- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification and HPLC Analysis of Cell Wall Muropeptides from Caulobacter crescentus

Published: Vol 9, Iss 21, Nov 5, 2019 DOI: 10.21769/BioProtoc.3421 Views: 6423
Reviewed by: Alexandros AlexandratosMelike ÇağlayanSteven Boeynaems
Original research article
The authors used this protocol in:

Apr 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






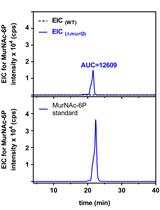

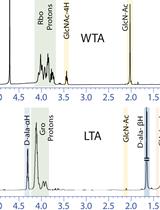
Abstract
The peptidoglycan sacculus, or cell wall, is what defines bacterial cell shape. Cell wall composition can be best characterized at the molecular level by digesting the peptidoglycan murein polymer into its muropeptide subunits and quantifying the abundance of muropeptides using high-pressure liquid chromatography. Certain features of the cell wall including muropeptide composition, glycan strand length, degree of crosslinking, type of crosslinking and other peptidoglycan modifications can be quantified using this approach. Well-established protocols provide us with highly-resolved and quantitatively reproducible chromatographic data, which can be used to investigate bacterial cell wall composition under a variety of environmental or genetic perturbations. The method described here enables the purification of muropeptide samples, their quantification by HPLC, and fraction collection for peak identification by mass spectrometry. Although the methods for peptidoglycan purification and HPLC analysis have been previously published, our method includes important details on how to re-equilibrate the column between runs to allow for automated analysis of multiple samples.
Keywords: LD-crosslinksBackground
Most bacteria are encased in a rigid mesh-like protective layer called peptidoglycan (PG) that is synthesized during cell growth and division. PG is a continuous polymer network made of glycan strands consisting of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) sugars linked by β (1→4) bonds. Peptide stems attached to the MurNAc sugars are the site of PG crosslinking. While the glycan sugars are largely-conserved in Gram-negative and Gram-positive species, the sugar backbone can be modified via N-deacetylation, N-glycolylation and O-acetylation. These modifications provide resistance to host defense mechanisms against infections, namely lysozyme resistance (Vollmer, 2008). By contrast, the compositions of the peptide strands vary significantly. During pentapeptide synthesis the bacterial family of Mur-ligases can introduce a variety of amino acids at each position of the pentapeptide stem. For example, at the third position of the pentapeptide most Gram-negative species incorporate meso-diaminopimelic acid (mDAP), while most Gram-positives insert L-lysine, although other variations have also been reported (Vollmer, 2008). Additionally, the peptide stem can be modified after synthesis. For instance, Corynebacteriales are reported to modify the third residue meso-Diaminopimelic acid via amidation once peptide synthesis has occurred (Levefaudes et al., 2015). Another aspect of peptidoglycan variation is the type of crosslinking used to connect multiple glycan strands (Schleifer and Kandler, 1972). Transpeptidation by DD-transpeptidases creates a 3-4 cross-linkage between third-position mDAP and fourth-position D-alanine. LD-transpeptidases, by contrast, form 3-3 crosslinks between two mDAP residues.
The sacculus, which provides the cell its architectural framework, also plays a large number of physiological roles in activating the immune system, aiding in colonization, evading antibiotics and host-responses, as well as inter-species recognition (Clarke and Weiser, 2011; Royet et al., 2011; Mesnage et al., 2014). Using high-resolution imaging (cryo-electron tomography and atomic force microscopy) and computational modeling, studies have begun to explain how cell wall composition effects the biophysical properties of cells (Tocheva et al., 2013; de Pedro and Cava, 2015). Genetic and cell biology approaches have demonstrated the role of LD-transpeptidation in mediating lysozyme resistance and bacterial virulence (Schleifer and Kandler, 1972; Schoonmaker et al., 2014; Stankeviciute et al., 2019). Thus, HPLC is a powerful tool to connect PG composition and structure to its physiological impact.
