- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

DNase I Chromatin Accessibility Analysis

Published: Vol 9, Iss 23, Dec 5, 2019 DOI: 10.21769/BioProtoc.3444 Views: 5803
Reviewed by: Alka MehraAswad KhadilkarManasi K. Mayekar
Original research article
The authors used this protocol in:

Mar 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Chromatin consists of compacted DNA in complex with proteins and contributes to the organization of DNA and its stability. Furthermore, chromatin plays key roles in regulating cellular processes such as DNA replication, transcription, DNA repair, and mitosis. Chromatin assumes more compact (inaccessible) or decondensed (accessible) conformations depending on the function that is being supported in the genome, either locally or globally. The activity of nucleases has been used previously to assess the accessibility of specific genomic regions in vitro, such as origins of replication at varying points in the cell cycle. Here, we provide an assay to determine the accessibility of specific human genomic regions (example used herein: Lamin B2 origin of DNA replication) by measuring the effect of DNase I nuclease on qPCR signal from the studied site. This assay provides a powerful method to interrogate the molecular mechanisms that regulate chromatin accessibility, and how these processes affect various cellular functions involving the human genome that require manipulation of chromatin conformation.
Keywords: Chromatin accessBackground
Studies aiming to elucidate the molecular underpinnings of genomic processes influenced by chromatin have utilized nuclease sensitivity to assess the accessibility of chromatin. This is a powerful concept for interrogating the regulation and timing of chromatin conformational changes that contribute to cellular processes such as DNA replication and transcription. For example, it has been shown that chromatin is more sensitive to micrococcal nuclease at two distinct sites within the regions encompassing origins of DNA replication in Chinese hamster ovary cells during G1 versus S-phase, suggesting that chromatin is more accessible at these origins in G1 phase (Pemov et al., 1998). However, the approach used in this latter study required laborious techniques, Southern blotting, large-scale and complex genomic DNA collections and restriction digests, and cumbersome gel-electrophoresis and membrane-transfer methods. In addition, the ability to interpret the results was limited, and only allowed one to observe changes to a single site in a larger locus, with no other changes in nuclease (chromatin) accessibility being detectable throughout the region. Older more classical techniques, such as nuclease-protection footprinting assays, can give more insight into specific loci in terms of chromatin changes. For example, the positioning or rearranging of nucleosomes can be detected by such techniques in a small region. However, these footprinting techniques also require laborious methods by the investigator, and are not amenable to asking questions regarding large-scale changes to chromatin accessibility in specific regions of interest in the genome.
To overcome many of the limitations of older techniques, our group developed a more simple and efficient method of detecting differences in large-scale chromatin accessibility for any site in the genome that an investigator wishes to assess. Our approach relies on the accessibility of DNase I to perform an initial limited digestion of genomic DNA in the context of chromatin prior to using such DNA as a substrate for an efficient quantitative real-time PCR (qPCR) analysis. No radioactivity or cumbersome gel-electrophoretic methods are required, and isolation of chromatin/DNA for nuclease digestion is straightforward. Analysis is not complicated, and the results are obtained using the rationale that the more DNase I can pre-digest the chromatin/DNA sample, the less substrate DNA there will be for qPCR relative to chromatin/DNA not exposed to DNase I (total uncut DNA). Less substrate DNA due to DNAse I digestion will require more PCR cycles to amplify, and this will be interpreted as indicating that the chromatin was more accessible for nuclease digestion. The investigator can manipulate the cells using various methods prior to isolating chromatin, allowing quick and efficient quantitative comparisons of chromatin accessibility changes to a specific region under different experimental conditions. The main limitations of our approach are knowing the genomic DNA sequences of the region under investigation, for primer design, and access to a real-time PCR machine.
In practice, our group has used this DNase I chromatin accessibility technique to assess changes at the human Lamin B2 origin of DNA replication in multiple studies. We demonstrated that chromatin at this origin was less accessible to DNase I (more condensed) in S phase (Wong et al., 2010) or in the absence of endogenous Myc protein specifically in late-G1 (Nepon-Sixt et al., 2019), since the qPCR signal from the Lamin B2 origin increased (required fewer cycles) under both of these conditions. While this assay has been utilized to determine chromatin accessibility at a specific origin of mammalian DNA replication under various conditions, chromatin accessibility at other genomic sites (e.g., promoters) can be evaluated by using suitable primers during qPCR designed against other sites of interest.
