- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Non-aqueous Fractionation (NAF) for Metabolite Analysis in Subcellular Compartments of Arabidopsis Leaf Tissues
Published: Vol 9, Iss 20, Oct 20, 2019 DOI: 10.21769/BioProtoc.3399 Views: 5658
Reviewed by: Ancheng HuangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The accurate determination of metabolite distribution in subcellular compartments is still challenging in plant science. Various methodologies, such as fluorescence resonance energy transfer-based technology, nuclear magnetic resonance spectroscopy and protoplast fractionation allow the study of metabolite compartmentation. However, large changes in metabolite levels occur during such procedures. Therefore, the non-aqueous fractionation (NAF) technique is currently the best method for the study of in-vivo metabolite distribution. Our protocol presents a detailed workflow including the NAF procedure and quantification of compartment-specific markers for three subcellular compartments: ADP glucose pyrophosphorylase (AGPase) as plastidic marker, phosphoenolpyruvate carboxylase (PEPC) as cytosolic marker, and nitrate and acid invertase as vacuolar markers.
Keywords: In-vivo metabolite distributionBackground
Metabolic activity and functionality require precise compartmentation of metabolism in eukaryotic cells. Despite knowing the spatial organization of metabolism within the plant cells, deciphering localization of the metabolites is still challenging due to metabolite redundancy, leakage, rapid turnover, futile cycling, and sophisticated transport systems and storage (Lunn, 2007; Klie et al., 2011; Sweetlove and Fernie, 2013). Several methods have been established to investigate metabolite compartmentation, among them non-aqueous fractionation (NAF) has been widely used for decades to determine the in-vivo subcellular metabolite distributions in eukaryotic cells. Firstly developed in animal sciences (Elbers et al., 1974), NAF was subsequently applied to plant leaf material from spinach (Gerhardt and Heldt, 1984 and 1987), bean (Sharkey and Vanderveer, 1989), maize (Weiner and Heldt, 1992) or barley (Winter et al., 1993). NAF is also successfully applied to rose petals (Yamada et al., 2009) and potato tubers (Farre et al., 2001). More recently, NAF has also been optimized to study metabolite compartmentation in the model plant Arabidopsis thaliana (Fettke et al., 2005; Krueger et al., 2009 and 2011; Klie et al., 2011; Arrivault et al., 2014; Shapiguzov et al., 2019). The NAF procedure prevents modification of metabolite levels (for example due to enzymatic activity) by having all steps performed at extremely low temperatures or under anhydrous conditions (Gerhardt and Heldt, 1984). After quenching plant material in liquid nitrogen, the frozen material is dehydrated by freeze drying, homogenized, and then applied to a gradient made of anhydrous organic solvents prior to centrifugation and subsequent collection of fractions. Subcellular compartments are separated on the basis of density in the centrifugation step and the fractions collected are enriched in organelles (such as chloroplasts, cytosol or vacuole; Gerhardt and Heldt, 1984; Lunn, 2006). By correlating the compartment-specific marker distributions with metabolite distributions over the gradient, it is possible to calculate compartment-specific metabolite distributions (Gerhardt and Heldt, 1984; Klie et al., 2011; Krueger et al., 2011). The software tool BestFit enables such calculations (Krueger et al., 2011 and 2014; Klie et al., 2011). Recently, a benchtop NAF method has been established and is adapted for a relatively small amount of tissue material (Fürtauer et al., 2016). This benchtop NAF method can be used as an alternative to the conventional NAF procedure. However, we still believe that the conventional NAF procedure is the method of choice for the determination of in vivo metabolite distribution among cellular compartments as (i) the plastidic and vacuolar compartments are more clearly separated on the density gradient used, and (ii) the higher amount of starting material leads to more material in each obtained fraction, giving the advantage to increase the number of sub-aliquots per fraction (thus increasing the range of measurements which can be done) and also to apply less sensitive quantification methods than mass spectrometry-based methods. A good example of the use of the conventional NAF procedure is a study performed by Arrivault et al. (2014) in which the distribution of about 1,000 proteins and 70 metabolites, including 22 phosphorylated intermediates in Arabidopsis thaliana rosette leaves were analyzed, using the conventional NAF combined with MS-based approaches.
The protocol presented here is based on the method described by Krueger et al. (2014) with minor modifications. In our method, we provide a complete workflow for the NAF procedure in Arabidopsis thaliana and detailed information for the analysis of compartment–specific marker enzymes and metabolites which can be assigned to three subcellular compartments: ADP glucose pyrophosphorylase (AGPase) as plastidic marker, phosphoenolpyruvate carboxylase (PEPC) as cytosolic marker and nitrate and acid invertase as vacuolar markers.
