS or HBSS without FBS. HBSS induces mTORC1 related pexophagy due to amino acid starvation. Both are supplemented with 2 μM E-64 and 0.5 mM leupeptin to prevent degradation of protein inside lysosomes and allow detection of mCherry signal. Incubate at 37 °C for 4-24 h. Incubation at 37 °C is accomplished through the use of a CO2 dependent incubator.
- Image acquisition
- For imaging, the cells should be imaged under the appropriate conditions (i.e., 37 °C and 5% CO2). If using a microscope system without CO2, the media should be replaced with appropriate CO2 independent media supplemented with 0.5 mM leupeptin and 2 μM E-64.
- Ensure microscope is powered on and warmed up to 37 °C before starting.
- For each set of experiments, calibrate settings such that the mean GFP and mCherry fluorescence intensities (IG/R) are approximately equal (i.e., IGFP/ImCherry = 1). Calibration will depend on the microscope used, however in short, the laser, gain and speed should be set to have the background close to zero intensity while most of the signal for the fluorophores are not saturated. The ratio of the mean intensity for RFP over GFP for each cell should be close to 1 for the control cells. Ensuring that GFP and mCherry signals are approximately equal allows for optimal detection of changes in the fluorescence intensity ratio upon pexophagy induction. For each trial, all conditions should be imaged on the same day. Either 3D z-sectioning or a single slice can be used for quantification but ensure to keep the imaging method consistent throughout trials. Refer to Figure 3 for sample images.
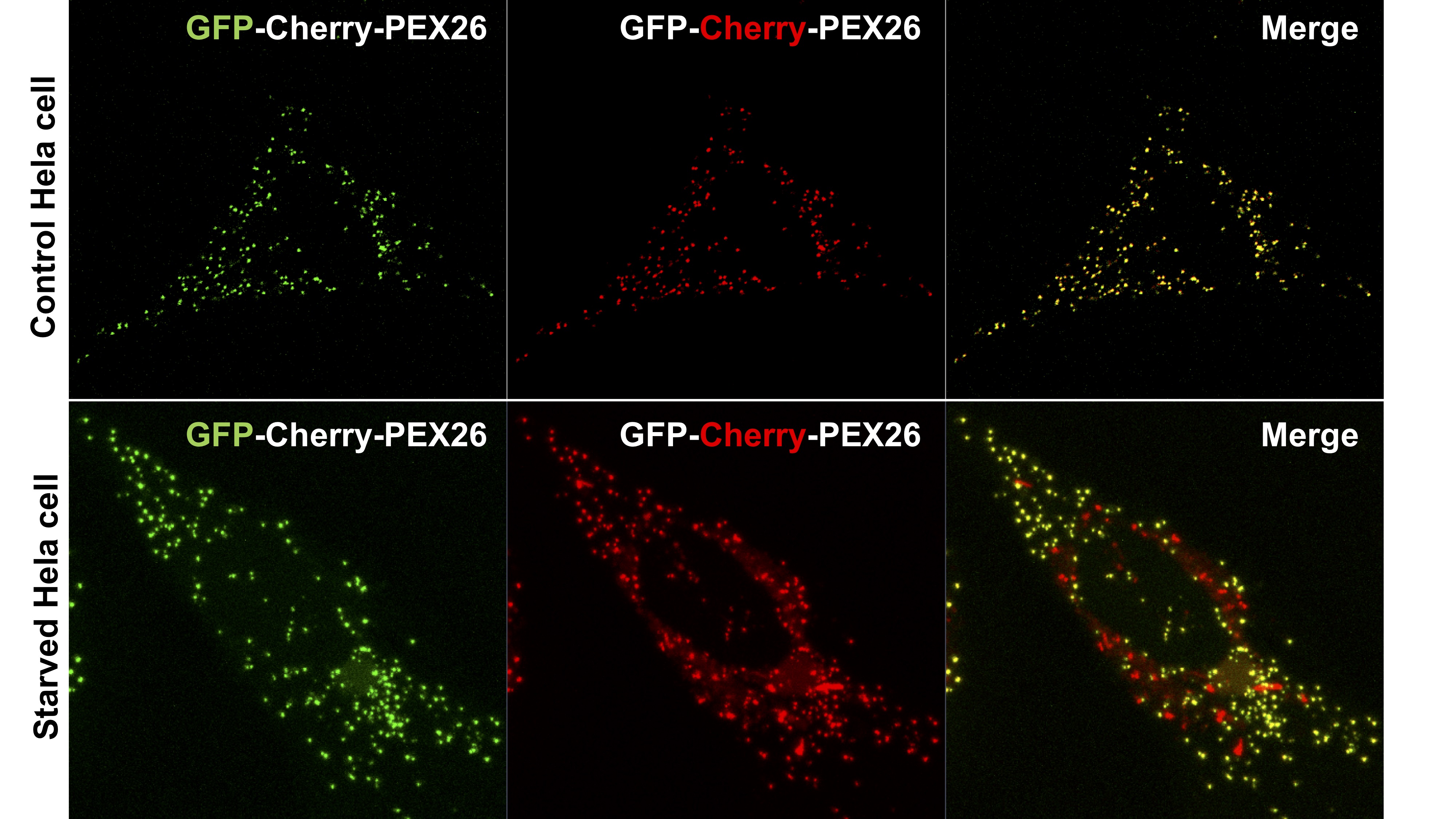
Figure 3. HeLa cells transfected with RG construct. Control cells have been incubated in DMEM supplemented with E-64 and leupeptin as above. Starved cells were grown in HBSS for 12 h before imaging, also supplemented with E-64 and Leupeptin. Both cells are expressing GFP-mCherry-PEX26TM. Control cell (Top) shows almost equal GFP and mCherry pixel intensities. Cells where pexophagy has been induced by amino acid starvation (Bottom) show increased 'red-only' signal indicating peroxisomes have been targeted to lysosomes. Note, an increase in ‘red-only’ signal is more easily observable as a decrease in green fluorescent intensity. Red fluorescent intensity should not change. Scale bar = 10 μm