Materials and Reagents
- 50 ml polypropylene conical tubes (Nunc, catalog number: 339653)
- Micro magnetic stir bar (Science ware, catalog number: F37121-0012)
- 2.0 ml Safe-lock microcentrifuge tubes (Eppendorf, catalog number: 022363352)
- 1.7 ml microcentrifuge tubes (VWR, catalog number: 87003-294)
- MColorpHast pH test strips (pH 0-6) (Millipore Sigma, catalog number: 1095310001)
- 1 ml syringe (Becton Dickinson, catalog number: 309597)
- Millex-GV syringe filter unit, 0.22 µm (Millipore Sigma, catalog number: SLGV033R)
- 12 x 32 mm screw neck HPLC vials (Waters, catalog number: 186000273)
- Bottle top vacuum filter (VWR, catalog number: 10040-468)
- Caulobacter crescentus strain NA1,000 (Laboratory strain, available upon request)
- Bactopeptone (BD Biosciences, catalog number: 211677)
- Yeast extract (BD Biosciences, catalog number: 212750)
- Magnesium sulfate heptahydrate (MgSO4·7H2O) (Fisher, catalog number: BP213)
- Calcium chloride dihydrate (CaCl2·2H2O) (Fisher, catalog number: BP510)
- Ethylenediaminetetraacetic acid (EDTA) (Fisher, catalog number: BP118)
- Zinc sulfate heptahydrate (ZnSO4·7H2O) (Millipore Sigma, catalog number: Z0251)
- Iron sulfate heptahydrate (FeSO4·7H2O) (ACROS Organics, catalog number: 423730050)
- Manganese sulfate hydrate (MnSO4·7H2O) (Millipore Sigma, catalog number: M7634)
- Copper (II) sulfate pentahydrate (CuSO4·5H2O) (Millipore Sigma, catalog number: 209918)
- Cobalt (II) nitrate hexahydrate (Co(NO3)2·6H2O) (Millipore Sigma, catalog number: 239267)
- Sodium tetraborate decahydrate (Na2B4O7·10H2O) (Millipore Sigma, catalog number: B9876)
- Sulfuric acid (Millipore Sigma, catalog number: 320501)
- Nitrilotriacetic acid (C6H9NO6) (Millipore Sigma, catalog number: N9877)
- Ammonium heptamolybdate tetrahydrate (NH4Mo7O24) (Millipore Sigma, catalog number: 09878)
- Sodium phosphate dibasic (Na2HPO4) (Fisher, catalog number: BP331)
- Potassium phosphate monobasic (KH2PO4) (Fisher, catalog number: P285)
- Imidazole (Millipore Sigma, catalog number: I2399)
- Glucose (Fisher, catalog number: D16500)
- Sodium glutamate (Millipore Sigma, catalog number: 49621)
- Ammonium chloride (NH4Cl) (Fisher, catalog number: A661)
- Sodium dodecyl sulfate (SDS) (Millipore Sigma, catalog number: L3771)
- Pronase E (VWR, catalog number: VE629)
- Tris base (Fisher, catalog number: BP152)
- Sodium chloride (NaCl) (Fisher, catalog number: BP358)
- Mutanolysin from Streptomyces globisporus ATCC 21553 (Millipore Sigma, catalog number: M9901)
- Phosphoric acid (H3PO4) (Millipore Sigma, catalog number: 79607)
- Sodium hydroxide (NaOH) (Fisher, catalog number: S318)
- Boric acid (H3BO3) (Millipore Sigma, catalog number: B6768)
- Sodium borohydride (NaBH4) (Millipore Sigma, catalog number: 452882)
- HPLC-grade methanol (Millipore Sigma, catalog number: 34860)
- Sodium azide (NaN3) (Fisher, catalog number: BP9221)
- Peptone-Yeast Extract (PYE) media (see Recipes)
- Hutner-Imidazole-Glucose-Glutamate (HIGG) growth media (see Recipes)
- Concentrated Hutner base (per 1 L) (see Recipes)
- 0.5 M Phosphate buffer, pH 7.0 (see Recipes)
- HIGG media (see Recipes)
- Pronase E buffer (see Recipes)
- 50 mM phosphate buffer (pH 4.9) (see Recipes)
- 500 mM borate buffer (pH 9) (see Recipes)
- HPLC Buffer A (see Recipes)
- HPLC Buffer B (see Recipes)
Equipment
- ELGA PURELAB Flex 3 water purification system (ELGA LabWater)
- Magnetic hot plate-stirrer (Thermo Scientific, catalog number: SP195025)
- Sorvall Lynx 6000 Centrifuge (Thermo Scientific)
- 50 ml conical tube centrifuge rotor (Thermo Scientific, catalog number: Fiberlite F14-14x50cy)
- pH meter (Fisher, catalog number: 13-636-AB150)
- Sorvall WX 80+ ultracentrifuge (Thermo Scientific, catalog number: 75000080)
- Swinging bucket rotor TH-641 (Thermo Scientific, catalog number: 54295)
- 14 x 89 mm thickwall polycarbonate tube for TH-641 rotor (Seton Scientific 2013)
- Fixed angle rotor T-1270 (Thermo Scientific, catalog number: 08259)
- 16 x 76 mm thickwall polycarbonate tube for T-1270 rotor (Seton Scientific 2004)
- Digital dry bath (Fisher, catalog number: 88-871-001)
- 6 x 1.5 ml block for dry bath (Fisher, catalog number: 88-871-103)
- 6 x 2.0 ml block for dry bath (Fisher, catalog number: 88-871-104)
- Microcentrifuge (Eppendorf, model: 5424)
- Glass culture tubes (13 x 100 mm) (Fisher, catalog number: 14-961-27)
- Microspatula (Fisher, catalog number: S50823)
- Vortexer (Fisher, catalog number: 02-215-414)
- Agilent 1260 Infinity II HPLC system including:
- Agilent 1260 Infinity II Quaternary pump
- Agilent 1260 Infinity II Multisampler
- Agilent 1260 Infinity II Multicolumn thermostat
- Agilent 1260 Infinity II Diode array detector (DAD)
- Agilent 1260 Infinity II Quaternary Pump
- Agilent 1260 Infinity II Analytical-scale fraction collector
- Hypersil ODS C18 HPLC column (3 µm particles, 4.6 mm diameter, 250 mm length) (Thermo Scientific, catalog number: 30103-254630)
- Uniguard guard cartridge holder (Thermo Scientific, catalog number: 850-00)
- Hypersil ODS C18 guard cartridge (3 µm particles, 4 mm diameter, 10 mm length) (Thermo Scientific, catalog number: 30103-014001)
- Vacufuge concentrator (Eppendorf, model: 022820109)
Software
- Matlab R2018b (Mathworks)
- Chromanalysis v. 1.0 (Desmarais et al., 2015)
Procedure
This procedure is largely based on previously described protocols (Glauner, 1988; Desmarais et al., 2014). We include additional details for muropeptide separation by HPLC as well as methods for running consecutive samples with column cleaning and re-equilibration steps.