Materials and Reagents
- NAF
- Plastic lids (Zinsser Analytic, catalog number: 3071400)
- 20 ml Polyvial–one per sample (Zinsser Analytic, catalog number: 3071400)
- Steel balls:
3 mm (Askubal, catalog number: 3 mm G100 1.4034)
5 mm (Askubal, catalog number: 5 mm G100 1.4034)
7 mm (Altmann, catalog number: KU.7G80-1.3541)
Note: We recommend using steel balls of three different diameters 3, 5, and 7 mm (two of each size in each 20 ml Polyvial). - Steel caps (custom made, please check Figure 1)
- 50 ml Falcon tubes–10 per sample (SARSTEDT, catalog number: 62.554.502)
- Paper tissue (KimWipes)
- pH-indicator paper (Merck, catalog number: 1.10962)
- Polyallomer centrifuge tubes (Beckman Coulter, catalog number: 326823)
- Nylon mesh filter 20 µm pore size (SEFAR Nitex®, catalog number: 03-20/14)
- 1.5 ml screw cap tubes–11 per sample (SARSTEDT, catalog number: 72.692)
- Activated molecular sieve 4 Å (Carl Roth, catalog number: 1318-02-1)
- Plastic Pasteur pipette (NeoLab, catalog number: 2600111)
- Glass pipettes (10 ml, 20 ml)
- Glass funnel
- Metal rack
- Arabidopsis thaliana leaf material
- Liquid nitrogen
- n-Heptane (Carl Roth, catalog number: 142-82-5)
- Tetrachloroethylene (Carl Roth, catalog number: 127-18-4)
- Liquid nitrogen (~5 L per sample)
Note: the required liquid nitrogen for harvesting (1 L), grinding (2 L), and aliquoting (2 L) samples suggested here is a rough estimate, since the amount of nitrogen needed will depend on several factors. Please consider that while planning your experiments.
- Measurement of metabolite markers
- 96 Deep-well plate (Sarstedt, catalog number: 82.1970.002)
- Steel balls (5 mm)
- Tubes holder for the Retsch mill (Retsch, catalog number: 22.008.0008)
- Ethanol (VMR Chemicals, catalog number: 20821.330)
- HEPES (CAS: 7365-45-9)
- NaOH (Carl Roth, catalog number: 6771.1)
- Potassium phosphate buffer 1 M, pH 7.5
- K2HPO4 (Sigma-Aldrich, CAS: 7758-11-4)
- KH2PO4 (Sigma-Aldrich, CAS: 7778-77-0)
- NADPH (Roche, CAS: 2646-71-1)
- Nitrate reductase (Roche, catalog number: 10981249001)
- Phenazine ethosulfate (PES) (Sigma-Aldrich, CAS: 10510-77-7)
- Sulfanilamide (Sigma-Aldrich, CAS: 63-74-1)
- N-(1-Naphthyl)ethylenediamine dihydrochloride (NNEDA) (Sigma-Aldrich, CAS: 1465-25-4)
- KNO3 (Sigma-Aldrich, CAS: 7757-79-1)
- Nitrate Standards (see Recipes)
0, 0.2, 0.4, 0.8, and 1.6 mM in 70% ETOH with 10 mM MES pH 5.9
- Measurement of enzyme markers
For the full description and order details of the reagents used in the measurements of enzyme activities, please see Supplemental Table 1.- PVPP
- Deionized water
- MgCl2
- EDTA
- EGTA
- Benzamidine
- ε-aminocapronic acid
- BSA (protease free)
- Leupeptin
- DTT
- PMSF
- Triton X-100
- Glycerol
- NaF
- 3-phosphoglycerate
- AGPase assay mix
- PPi
- Glycerokinase
- ADP-glucose
- GPOX
- GDH
- NaHCO3
- PEP
- Malate dehydrogenase
- MTT
- Alcohol Dehydrogenase
- Acid invertase assay Buffer
- Glucose oxidase
- Horse Radish Peroxidase
- Amplex Red
- DMSO
- Nitrate standards (see Recipes)
- Extraction buffers (see Recipes)
- Extraction Buffer 10x
- Extraction Buffer 1x
- AGPase activity (see Recipes)
- AGPase assay buffer
- AGPase assay mix
- AGPase determination mix
- PEPC activity (see Recipes)
- PEPC assay Buffer
- PEPC assay Mix
- PEPC determination mix
- PEPC assay Buffer
- Acid Invertase activity (see Recipes)
- Acid invertase assay Buffer
- Acid invertase Assay Mix
- Acid invertase Determination mix
Equipment
- NAF
- Capper/decapper machine (custom made, see Figure 1)
- Retsch mill (Retsch®, model: MM400)
- Freeze dryer (Christ, model: Alpha 2-4)
- Sonicator (BANDELIN SONOPULS HD 2070)
- Peristaltic gradient pump (Bio-Rad Econo Gradient Pump, catalog number: 731-9002)
- Gradient mixer (Bio-Rad Gradient mixer, catalog number: 731-8323)
- Centrifuge (Beckman Coulter, model: Allegra X-15R)
- Rotor (Beckman Coulter, model: SX4750A)
- Ultracentrifuge (Beckman Coulter, model: OptimaTM L80 XP)