Data analysis
Note: Please refer to Figure 3 for a detailed spreadsheet.
- Using Volocity software or its equivalent, first determine the cell area by drawing a region of interest (ROI) around an individual cell expressing the RG construct (Figure 4A).
- Next, identify peroxisomes by using the mCherry signal only (Figure 4B, column 1). In Volocity, this can be done in several different ways, however for most images with very high signal to noise in the ROI is the easiest. Most other imaging analysis software with similar algorithms can be used to find objects within Z-stack images. Measure the mean fluorescent intensity of GFP and mCherry signal per puncta as well as the volume of each puncta, and input into a spreadsheet (Figure 4B, columns 2-4).
- Next, identify the proportion of lysosomally targeted peroxisomes (“red only” peroxisomes) by calculating the ratio of mCherry to GFP fluorescence per puncta (Figure 4B, column 5). A puncta is considered ‘red-only’ if the mean mCherry fluorescent signal is at least 3 times higher than the mean GFP fluorescent signal. This is denoted in the spreadsheet as a value of 1 (lysosomally targeted) or 0 (not lysosomally targeted) (Figure 4B, column 6A). Using this information, the volume of lysosomally targeted peroxisomes per cell can be tabulated efficiently by Excel (Figure 4B, column 6B).
- Add the volumes of all lysosomally targeted peroxisomes per cell (Figure 4B, column 7). Identify the area of all peroxisomes in the cell by adding the volume of all mCherry positive puncta (Figure 4B, column 8). To identify the proportion of lysosomally targeted peroxisomes per cell, divide the volume of the lysosomally targeted peroxisomes by the total peroxisome volume (Figure 4B, column 9).
- Repeat this process for every cell, quantifying at least 50 cells per trial. Calculate an average of proportion of lysosomally targeted puncta per tested condition and plot on a graph.
- Once a minimum of 3 independent trials has been completed, perform statistical analysis and graph. For a sample graph, refer to Figure 4C.