- Inoculation and growth of bacterial cultures
- Inoculate 2.5 ml PYE media with a single colony of Caulobacter crescentus from an agar plate or directly from a freeze-down culture and grow overnight at 30 °C with shaking at 200 rpm.
- The next day, back-dilute the culture into 500 ml of HIGG media (containing the desired phosphate concentration). Incubate this culture for 48 h at 30 °C with 200 rpm shaking.
- Muropeptide purification
Day 1–Cell lysis- Bring 700 ml of water to a boil in a glass beaker on a stirrer-hot plate set to 450 °C. For each culture to be processed, prepare a 50 ml conical tube containing 6 ml of 6% (w/v) SDS and a micro stir bar, place the tube in the boiling water, and turn on the magnetic stirrer to 300 rpm (Figure 1).

Figure 1. Cell lysis. Cells in 4% SDS are boiled with stirring for 3 h. The arrow indicates the micro stir bar. - Harvest the cells after 48 h by centrifugation at 12,000 x g for 10 min (4 °C). Decant the supernatant. The starved cells are very buoyant and some of the pellet will slip away.
- Resuspend the pellet in 3 ml of HIGG media and quickly transfer to the preheated 50 ml conical tubes to yield a final concentration of 4% SDS (w/v).
- Boil the samples for 3 h with continuous stirring, periodically adding water to the beaker when necessary. Do not remove from heat too early as this step is critical for complete cell lysis as well as the inactivation of cell wall altering enzymes. After 3 h, turn off the heat and continue to stir the lysed cultures overnight.
Day 2–Enzymatic digestions- Check whether the SDS has precipitated overnight; if so, boil the sample for one hour to redissolve the SDS. Collect the sacculi by centrifugation at 276,000 x g for 1 h in a TH-641 rotor. Spin at 20 °C to avoid SDS precipitation. Resuspend the pellet in 6.5 ml ultrapure water and centrifuge the sample at 329,738 x g for 30 min (20 °C) in a T-1270 rotor.
- Repeat this washing two times. If bubbles are still seen in the sample, add an extra wash to fully remove SDS (Figure 2).

Figure 2. SDS removal. Samples washed in ultra-pure water should be transparent (left tube) and have lost the sudsy appearance with multiple washes (right tube). - Prepare 1 mg/ml Pronase E in 10 mM Tris-HCl (pH 7.2) + 0.06% w/v NaCl. Activate the Pronase E by heating the stock solution at 60 °C in a heat block for a minimum of 30 min (Figure 3).