Materials and Reagents
- Tissue culture plates (Sarstedt, catalog number: 83.3902)
- Cell lifter (Fisher, catalog number: 8100240)
- Microtubes (Axygen, catalog number: MCT-150-C)
- Multiplate PCR Plates, 96-well, clear (Bio-Rad, catalog number: MLL9601)
- Microseal ‘B’ seal Seals (Bio-Rad, catalog number: MSB1001)
- Human cell line (e.g., HaCaT–immortalized human keratinocytes)
- Appropriate tissue culture medium (e.g., Dulbecco’s modification of Eagle’s medium + 10% fetal bovine serum)
- Aprotinin (Fisher, catalog number: 61-637-010MG)
- Leupeptin (Fisher, catalog number: 50550413)
- PMSF (RPI Corp., catalog number: P20270-5.0)
- DNase I Enzyme (Promega, catalog number: M610A)
- EDTA (Fisher, catalog number: BP120-500)
- EGTA (Calbiochem, catalog number: 4100-50GM)
- SDS (Sigma-Aldrich, catalog number: L5750-5KG)
- NaCl (Fisher, catalog number: BP358-10)
- RNase A (Sigma-Aldrich, catalog number: R4875-100MG)
- Proteinase K (Fisher, catalog number: BP1700-100)
- Sterile deionized H2O
- Chloroform (Fisher, catalog number: C298-500)
- EtOH (Pharmco-Aaper, catalog number: 111ACS200)
- 5x Colorless GoTaq Reaction Buffer (Promega, catalog number: M792A)
- 10 mM dNTPs (Promega, catalog number: U151B)
- GoTaq DNA Polymerase (Promega, catalog number: M300B)
- Forward Primer (see Procedure)
- Reverse Primer (see Procedure)
- FAM-labeled probe (see Procedure)
- KCl (Fisher, catalog number: P217-500)
- Na2HPO4 (Fisher, catalog number: S374-500)
- KH2PO4 (Fisher, catalog number: P285-500)
- NaOH (Fisher, catalog number: BP359-500)
- MgCl2 (Sigma-Aldrich, catalog number: M0250-1KG)
- CaCl2 (Fisher, catalog number: BP510-500)
- Sucrose (Fisher, catalog number: BP220-1)
- Triton X-100 (Fisher, catalog number: BP151-100)
- Phenol crystals (Fisher, catalog number: A92-100)
- 8-hydroxyquinoline (Fisher, catalog number: AC121900250)
- Tris Base (Fisher, catalog number: BP152-5)
- 1x PBS Solution, kept in 4 °C fridge (called ice cold) (see Recipes)
- Lysis Buffer (see Recipes)
- DNase Buffer (see Recipes)
- Tris-saturated Phenol (see Recipes)
- TE Buffer (see Recipes)
Equipment
- Pipettes (VWR)
- Centrifuge (Eppendorf mini-centrifuge, model: 5424, catalog number: 022620401)
Note: All “rpm” listed below also include RCF in g, for those who need to convert for use in a different model of centrifuge. - Isotemp Water Bath (Fisher, catalog number: 2223)
- Bioruptor 300 Plus Sonication Device w/ Water cooler system (Diagenode, catalog numbers: B01020001 and B02010003)
- CFX96 Real-Time PCR Detection System including C1000 Thermal Cycler Chassis (Bio-Rad, catalog number: 185-5096)
- Autoclave (Steris, model: SG-120 Scientific Gravity Sterilizer)
Software
- MS Excel for Mac 2011 Version 14.6.0
- CFX Manager Version 3.1 (Bio-Rad, catalog number: 184-5000)
Procedure
- Genomic DNA collection
- Add protease inhibitors fresh to Lysis Buffer and DNase Buffer to final concentrations: 5 µg/ml Aprotinin, 5 µg/ml Leupeptin, and 1 mM PMSF. Store buffers on ice.
- Collect human cell line from 1 x 10 cm plate (per experimental condition; ~5 x 106 cells/plate):
- Aspirate cell culture medium.
- Wash cells with 4 ml of ice cold 1x PBS solution and aspirate. Repeat once.
- Add 500 µl ice cold 1x PBS to the plate.
- Scrape cells with cell lifter and collect cell suspension into a microtube and keep on ice.