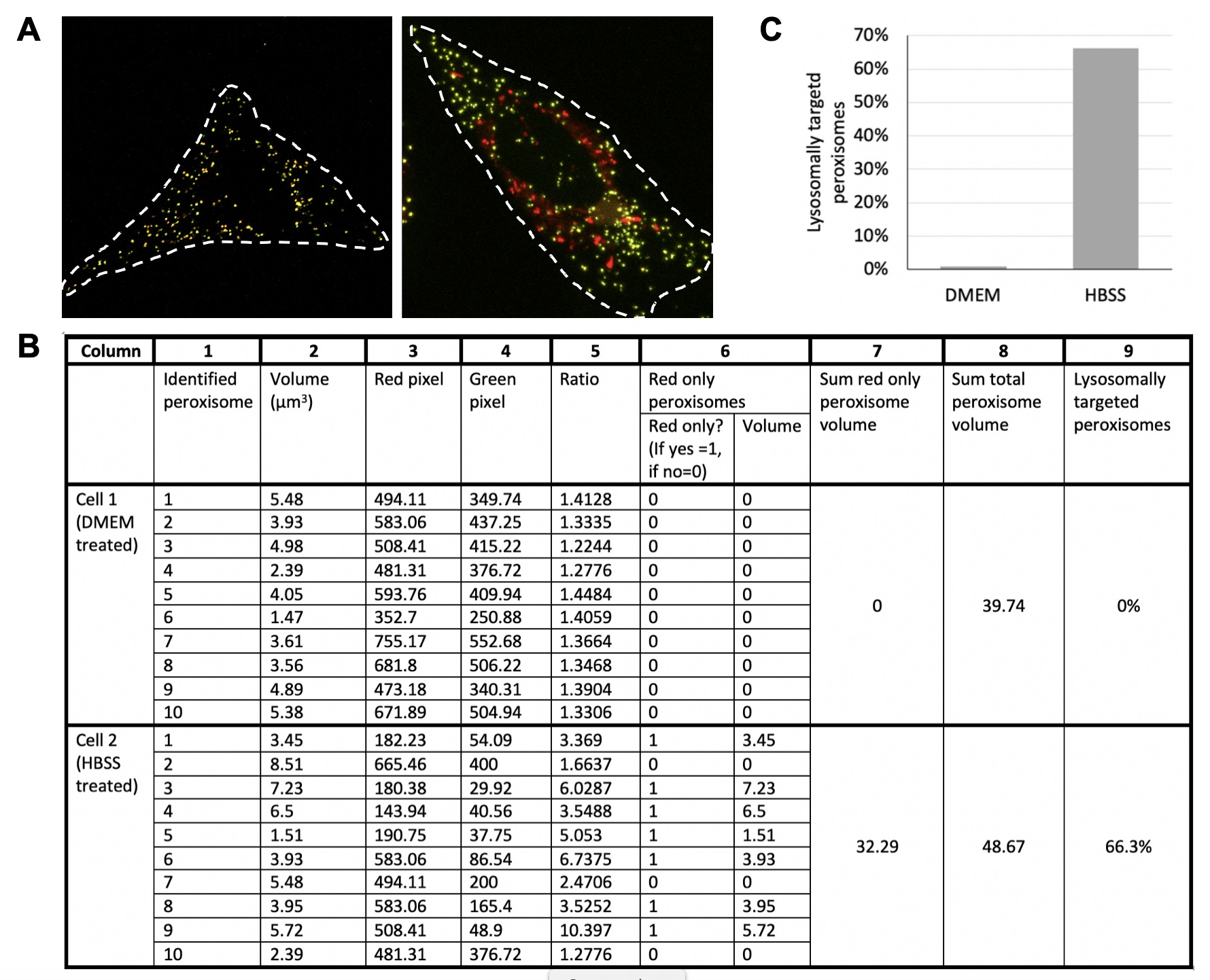
Figure 4. Sample cell ROI generation and calculation spreadsheet. A. ROI generation for cells imaged in Figure 2 (left–cell incubated for 12 h in DMEM, right–cell incubated for 12 h in HBSS). ROI is denoted by the white dotted line. Merged images are shown. B. Sample calculation spreadsheet. Quantification of 10 peroxisomes from each cell imaged in A is shown. Cell 1 was incubated in DMEM (control), while Cell 2 was incubated in HBSS (pexophagy induced) for 12 h. Please refer to the protocol for detailed column explanations. Note, red only peroxisomes (column 6) are those peroxisomes which have a mean red fluorescent intensity at least 3 times greater than the mean green fluorescent intensity. C. Sample data from B graphed.