- Swinging bucket rotor (Beckman Coulter, model: SW32Ti)
- Desiccator
- Vacuum pump
- Measurement of metabolite markers
- Thermomixer (Eppendorf, model: Compact 5350)
- Centrifuge (Eppendorf, model: 5430)
- Microplate Spectrophotometer (BioTeK®, Epoch 2)
- Measurement of enzyme markers
- Microplate Spectrophotometer (BioTeK®, ELx808)
- Microplate Fluorescence reader (BioTeK®, Synergy HT)
Software
- BestFit (Klie et al., 2011; http://www.csbdb.de/csbdb/bestfit/bestfit.html)
Procedure
- Harvesting, grinding and preparation of leaf material
- Harvest Arabidopsis thaliana leaf material and place into a 20 ml Polyvial (containing steel balls of three different diameters 3, 5, and 7 mm, two of each size in each 20 ml Polyvial) pre-cooled in liquid N2 and snap-freeze in liquid N2.
Note: If the metabolites of interest have extremely fast turn-over time, plant material should be harvested by cutting rosettes and quenching them instantaneously in a bath of liquid N2 under the prevailing irradiance. The quenched material is then transferred into a 20 ml Polyvial (containing steel balls) pre-cooled in liquid N2. For an optimal grinding process, fill the vial to a maximum of 2/3 of its volume with the plant material. For NAF, 4 g of material is necessary, but it is recommended to harvest slightly more in order to have extra material to perform additional analysis in this material (such as quantification of metabolites, see section Data analysis–4). - For homogenization, replace the plastic lids of the Polyvials by steel caps and place the Polyvials containing the leaf material into a pre-cooled steel grinding adapter for the Retsch mill (Figures 1A-1D).
Note: Our grinding adapters and steel caps are custom-made for the 20 ml Polyvials. Alternatively, the grinding of the leaf material can be performed by using a pre-cooled mortar and pestle. Make sure that the leaf material is kept frozen during the whole procedure. - Homogenize the tissue for 1 min at 25 Hz (two times) in the Retsch mill (Figures 1E and 1F).
Note: Make sure that the ground tissue is very well homogenized (thin powder). We suggest grinding the leaf material twice for 1 min each at 25 Hz. Cool down the vials and adapters in between with liquid N2 to avoid thawing of the material.
Figure 1. Grinding procedure. A. Cool down the Polyvials, steel grinding adapter, and steel caps in liquid nitrogen. B. Remove the plastic lid of the Polyvials and place the steel cap frozen. C. Use a capper/decapper machine (custom-made) to close the vial with the steel cap. D. Closed vial. E. Place the Polyvial inside steel grinding adapter. F. Place the grinding adapter in the Retsch mill and proceed as in Step A3. After grinding use the same capper/decapper machine for replacing the steel caps for the plastic lids. The samples can be stored at -80 °C or proceed to Step A4. Note that to avoid overheating of the vial, steel caps and grinding adapter deep them in liquid nitrogen or make use of dried ice while preparing a second sample. - Weigh ~4 g of frozen tissue into a 50 ml Falcon tube pre-cooled in liquid N2.The samples can be stored at -80 °C for up to three months. Prepare as many Falcon tubes as necessary (each one will be used for one NAF).
Note: Label the Falcon tubes and their corresponding lids. Record the weights of the empty Falcon tubes (with lids) and the precise amount of plant material weighed. - Freeze dry the homogenate. To avoid losing leaf material during this process, remove the lid and cover the Falcon tubes with paper tissue (KimWipes) fixed with an elastic band. Place the tubes in a metal rack into the freeze drier at 0.02 bar and -80 °C for 5 days. Close the tubes with their corresponding lids immediately after removing them from the freeze drier. The dried material can be stored under vacuum in a desiccator protected from light and humidity up to 6 months.