- Centrifuge cells for 5 s at 10,000 rpm (~9,400 x g) to pellet, then carefully remove and discard the 1x PBS supernatant with a pipette. Should be performed in a cold room.
- Resuspend cell pellet in 300 µl Lysis Buffer with protease inhibitors, and incubate on ice for 5 min. This step creates the chromatin/DNA to be analyzed below.
- Centrifuge lysate at 4,000 rpm (~1,500 x g) for 5 min at 4 °C.
- Carefully remove and discard supernatant with a pipette, and then resuspend DNA pellet in 300 µl DNase Buffer with protease inhibitors.
- Centrifuge lysate at 4,000 rpm (~1,500 x g) for 5 min at 4 °C.
- Resuspend DNA pellet in 300 µl DNase Buffer with protease inhibitors. No need to quantify the DNA.
- Aliquot 100 µl of the lysate to each of two separate microtubes (one for “DNase I”, one for “No DNase I”). Note here that equal amounts of chromatin/DNA for each condition are being assessed, and normalization to uncut input DNA (i.e., No DNase I) will be used in the Data Analysis below. Be accurate in your pipetting. The DNA is not viscous here because these are chromatin samples, where DNA is still on nucleosomes, etc. The Lysis Buffer does not disrupt the nuclear structure (on purpose) so that chromatin can be interrogated by the investigator using DNAse I in Procedure B of the protocol.
- DNase I reaction and substrate preparation (per experimental condition)
- Add 3 units (3 µl) DNase I Enzyme to one of the microtubes only (‘DNase I’), mix by tapping, and incubate both microtubes at room temperature for 7-8 min.
Note: Incubation time is determined by trial and error (empirically) using distinct 10 cm plates of cells to test specific incubation durations for DNAse I exposure. Too short can limit nuclease effectiveness, and too long can tip activity beyond a linear range of detectable differences between experimental conditions. Chromatin will vary greatly for different loci, and DNase I will have different effectiveness at different sites, which makes it very difficult to pre-determine the DNase I duration for chromatin analysis with an optimization step on naked total input DNA. As an example, for the results obtained in Nepon-Sixt et al. (2019) (graphs in Figure 8c&d) we determined by empirical testing that 7 min of DNase I produced an acceptable partial digestion of chromatin that was not under-digested, nor exhaustive, to allow DNase I sensitivity differences to be measured for the Lamin B2 locus under the experimental conditions that were assessed. Your empirical testing would follow this same protocol to perform initial determinations of DNase I time durations needed for your particular chromatin site under investigation. - Add 4.25 µl of 0.25 M EDTA pH 8.0 and 2.1 µl of 100 mM EGTA to each microtube (10 mM and 2 mM final concentrations, respectively), mix by tapping, and incubate in a 65 °C water bath for 10 min to stop reaction.
- Add 5.75 µl of 10% SDS (0.5% final concentration) to each tube, and mix.
- Lightly sonicate with Bioruptor 300 for an 8 min duration, 30 s on/30 s off.
- Add 20 µl of 2.5 M NaCl (~500 mM final concentration) to each microtube.
- Add 1 µl of 10 mg/ml RNase A (50 µg/ml final concentration) to each microtube, mix by pipetting, and incubate in a 37 °C water bath for 30 min.
- Add 3 µl of 10 mg/ml Proteinase K (250 µg/ml final concentration) to each microtube, mix by pipetting, and incubate in a 42 °C water bath overnight.
- Quick spin microtubes in centrifuge to remove condensation from the lid.
- Add ~350 µl of sterile deionized H2O (dH2O) to each tube, and purify DNA with phenol/chloroform extractions:
- Add 250 µl Tris-saturated phenol and 250 µl chloroform to each microtube, and invert 8-10 times to mix well.
- Centrifuge tubes at 12,000 rpm (~13,500 x g) for 5 min.
- Move aqueous layer (top layer) to new microtubes, respectively.
- Add 500 µl chloroform to each tube.
- Centrifuge tubes at 12,000 rpm (~13,500 x g) for 5 min.
- Move aqueous layer (top layer) to new microtubes, respectively.
- Fill tubes with ice cold 100% EtOH, shake, and incubate at -20 °C for 1 h.
- Centrifuge tubes at 12,000 rpm (~13,500 x g) at 4 °C for 25 min to pellet DNA.