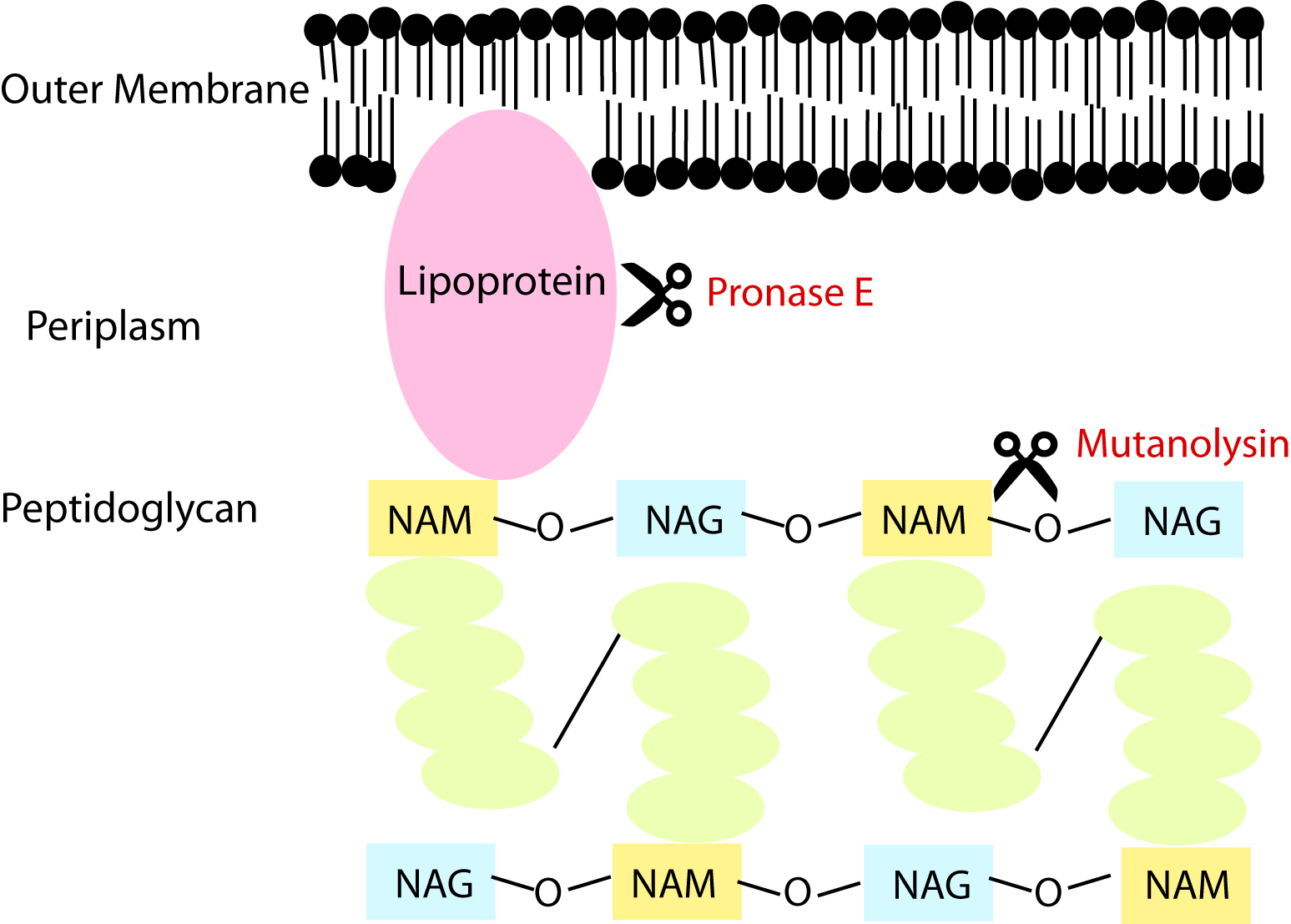
Figure 3. Enzymatic digestions. The cell envelope is digested with Pronase E to eliminate lipoproteins linking the outer membrane to the peptidoglycan. The intact sacculus is digested into muropeptide fragments that cleave along the glycan backbone. - Resuspend each sample pellet in 900 µl of 10 mM Tris-HCl (pH 7.2) + 0.06% w/v NaCl and transfer to a safe-lock microcentrifuge tube. Add 100 µl of the activated Pronase E to the tube and incubate for a minimum of 2 h on a 60 °C heat block. Pronase E is a protease that will digest lipoproteins associated with the peptidoglycan.
- Add 200 µl of 6% SDS (w/v) to the digested samples and boil them in a 100 °C dry bath for 30 min to stop the proteolysis.
- Resuspend the sacculi in 6.5 ml ultrapure water and centrifuge them at 329,738 x g for 30 min (20 °C) in a T-1270 rotor to wash out the SDS. Repeat the washing two times as before (or as long as necessary to eliminate any bubbles).
- Prepare 1 mg/ml mutanolysin stocks in 50 mM phosphate buffer (pH 4.9), aliquot 40-50 µl into microcentrifuge tubes, and store the tubes at -20 °C.
- After the last wash, resuspend the peptidoglycan pellet in 75-200 µl of 50 mM sodium phosphate buffer (pH 4.9) and transfer the sample to a 1.7 ml microcentrifuge tube. This volume will depend on how much peptidoglycan material is in the sample. For example, sacculi from a 500 ml C. crescentus culture at OD660 of 0.8-1.0 were resuspended in 200 µl. Add mutanolysin to the sample to a final concentration of 40 µg/ml and digest overnight in a 37 °C dry bath (at least 10 h).
Day 3–Sample preparation for HPLC- Inactivate the muramidase by boiling the samples in a 100 °C dry bath for 5 min.
- Remove undigested material by centrifugation at 16,000 x g at room temperature for 10 min. Carefully transfer the supernatant into a 13 x 100 mm glass tube without disturbing the pellet.
- Add 500 mM Borate buffer (pH 9) to the supernatant to a final concentration of 100 mM.
- Add approximately half a micro-spatula amount (~ 6 mg) of sodium borohydride into each sample (Figure 4). Vigorous bubbling should be seen immediately, after which small bubbles will continue to rise in the solution. Reduce the muropeptides at room temperature for a minimum of 30 min. Vortex the tube gently to ensure complete mixing of the sodium borohydride.