Notes
- This experiment must be imaged live, as fixation may alter the mCherry and GFP pixel intensity.
- If additional staining is required, for example to determine if a construct is expressed, cells may be fixed following imaging, stained normally and re-imaged, to determine what proportion of cells expressing the RG construct are also expressing your construct of interest.
- Laser intensity or gain must not be altered between images or samples within the same trial.
Acknowledgments
Funding Sources: This work was supported by the Canada Institute of Health Research grants (PJT156196 to P.K. Kim and MOP-130281 to G.A. McQuibban) and Natural Sciences and Engineering Research Council of Canada Discovery grants (RGPIN-2015-04077 to P.K. Kim and RGPIN-2015-05969 to G.A. McQuibban). This protocol was adapted from our previous work (Deosaran et al., 2013; Wang et al., 2015; Riccio et al., 2019).
Competing interests
There are no significant competing financial, professional, or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
References
- Allen, G. F., Toth, R., James, J. and Ganley, I. G. (2013). Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep 14(12): 1127-1135.
- Deosaran, E., Larsen, K. B., Hua, R., Sargent, G., Wang, Y., Kim, S., Lamark, T., Jauregui M., Law, K., Lippincott-Schwartz, J., Brech, A., Johansen, T. and Kim, P. K. (2013). NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 126(Pt 4): 939-52.
- Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12(1): 1-222.
- Kim, S. J., Syed, G. H., Khan, M., Chiu, W. W., Sohail, M. A., Gish, R. G. and Siddiqui, A. (2014). Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA 111(17): 6413-6418.
- Lahiri, V., Hawkins, W. D. and Klionsky, D. J. (2019). Watch what you (self-) eat: Autophagic mechanisms that modulate metabolism. Cell Metab 29(4): 803-826.
- Morishita, H. and Mizushima, N. (2019). Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol 35: 453-475.
- Nazarko, T. Y., Ozeki, K., Till, A., Ramakrishnan, G., Lotfi, P., Yan, M. and Subramani, S. (2014) Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J Cell Biol 204(4): 541-557.
- Pankiv, S., Clausen, T. H., Lamark, T., Brench, A., Bruun, J. A., Outzen, H., Overvatn, A., Bjorkoy, G. and Johansen, T. (2007). p62/SQSTM1 binds directly to ATG8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282(33): 24131-24145.
- Riccio, V., Demers, N., Hua, R., Vissa, M., Cheng, D. T., Strilchuk, A. W., Wang, Y., McQuibban, G. A. and Kim, P. K. (2019). Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J Cell Biol 218(3): 798-807.
- Wang, Y., Serricchio, M., Jauregui, M., Shanbhag, R., Stoltz, T., Di Paolo, C. T., Kim, P. K. and McQuibban, G. A. (2015). Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11(4): 595-606.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Riccio, V., Vissa, M., McQuibban, A. and Kim, P. K. (2019). Lysosome Targeting RedGreen-assay: Selective Autophagy Sensing Assay for Mammalian Cells. Bio-protocol 9(24): e3455. DOI: 10.21769/BioProtoc.3455.
- Riccio, V., Demers, N., Hua, R., Vissa, M., Cheng, D. T., Strilchuk, A. W., Wang, Y., McQuibban, G. A. and Kim, P. K. (2019). Deubiquitinating enzyme USP30 maintains basal peroxisome abundance by regulating pexophagy. J Cell Biol 218(3): 798-807.
Category
Immunology > Immune cell function > Macrophage
Microbiology > Microbe-host interactions > Bacterium
Neuroscience > Cellular mechanisms > Synaptic physiology
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.