Note: In order to avoid thawing, place the Falcon tubes in a polystyrene box containing dry ice and replace the lids by the paper tissue while the Falcon tubes are in the box. - Determine the plant material dry weight by weighing the closed Falcon tubes after the freeze drying process.
- Harvest Arabidopsis thaliana leaf material and place into a 20 ml Polyvial (containing steel balls of three different diameters 3, 5, and 7 mm, two of each size in each 20 ml Polyvial) pre-cooled in liquid N2 and snap-freeze in liquid N2.
- Preparation of solutions and filters
- Prepare the solutions for the NAF three days before starting the fractionation.
- Add the activated molecular sieve 4 Å to the Heptane (C7H16) and Tetrachloroethylene (C2Cl4) solutions to ensure that no residual water is present.
- Prepare a mixture of Tetrachloroethylene/Heptane 66:34 (v/v); density = 1.3 g cm-3. Add the activated molecular sieve 4 Å and store the mixture protected from light in a brown glass bottle.
Note: We usually use the ~100 ml of activated molecular sieve in a 1 L bottle. For 10 samples it is needed: a) 1 L of the Tetrachloroethylene/Heptane mixture 66:34 (v/v): 660 ml of Tetrachloroethylene + 340 ml of Heptane, b) 2 L of Heptane, c) 500 ml of Tetrachloroethylene. - Before use always check the pH of the solutions with a pH paper, especially Tetrachloroethylene. pH should be neutral. If Tetrachloroethylene solution turns acidic, the dried leaf material will become brown during gradient centrifugation and the NAF will fail.
- Cut 15 cm x 15 cm the nylon filters (one per NAF), weigh them and store them in clean 50 ml Falcon tubes.
- Fractionation procedure
Note: The whole procedure should be performed under a fume hood. Only dried aliquots (after Step C17) can be processed on regular bench. For pouring solutions, use glass pipettes.- Resuspend the dried leaf material in 20 ml of the Tetrachloroethylene/Heptane = 66:34 (v/v) mixture.
- Ultrasonicate the suspension for 2 min, with 6 x 10 cycles, 65% of power (Figure 2). To avoid overheating the suspension during the sonication process, keep the Falcon tube in an ice bath.
Note: Place the sonicator tip inside the solution, but avoid touching the tube wall. Make sure the sonication is successful. This is a critical step in the fractionation, since insufficient sonication can produce aggregates that do not pass through nylon mesh.
Figure 2. Setup for the ultrasonication procedure. Note that to avoid overheating the suspension during the process, the Falcon tube should be kept in an ice bath. - Fold a nylon mesh into a glass funnel placed on top of a 50 ml Falcon tube and pour the suspension through the nylon mesh. Wash the nylon mesh 3 x with 10 ml of heptane.
Note: The first two washing steps are performed by slowly pouring heptane on the nylon mesh and collecting the flow through in the Falcon tube below. For the third washing step, remove all the residual solution in the nylon mesh by squeezing it (while wearing adequate gloves). Place the nylon mesh back in a 50 ml Falcon tube and continue at Step C16. Be careful that all liquid go through the nylon mesh. If it´s the case, filter the whole obtained mixture again through a fresh nylon mesh. - Centrifuge for 10 min at 3,200 x g and 4 °C, using a swing-out rotor. Discard the supernatant (by pouring it into a waste glass bottle) and resuspend the pellet in 5 ml of Tetrachloroethylene/Heptane = 66:34 (v/v) mixture.
- Transfer 100 µl of the well homogenized resuspended pellet into a 1.5 ml screw cap tube. This aliquot is named fraction 0 (F0) and corresponds to the unfractionated material. Prepare three aliquots, add 900 µl of C7H16 to each, and keep protected from the light and continue at Step C15.
Note: Use a cut 200 µl pipette tip to collect F0. Homogenize well the mixture before taking each aliquot. - Prepare the NAF gradient (Figure 3A): Linear gradient (30 ml, density from 1.43 to 1.62 g cm-3)
- With a Pasteur pipette fill the 50 ml Falcon tube A with Tetrachloroethylene/Heptane mixture and the 50 ml Falcon tube B with Tetrachloroethylene.
- The dispensing needle is placed above a waste container.