- Discard supernatant and dry DNA pellets by inverting microtubes in a rack and incubating at room temperature for 10 min.
- Resuspend each DNA pellet in 50 µl TE Buffer to make DNA substrates. Again, no need to quantify DNA, but care must be taken in pipetting accuracy.
- Add 3 units (3 µl) DNase I Enzyme to one of the microtubes only (‘DNase I’), mix by tapping, and incubate both microtubes at room temperature for 7-8 min.
- qPCR analysis of DNA substrates
- Create master mix for qPCR analysis containing the following (enough for quadruplicate 10 µl reactions per experimental condition, each with ‘DNase I’ and ‘No DNase I’ respectively):
- Sterile deionized H2O: 35 µl
- 5x Colorless GoTaq Reaction Buffer:10 µl
- 10 mM dNTPs:1 µl
- GoTaq Polymerase: 1 µl
- Forward Primer (10 µM):1 µl (e.g., sequence used for Lamin B2 ori accessibility analysis: 5′-GCTACACTAGCCAGTGACCTT-3′)
- Reverse Primer (10 µM): 1 µl (e.g., sequence for used for Lamin B2 ori accessibility analysis: 5′-GTTCTGCCTCTGAGTTTATTCCTG-3′)
- FAM-labeled Probe (10 µM): 0.5 µl (e.g., sequence used for Lamin B2 ori accessibility analysis: 5′-/56-FAM/-TTAGACATCCGCTTCATTAGGGCAGAGG-/3BHQ_1/-3′)
- Aliquot 49.5 µl master mixes into two microtubes each.
- Add 0.5 µl ‘DNase I’ DNA substrate to one tube, and 0.5 µl ‘No DNase I’ DNA substrate to the other, for each experimental condition, and mix well.
- Aliquot 10 µl of each reaction mix into 4 wells of a 96-well multiplate.
Note: Try to avoid using the outer wells on the edge of the 96-well plate, if possible. In our experience use of the outer regions can lead to non-reproducible variability in results, likely due to temperature issues. - Cover the 96-well plate with seal (Microseal ‘B’ Seals) and press to secure.
- Put the 96-well plate in CFX96 Real-Time PCR Detection System and run the following program:
- 95 °C for 10 min
- 95 °C for 15 s
- 60 °C for 1 min
- Repeat steps b and c 40 x
- Analyze qPCR results with CFX Manager to obtain quantitation cycle (Ct) values using FAM.
- Create master mix for qPCR analysis containing the following (enough for quadruplicate 10 µl reactions per experimental condition, each with ‘DNase I’ and ‘No DNase I’ respectively):
Data analysis
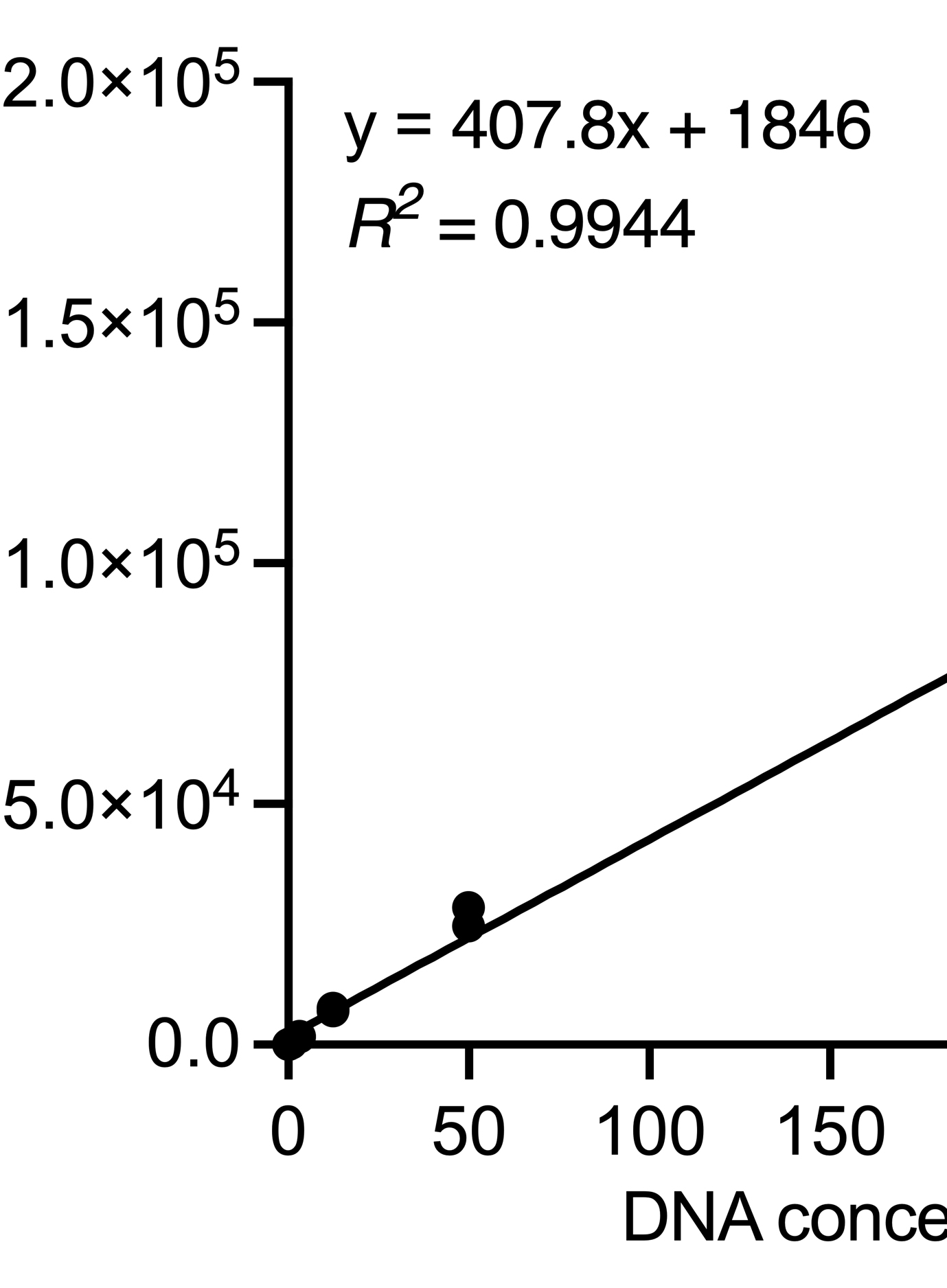