Caution: If too much sodium borohydride is added, it will precipitate during the pH titration and this will ruin the muropeptide purification.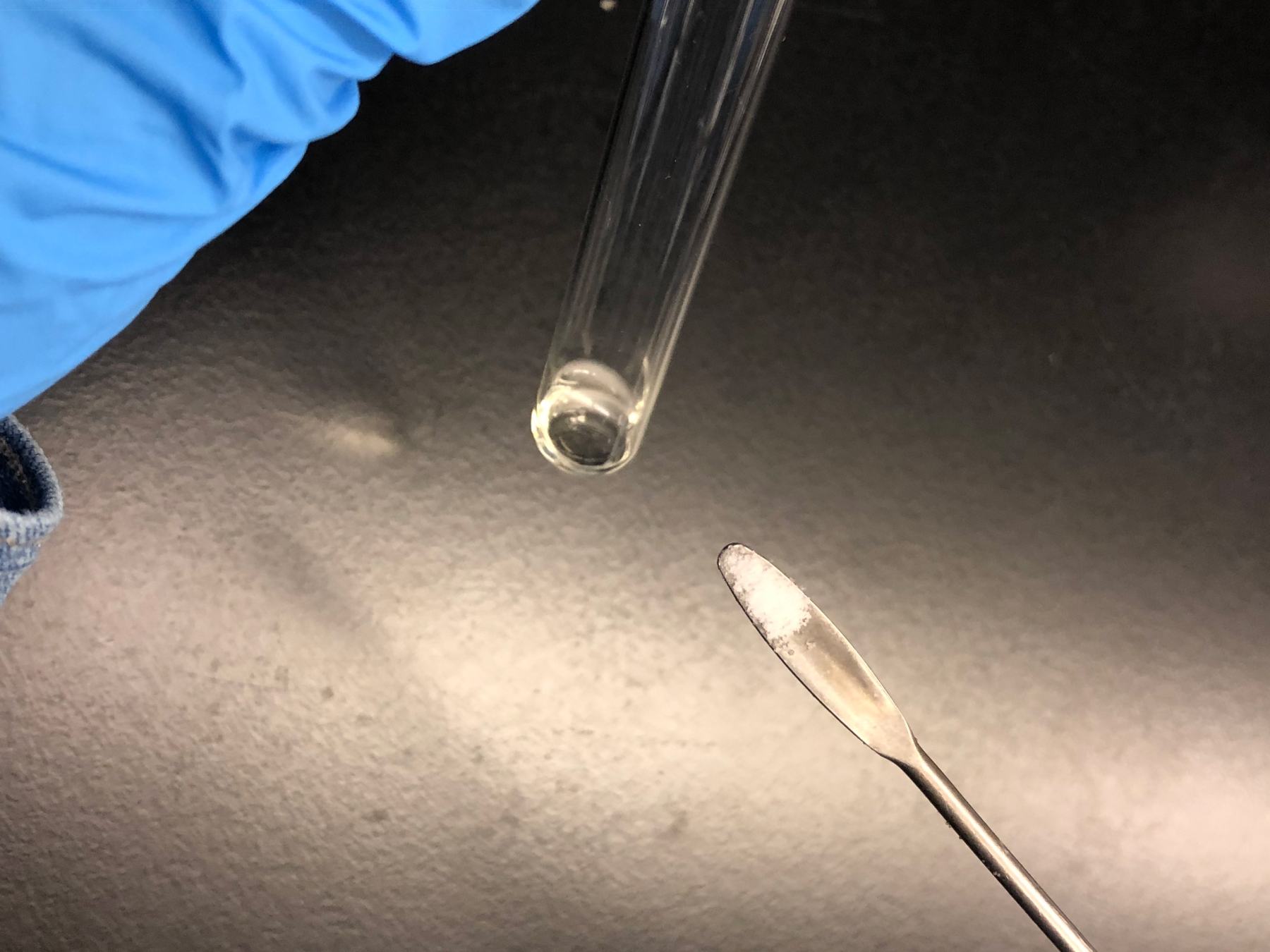
Figure 4. Reduction of muropeptides with sodium borohydride. Approximately 0.6 mg of sodium borohydride (half a micro-spatula) is added to the sample for muropeptide reduction. - Acidify the reduced muropeptides to pH 3.0-4.0 (the isoelectric point of the muropeptides is approximately 3.5). Start by adding 15 µl of 50% phosphoric acid (vigorous bubbling will be seen) and gently vortex the tube to ensure thorough mixing. Test the pH by placing a 1 µl drop of sample onto the pH indicator paper. Continue to add phosphoric acid in increments of 5 µl until the pH is 5.5-6.0 (typically 1-2 additions). Continue adding phosphoric acid in increments of 1 µl until reaching a pH between 3.0 and 4.0.
- Transfer the samples to a 1 ml syringe and filter them through a Millex-GV syringe filter directly into a 12 x 32 mm HPLC vial. Samples can be stored long-term at -20 °C.
- Bring 700 ml of water to a boil in a glass beaker on a stirrer-hot plate set to 450 °C. For each culture to be processed, prepare a 50 ml conical tube containing 6 ml of 6% (w/v) SDS and a micro stir bar, place the tube in the boiling water, and turn on the magnetic stirrer to 300 rpm (Figure 1).
- HPLC set-up and sample analysis
- While directing flow into the waste container, wash all 4 pump lines of the Agilent HPLC system with ultra-pure water for a minimum of 10 min. Close the waste valve and wash the internal tubing by flushing with ultra-pure water for an additional 10 min.
- Connect pump A with Solvent A (Recipe 9), pump B with Solvent B (Recipe 9), pump C with HPLC-grade methanol and pump D with ultra-pure water. While directing flow to the waste container, flush each line with its respective solvent.
- Set the temperature of the column incubator to 55 °C. Close the waste valve and set the flow-rate to 0.5 ml/min from Line D (100% Ultra-pure water).