Note: Ensure that there are no air bubbles in any tubing by purging thoroughly the whole system. - Set the peristaltic pump at a flow rate of 1.15 ml min-1 and the following program:
Notes:Step ml Solvent A Solvent B 1* 0 70% 30% 2** 3 70% 30% 3 28 0% 100% 4 33 0% 100% - *After 3 ml, pause the program. This step is used to ensure that the first mixture reaches the end of the dispensing needle before starting the actual gradient.
- **Remove the waste container and place the Polyallomer tube at this step. The needle should be placed slightly over the bottom of the Polyallomer tube and slightly touching the side of the tube (Figure 3B). Re-start the program.
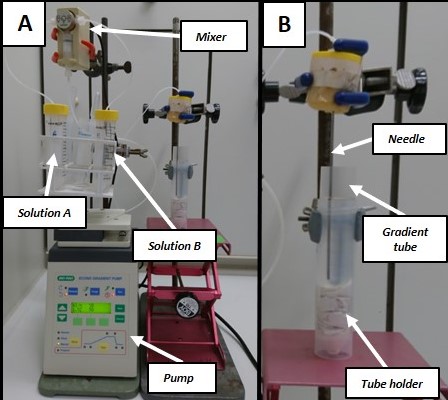
Figure 3. Setup of the gradient pump and the mixer. A. Overview of the setup for preparing the NAF gradient. Solution A (Tetrachloroethylene/Heptane mixture) and solution B (Tetrachloroethylene). B. Detail of the bottom-up preparation of the gradient. Tube holder is a 50 ml falcon tube with paper support on the bottom to help holding the gradient tube.
- Take the needle carefully out from the gradient and place the Polyallomer tube in a rack. Apply the suspension from Step C4 to the gradient, very carefully with a Pasteur pipette. Touch the wall of the tube with the pipette slightly above the gradient and release carefully to avoid disturbing the gradient.
- Place the gradients in the appropriate tube buckets from the ultracentrifuge. Verify their weights and to counterbalance, adjust the weight by adding 100% C2Cl4.
- Centrifuge for 50 min at 5,000 x g, and 4 °C using a swinging bucket rotor.
Note: We always use the Ultracentrifuge Beckman Coulter OptimaTM L80 XP and Swinging bucket rotor Beckman Coulter SW32Ti. Set the acceleration/deceleration on three. - Place the gradient tubes in a rack and mark eight fractions. Figure 4 illustrates how we usually separate the fractions based on the color.

Figure 4. NAF gradient after centrifugation. Usually, we separate eight fractions based on their color. The dark green layer is enriched in chloroplast and usually divided into two fractions (F2 and F3) to avoid metabolite overloading. Note that fraction 8 is a pellet, which is the vacuolar-enriched fraction. - Transfer carefully each fraction into clearly labeled clean 50 ml Falcon tube by using Pasteur pipette. The fractions are collected starting from the upper part (F1) to the bottom (F8). After F7 is removed, the pellet is resuspended in 20 ml heptane and the mixture transferred to a clean 50 ml Falcon tube. Use a new pipette for each fraction.
Note: Transferring the fractions is a critical point for the method, since at this step cross contamination of the fractions should be avoided as much as possible. Therefore, always transfer the fractions from the very top of each layer. - Add heptane up to 20 ml to the Falcon tubes containing F1 to F8 and mix carefully. Centrifuge all Falcon tubes for 10 min at 3,200 x g and at room temperature.
- Discard the supernatant (by carefully pouring it into a waste glass bottle) and resuspend the pellet with 7 ml of Heptane.
Note: The volume depends on how many aliquots you will need for your analysis. We usually prepare seven sub-aliquots per fraction. This number is enough for various metabolite analysis and determination of the compartment-specific markers. - Transfer 6 x 1 ml from each suspension into 1.5 ml screw caps tubes. The pellet can easily re-aggregate at this step. Keep mixing by hand the solution while transferring into the tubes in order to obtain homogeneous aliquots from the same fraction.
Note: Use a cut 1000 µl pipette tip to collect sub-aliquots from each fraction. As adding 7 ml generally does not allow to obtain seven aliquots of 1 ml, we generally take 0.8 ml for the seventh aliquot and clearly label the corresponding tube. - Centrifuge for 10 min at 14,000 x g and at room temperature. Discard excess of supernatant (c.a. 900 µl) carefully, using a 1000 µl pipette tip. Process the F0 aliquots obtained in Step C5 in the same manner.
- Place the screw caps tubes containing the sub-aliquots open in racks inside of a desiccator connected to a vacuum pump. Also, place the open Falcon tubes containing the filters from Step C3. Keep the aliquots under vacuum and protected from light overnight.