For each experimental condition, analyze results according to the formula: 100/2Ct(DNase I)−Ct(no DNase I), which normalizes the data to input DNA (‘no DNase I’). This is performed separately for each experimental condition that the investigator chooses to study. The changes in the ability of DNase I to digest DNA contained in chromatin are then plotted on graphs to determine if a tested experimental condition rendered a locus more sensitive to DNase I digestion (and is therefore interpreted as being more open in chromatin accessibility). Examples of how the final data will appear in a graph can be seen Figure 8c&d in Nepon-Sixt et al., (2019). Using our experiment from Figure 8c in ref. 1 as an example, we next describe how to mathematically analyze the data obtained using our tested experimental conditions, allowing graphical plotting of the final results.
As an example for comparing a single locus for two experimental conditions [e.g., with normal Myc expression (si-Control; e.g., Luciferase control, si-Luc), and with Myc si-RNA suppression; see ref. 1, Figure 8c, left pair of bars]: If one obtains Ct values of 24 for ‘no DNase I’ and 26 for the ‘DNase I’ sample, then the calculation would be 100/226-24 = 25 [note, the whole numbers chosen here are not the exact same as what we obtained for Figure 8c in ref. 1, but are used here for simplicity in illustrating how to perform the mathematics needed]. This means that using the substrate with DNase I treatment gave 25% of the signal relative to the sample without DNase I under the one experimental condition (e.g., with normal Myc; si-Luc/Control). This is also referred to as ‘25% untreated input chromatin/DNA’ as shown on the Y-axis in Fig 8c in ref. 1. This is then performed on the samples from the other experimental condition (e.g., with Myc si-RNA exposure and loss of Myc expression; si-Myc in Figure 8c from ref. 1). If for example the latter gives Ct values of 24 for ‘no DNase I’ and 25 for the ‘DNase I’ sample, then the calculation would be 100/225-24 = 50. This means that using the substrate with DNase I treatment gave 50% of the signal relative to the sample without DNase I for this experimental condition (e.g., loss of Myc, si-Myc). The interpretation would be that loss of Myc expression by si-RNA renders the locus tested to be less sensitive to DNase I digestion (relative to si-Luc control), and therefore more compact from a chromatin standpoint (less accessible to the added DNase I enzyme). Thus, loss of Myc expression caused the locus to become more condensed in its chromatin state. Such data are then plotted on graphs after analyzing technical replicates (separate samples analyzed from same day and plating of cells). Further validity to the interpretations will need to be achieved with biological replicates giving the same results (samples analyzed from different plating of cells collected on a different day).