- Prepare the HPLC column by first attaching the Uniguard column containing a guard cartridge. Attach the column to the HPLC system by first allowing 5-6 drops of water to go into the upstream end of the column. Firmly tighten the upstream connector and wait for the water to drip from the downstream end before connecting the other end of the column.
- Wash the column with ultrapure water for 20 min and ensure that the column pressure does not exceed 150 bar. Set the DAD detector to collect the absorbance at 205 nm.
- Equilibrate the column with Solvent A at 0.5 ml/min for 50 min. The column pressure and UV absorbance will stabilize during this step.
- Prepare the HPLC samples as follows. Place a 100 µl insert into a clean HPLC vial and add 50-90 µl of sample to the insert. Prepare 2 vials with ultrapure water which will be used to blank the system as well as during cleanup. Arrange the vials in the autosampler unit.
- If collecting peaks for further analysis by mass spectrometry, enable time-based fraction collection to collect the desired peaks. Place the appropriate number of collection tubes in the fraction collector according to the number of collected peaks. Each tube will collect approximately 100-450 µl of sample depending on the duration of the fraction collection window (10-50 s). Since the elution time can vary slightly from run to run, we recommend collecting extra fractions before and after the expected elution time to ensure recovery of the desired peak.
- The HPLC method consists of a 135 min linear gradient (0.5 ml/min) from 100% Solvent A to 100% Solvent B. To re-equilibrate the column between samples, continue flowing 100% Solvent B (0.5 ml/min) for 5 min, followed by a 25 min linear gradient to 100% Solvent A, and finally an additional 25 min of 100% Solvent A. At the end of this washing protocol, the pressure and absorbance at 205 nm should be stabilized.
- Prior to running muropeptide samples, inject 50 µl of ultra-pure water into the HPLC system to serve as a blank for background subtraction. For the muropeptide samples, inject 40-80 µl; be certain not to inject more than the total volume of the sample to ensure that an air bubble is not introduced into the HPLC system.
- After the last sample has completed, wash the system and the column by injecting 20 µl of ultra-pure water and running the following washing method. Begin with a 30 min flush with 100% water (0.2 ml/min), followed by a 10 min linear gradient to 100% methanol. Wash with 100% methanol for 60 min followed by a 10 min linear gradient to 10% methanol/90% water. Flush the column with 10% methanol/90% water for an additional 10 min before removing the column for storage.
- If fractions were collected, combine the fractions containing the peak of interest and remove the solvent using a vacufuge concentrator. Set the vent mode to ‘aqueous’ and spin the samples in the vacufuge concentrator until dry, typically 30-60 min at 30 °C. A small translucent, white pellet may be seen on the bottom of the tube. These samples are suitable for LC/MS analysis.
- The HPLC trace can be quantified using the Matlab-based Chromanalysis software as previously described (Desmarais et al., 2015).
Data analysis
The protocol above can be used to compare the muropeptide compositions of bacteria growing in different nutrient environments. For C. crescentus, phosphate starvation leads to an increase in LD-transpeptidation (Figure 5). 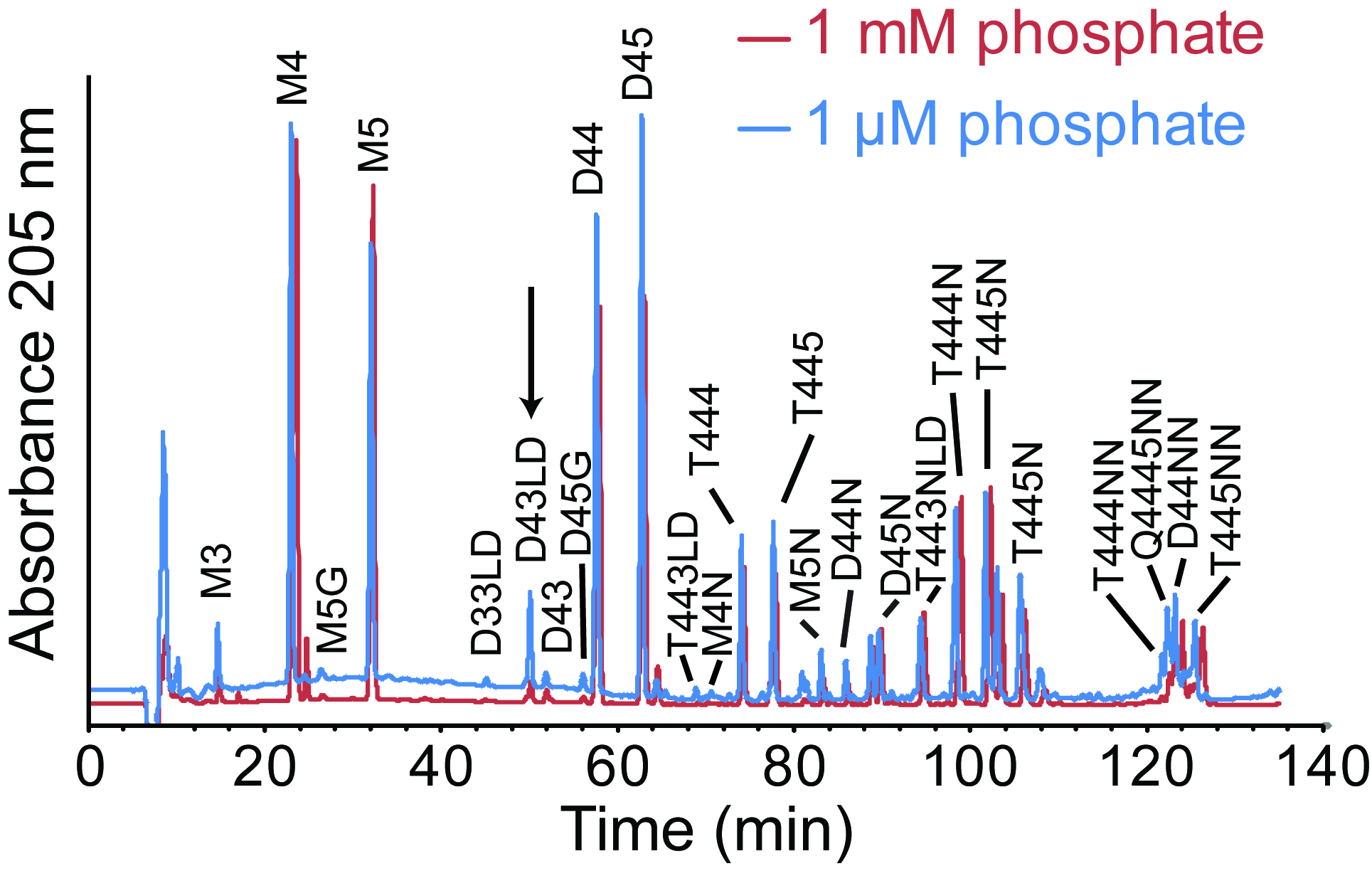