- Close the tubes and store them at -80 °C until further process.
- Weigh the dried filters.
- Measurements of metabolite markers
- Extraction
- Add to the dried aliquots a steel ball and 250 µl 80% ethanol/10 mM HEPES pH 7 to the samples.
- Place the tubes in a cooled (4 °C) holder for the ball mill and disrupt the pellet for 1 min at 25 Hz.
- Incubate in a thermomixer for 30 min at 95 °C and 1000 rpm.
- Centrifuge for 10 min at 20,800 x g and 4 °C.
- Transfer the supernatant to a 96-deep well plate and keep it protected from light and on ice.
- Add to the Pellet 150 µl 80% ethanol/10 mM HEPES pH 7.
- Incubate in a thermomixer for 30 min at 95 °C 1000 rpm.
- Centrifuge for 10 min at 20,800 x g and 4 °C.
- Transfer and combine with the previous supernatant.
- Add to the Pellet 250 µl 50% ethanol/10 mM HEPES pH 7.
- Incubate in a thermomixer for 30 min at 95 °C and 1,000 rpm.
- Centrifuge for 10 min at 20,800 x g and 4 °C.
- Transfer and combine with the previous two supernatants. It can be stored at -20 °C until further use.
- Nitrate (vacuolar marker)
Nitrate content is determined following Cross et al. (2006). Nitrate can be used as a vacuolar marker in addition or alternatively to Acid Invertase.
Note: Nitrite content must be determined and subtracted from the Nitrate values. For measuring nitrite content, nitrate reductase is replaced by water in the assay mix.
Solutions: For storage recommendations, the solutions for nitrate measurement can be stored as follow:
Nitrate Standards: 0, 0.2, 0.4, 0.8, and 1.6 mM in 70% ETOH with 10 mM MES pH 5.9Solution: Storage temperature Potassium phosphate buffer 1 M pH 7.5 -20 °C NADPH 50 mM in NaOH 20 mM -80 °C Nitrate reductase (5 U ml-1 in Potassium phosphate buffer 0.1 M) -80 °C PES 0.25 mM -80 °C Sulfanilamide 1% (w/v) in phosphoric acid 5% 4 °C NNEDA 0.02% (w/v) 4 °C Nitrate standards -80 °C
Protocol:- For one reaction, dispense in a 96-well plate 95 µl of the assay mix:
10.0 µl phosphate buffer
0.5 µl NADPH
2.0 µl Nitrate reductase
82.5 µl deionized water - Add 5 µl of standard or extract (1 to 10 diluted extract).
- Mix and incubate for 30 min at 25 °C, protected from light.
- Add 15 µl of PES.
- Mix and incubate for 20 min at 25 °C, protected from light.
- Add 50 µl of Sulphanilamide and 50 µl of NNEDA.
- Mix and incubate for 10 min at 25 °C, protected from light.
- Read the plate at 540 nm in a microplate reader.
- For one reaction, dispense in a 96-well plate 95 µl of the assay mix:
- Extraction
- Measurements of enzyme markers
- Enzyme extraction
- Add to the dried extract ~2 mg of PVPP (one little spoon), one steel ball (5 mm diameter), and 500 µl of buffer 1x (see Recipe 1b). Keep the samples on ice.
- Place the tubes in a cooled (4 °C) holder for the ball mill and disrupt the pellet for 1 min at 25 Hz. Make sure that the entire pellet is dissolved. If not, repeat the pellet disruption procedure.
- Centrifuge for 10 min at 20,800 x g and 4 °C. Transfer the enzyme extract (supernatant) to a 96 deep-well plate. Keep on ice and proceed to the enzyme assays. If required, the supernatant can be stored at -80 °C.
- Enzyme assays
All the three enzyme markers are measured according to Gibon et al. (2004).- AGPase activity (plastid marker)
- Dispense in a 96-well plate placed on ice:
14 µl of AGPase assay mix (see Recipe B2b)
6 µl of standard or extract. - Mix and incubate for 20 min at 25 °C.
- Add 20 µl of 0.5 M HCl in 100 mM Tricine/KOH pH 8.
- Mix and incubate for 10 min at room temperature.
- Add 20 µl of 0.5 M NaOH.
- Mix, add 50 µl of AGPase Determination mix (see Recipe B2c).
- Mix, spin down, and read at wavelength of 340 nm and 30 °C until the maximum rate of reaction stabilizes.