Notes
- If an investigator wants to normalize their data for a locus in question to an internal control/comparison, such as a gene or promoter that is known to have no chromatin changes between the conditions tested (will need to be verified first in separate testing), then separate analyses would need to be performed on that locus (internal control/comparison) and the locus in question. The changes between the results of the locus in question will then be normalized to any changes in the presumptively non-changing control/comparison locus. However, it is important to understand that many loci will change when one subjects cells to various conditions (such as global effects due to drugs, or si-RNA loss of important transcription factors), so it is perhaps better to use biological and technical replicate testing to achieve statistically valid interpretations.
- This approach is not designed to compare the same locus in two different cell types, but instead has been validated for comparing chromatin changes occurring to one locus in the same cell type that is exposed to different experimental conditions (see Nepon-Sixt et al. [2019] for examples).
Recipes
- 1x PBS Solution; kept in 4 °C fridge (called ice cold)
Make 10x PBS solution (below) and dilute 1:10 to 1x
80 g of NaCl (1.4 M)
2 g of KCl (26.8 mM)
14.4 g of Na2HPO4 (101.4 mM)
2.4 g of KH2PO4 (17.6 mM)
Add sterile deionized H2O (dH2O, used below) to 800 ml
Adjust pH to 7.4 by 10 N NaOH
Add dH2O to 1 L
Autoclave and cool to room temperature - Lysis Buffer
10 mM Tris pH 7.5
5 mM MgCl2
1 mM CaCl2
10 mM KCl
300 mM Sucrose
0.1% Triton X-100 - DNase Buffer
10 mM Tris pH 7.5
5 mM MgCl2
1 mM CaCl2
10 mM KCl
300 mM Sucrose - Tris-Saturated Phenol (can use commercially available Tris-Phenol if desired, but our method here allows investigator control over phenol preps and saturation levels)
- Dissolve 100 g phenol crystals with 100 ml dH2O in 37 °C water bath
- Add 0.1 g 8-hydroxyquinoline (0.1% w/vol) and mix by shaking
- Saturate aliquots with 1:1 volume of 0.5 M Tris pH 8.0 by shaking and allow separation. Discard Tris solution as phenol waste (Repeat 1 x)
- Saturate with 1:1 volume of 0.2 M Tris pH 8.0 by shaking and allow separation. Discard most Tris solution as phenol waste, leaving a small layer
- TE Buffer pH 7.5
10 mM Tris pH 7.5
1 mM EDTA pH 8.0
Acknowledgments
This protocol was adapted from Nepon-Sixt et al. (2019). The Alexandrow lab was supported by NIH grants R01-CA130865 and R21-CA155393.
Competing interests
All authors acknowledge that they have no competing financial or non-financial interests in the development of this manuscript.
References
- Nepon-Sixt, B. S., Bryant, V. L. and Alexandrow, M. G. (2019). Myc-driven chromatin accessibility regulates Cdc45 assembly into CMG helicases. Commun Biol 2: 110.
- Pemov, A., Bavykin, S. and Hamlin, J. L. (1998). Attachment to the nuclear matrix mediates specific alterations in chromatin structure. Proc Natl Acad Sci U S A 95(25): 14757-14762.
- Wong, P. G., Glozak, M. A., Cao, T. V., Vaziri, C., Seto, E. and Alexandrow, M. (2010). Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle 9(21): 4351-4363.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nepon-Sixt, B. S. and Alexandrow, M. G. (2019). DNase I Chromatin Accessibility Analysis. Bio-protocol 9(23): e3444. DOI: 10.21769/BioProtoc.3444.
Category
Cancer Biology > Oncogenesis > Ex vivo tissue culture model
Molecular Biology > DNA > Chromatin accessibility
Molecular Biology > DNA > DNA quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.