Figure 5. Muropeptide analysis of C. crescentus grown in different phosphate concentrations. Muropeptides were purified from Caulobacter crescentus grown in HIGG media with 1 mM or 1 µM phosphate and analyzed via HPLC. The arrow indicates a peak uniquely present in the 1 µM phosphate chromatogram. Major peaks were labeled based on previous analysis of C. crescentus muropeptides (Takacs et al., 2010). LD-crosslinks were labeled based on previous analysis of E. coli muropeptides (Glauner, 1988) and confirmed by LC/MS. This figure is reprinted with permission from John Wiley and Sons (Stankeviciute et al., 2019).
Notes
- The protocol for operating the HPLC system may vary depending on your particular instrumentation. For an overview of HPLC operation, we recommend the article and accompanying video from the JoVE Science Education Database (https://www.jove.com/science-education/10156/high-performance-liquid-chromatography-hplc).
- Prior to peak quantification, perform background subtraction using the blank-run (water sample).
Recipes
- Peptone-Yeast Extract (PYE) media (1 L)
2.0 g peptone
1.0 g yeast extract
1 mM MgSO4
0.5 mM CaCl2 - Hutner-Imidazole-Glucose-Glutamate (HIGG) media (Poindexter, 1978)
There are several stock solutions required for this media:- Metals “44” (per 100 ml)
1.3 ml 0.5 M disodium EDTA
1,095.0 mg ZnSO4·7H2O
500.0 mg FeSO4·7H2O
154.0 mg MnSO4·H2O
39.2 mg CuSO4·5H2O
24.8 mg Co(NO3)2·6H2O
17.7 mg Na2B4O7·10H2O
Bring volume to 100 ml with distilled water
Add 2-3 drops of sulfuric acid to prevent precipitation - Concentrated Hutner base (per 1 L)
10.0 g Nitrilotriacetic acid
29.59 g MgSO4·7H2O
3.335 g CaCl2·2H2O
9.25 mg (NH4)6Mo7O24·4H2O
99.0 mg FeSO4·7H2O
50.0 ml Metals “44”
Distilled water to 1,000 ml- Before adding all reagents, dissolve nitrilotriacetic acid and neutralize with KOH; approximately 5.6 g KOH will be required
- Add the rest of the components and adjust the pH to 6.6-6.8 before adjusting the volume
- Bring the solution to 1,000 ml with distilled water
- Sterilize the solution by filtration (do not autoclave)
- 0.5 M Phosphate buffer, pH 7.0
Mix 61 ml of 0.5 M Na2HPO4 with 39 ml of 0.5 M KH2PO4 and sterilize by autoclaving - 1 M imidazole, pH 7.0 (autoclaved)
- 20% (w/v) glucose (sterile filtered)
- 20% (w/v) sodium glutamate (sterile filtered)
- 50 mM CaCl2 (autoclaved)
- 1 M NH4Cl (autoclaved)
5 mM imidazole, pH 7.0
2% (v/v) Hutner’s base
1 mM CaCl2
0.15% (w/v) glucose
0.15% (w/v) sodium glutamate
8.9 mM NH4Cl
1 μM-1,000 μM phosphate - Metals “44” (per 100 ml)
- Pronase E buffer
10 mM Tris, pH 7.2
0.06% (w/v) NaCl - 50 mM phosphate buffer, pH 4.9 (100 ml)
- Dissolve 0.2 g NaOH in ~90 ml water
- Adjust the pH of the solution to 4.9 with phosphoric acid
- Bring the total volume to 100 ml with water
- 500 mM borate buffer, pH 9 (500 ml)
- Dissolve 15.46 g boric acid in ~480 ml water
- Adjust the pH to 9.0 with NaOH
- Bring the total volume to 500 ml with water
- HPLC buffers
Buffer A: 50 mM NaPO4 pH 4.35, 0.4% NaN3- Dissolve 2.0 g NaOH into ~950 ml ultrapure water
- Adjust the pH to 4.35 with phosphoric acid
- Add 200 µl 2% (w/v) NaN3
- Bring the total volume to 1,000 ml with ultrapure water
- Re-adjust the pH to 4.35
- Sterile filter before use
- Dissolve 3.0 g NaOH into ~800 ml ultrapure water
- Adjust the pH to 4.95 with phosphoric acid
- Bring the total volume to 850 ml with ultrapure water
- Add 150 ml HPLC-grade methanol
- Re-adjust the pH to 4.95
- Sterile filter before use
Acknowledgments
We thank Amanda Miguel and K.C. Huang (Stanford University) for their guidance in muropeptide purification. We have generally adapted their protocol for UPLC-based separation (Desmarais et al., 2014) for use on an HPLC system. This work was supported by a grant from the National Science Foundation (MCB-1553004) to E.A.K.