- Dispense in a 96-well plate placed on ice:
- PEPC (Cytosol marker)
- Dispense in a 96-well plate placed on ice.
18 µl PEPC Assay Mix (see Recipe B3b)
2 µl of standard or extract - Mix and incubate for 20 min at 25 °C.
- Add 20 µl of 0.5 M HCl in 100 mM Tricine/KOH pH 9.
- Mix, spin down, and incubate at 95 °C for 5 min. Cool and spin down. Keep the plate on ice.
- Add 20 µl 0.5 M NaOH.
- Mix, add 45 µl of PEPC Determination mix (see Recipe 3c, protect from light).
- Add 5 µl of 4 mM PES (protect from light).
- Mix and read at wavelength of 570 nm and 30 °C until the maximum rate of reaction stabilizes.
- Dispense in a 96-well plate placed on ice.
- Acid Invertase (Vacuole marker)
- Dispense in a black 96-well microplate:
10 µl Acid invertase Assay mix (see Recipe B4b)
5 µl of standard or enzyme extract - Mix and incubate at 25 °C for 5 and 40 min.
- Add 10 µl 0.5 M NaOH.
- Mix and incubate at R.T. for 10 min.
- Add 10 µl 0.5 M HCl in 100 mM Tricine/KOH pH 8.
- Mix and add 50 µl of Acid invertase Determination mix (see Recipe B4c).
- Mix and read the fluorescence for 10-15 min using the following setting:
Excitation at 530 nm, emission at 590 nm, temperature: 30 °C, sensitivity at 25. Use the rate of reaction for calculations. We use the Synergy fluorescence meter.
- Dispense in a black 96-well microplate:
- AGPase activity (plastid marker)
- Enzyme extraction
Data analysis
- For evaluation of subcellular metabolite distributions, we recommend the statistical software BestFit. The BestFit is a C-language command line tool that allows calculation and evaluation of subcellular distributions from NAF data. The software requires; (i) the distribution of specific markers for each subcellular compartment analyzed throughout the different fractions (Figure 5) and (ii) the distribution of the metabolites of interest throughout the different fractions.
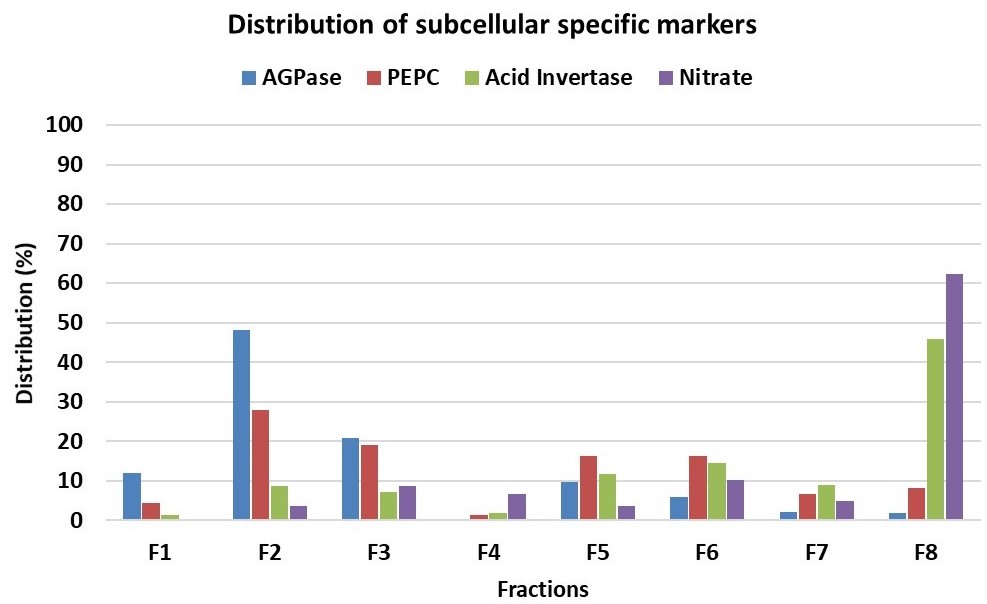
Figure 5. Distribution in percentage of subcellular compartment-specific markers throughout the gradient showed in Figure 4. ADP glucose pyrophosphorylase (AGPase) as plastidic marker, phosphoenolpyruvate carboxylase (PEPC) as cytosolic marker and nitrate and acid invertase as vacuolar markers. - For the values of either markers and metabolites of interest in the input file, we recommend using percentage, although absolute measurements can also be used (note that in this case values must be ≥ 0).