Competing interests
The authors have no competing interests to declare.
References
- Clarke, T. B. and Weiser, J. N. (2011). Intracellular sensors of extracellular bacteria. Immunol Rev 243(1): 9-25.
- de Pedro, M. A. and Cava, F. (2015). Structural constraints and dynamics of bacterial cell wall architecture. Front Microbiol 6: 449.
- Desmarais, S. M., Cava, F., de Pedro, M. A. and Huang, K. C. (2014). Isolation and preparation of bacterial cell walls for compositional analysis by ultra performance liquid chromatography. J Vis Exp 83: e51183.
- Desmarais, S. M., Tropini, C., Miguel, A., Cava, F., Monds, R. D., de Pedro, M. A. and Huang, K. C. (2015). High-throughput, highly sensitive analyses of bacterial morphogenesis using ultra performance liquid chromatography. J Biol Chem 290(52): 31090-100.
- Glauner, B. (1988). Separation and quantification of muropeptides with high-performance liquid chromatography. Anal Biochem 172(2): 451-64.
- Levefaudes, M., Patin, D., de Sousa-d'Auria, C., Chami, M., Blanot, D., Herve, M., Arthur, M., Houssin, C. and Mengin-Lecreulx, D. (2015). Diaminopimelic acid amidation in Corynebacteriales: new insights into the role of LtsA in peptidoglycan modification. J Biol Chem 290(21): 13079-94.
- Mesnage, S., Dellarole, M., Baxter, N. J., Rouget, J. B., Dimitrov, J. D., Wang, N., Fujimoto, Y., Hounslow, A. M., Lacroix-Desmazes, S., Fukase, K., Foster, S. J. and Williamson, M. P. (2014). Molecular basis for bacterial peptidoglycan recognition by LysM domains. Nat Commun 5: 4269.
- Poindexter, J. S. (1978). Selection for nonbuoyant morphological mutants of Caulobacter crescentus. J Bacteriol 135(3): 1141-5.
- Royet, J., Gupta, D. and Dziarski, R. (2011). Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nat Rev Immunol 11(12): 837-51.
- Schleifer, K. H. and Kandler, O. (1972). Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36(4): 407-77.
- Schoonmaker, M. K., Bishai, W. R. and Lamichhane, G. (2014). Nonclassical transpeptidases of Mycobacterium tuberculosis alter cell size, morphology, the cytosolic matrix, protein localization, virulence, and resistance to beta-lactams. J Bacteriol 196(7): 1394-402.
- Stankeviciute, G., Miguel, A. V., Radkov, A., Chou, S., Huang, K. C. and Klein, E. A. (2019). Differential modes of crosslinking establish spatially distinct regions of peptidoglycan in Caulobacter crescentus. Mol Microbiol 111(4): 995-1008.
- Takacs, C. N., Poggio, S., Charbon, G., Pucheault, M., Vollmer, W. and Jacobs-Wagner, C. (2010). MreB drives de novo rod morphogenesis in Caulobacter crescentus via remodeling of the cell wall. J Bacteriol 192(6): 1671-84.
- Tocheva, E. I., Lopez-Garrido, J., Hughes, H. V., Fredlund, J., Kuru, E., Vannieuwenhze, M. S., Brun, Y. V., Pogliano, K. and Jensen, G. J. (2013). Peptidoglycan transformations during Bacillus subtilis sporulation. Mol Microbiol 88(4): 673-86.
- Vollmer, W. (2008). Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol Rev 32(2): 287-306.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Stankeviciute, G. and Klein, E. A. (2019). Purification and HPLC Analysis of Cell Wall Muropeptides from Caulobacter crescentus. Bio-protocol 9(21): e3421. DOI: 10.21769/BioProtoc.3421.
Category
Microbiology > Microbial physiology > Cell wall
Biochemistry > Carbohydrate > Peptidoglycan
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.