- For more details on how to use BestFit, we suggest reading the documentation pdf file within the BestFit Folder (http://www.csbdb.de/csbdb/bestfit/bestfit.html). Alternatively, you can also find details regarding data input and use of BestFit in Krueger et al. (2014).
- BestFit outputs the subcellular-distribution of the metabolites of interest in percentage. In order to calculate which amounts this represents, metabolites are also measured in ground material not processed through NAF (see Note in A1). Various recovery calculations can be done by using the following information: (i) material fresh weight used for NAF (determined in step A4), (ii) corresponding dry weight (determined in step A6), (iii) how much material was applied to the gradient (determined in step C18 by weighing the residual material in the filter), (iv) F0 obtained step C6. We recommend always recording these data and collecting F0 for each NAF.
Recipes
- Measurement of metabolite markers
- Nitrate standards
0, 0.2, 0.4, 0.8, and 1.6 mM in 70% ETOH with 10 mM MES pH 5.9
- Nitrate standards
- Measurement of enzyme markers
- Extraction buffers
- Extraction Buffer 10x
500 mM HEPES
100 mM MgCl2
10 mM EDTA
10 mM EGTA
10 mM Benzamidine
10 mM ε-aminocapronic acid
2.5% w/v BSA (protease-free)
Bring volume to c.a. 40 ml, adjust pH to 7.5 with KOH 10 M, and then adjust the volume to 50 ml
The extraction buffer 10x can be stored at -20 °C
Prepare the following solutions and store accordingly:
Leupeptin 2 mM (100x), freeze in liquid nitrogen and then store at -80 °C
DTT 500 mM (1,000x), freeze in liquid nitrogen and then store at -80 °C
PMSF 100 mM in isopropanol (100x), store at 4 °C, protect from light, stable for 1 month
Triton X-100 10% v/v (10x), store at RT
Glycerol (87% v/v), store at RT - Extraction Buffer 1x
Prepare only prior the extraction combining as the following:[Final] Reagent [stock] For 1,000 µl 1x Buffer 10x (see Recipe 1a) 10x 100 µl 10 µM Leupeptin 2 mM 5 µl 1 mM DTT 500 mM 2 µl 1 mM PMSF 100 mM in isopropanol 10 µl 1% Triton X-100 10% v/v 100 µl 20% Glycerol 87% (v/v) 230 µl H2O 553 µl
- Extraction Buffer 10x
- AGPase activity
- AGPase assay buffer (can be stored at -20 °C)
0.25 M HEPES/KOH pH 7.5
7.5 mM NaF
25 mM MgCl2
5 mM 3-phosphoglycerate
0.25% (v/v) Triton X-100 - AGPase assay mix
7.6 µl water
4 µl AGPase assay buffer (see Recipes B2a)
2 µl 20 mM PPi
0.2 µl Glycerokinase (200 U ml-1 in 200 mM Tricine/KOH pH 8, 10 mM MgCl2)
0.2 µl ADP-glucose 0 mM (Blank) or 100 mM (maximum) - AGPase determination mix
37.8 µl water
10 µl 1 M Tricine/KOH pH 8
0.2 µl 1 M MgCl2
0.5 µl GDH (200 U ml-1 in 200 mM Tricine/KOH pH 8, 10 mM MgCl2)
0.5 µl GPOX (500 U ml-1 in 200 mM Tricine/KOH pH 8, 10 mM MgCl2)
1 µl 66 mM NADH
- AGPase assay buffer (can be stored at -20 °C)
- PEPC activity
- PEPC assay Buffer (can be stored at -20 °C)
0.5 M Tricine/KOH pH 8
100 mM MgCl2
50 mM NaHCO3
0.25% Triton X-100 - PEPC assay Mix
11.4 µl H2O
4 µl PEPC assay Buffer (see Recipe B3a)
2 µl 0 (Blank) or 20 mM PEP (maximum)
0.4 µl 5 mM NADH
0.2 µl Malate dehydrogenase (100 U ml-1 in 200 mM Tricine/KOH pH 8, 10 mM MgCl2) - PEPC determination mix (protect from light)
18 µl water
10 µl 1M Tricine/KOH pH 9.0
10 µl 10 mM MTT
4 µl 200 mM EDTA
2 µl 50% Ethanol
1 µl Alcohol Dehydrogenase (2,000 U ml-1 in 200 mM Tricine/KOH pH 9, 10 mM MgCl2)
- PEPC assay Buffer (can be stored at -20 °C)
- Acid Invertase activity
- Acid invertase assay Buffer (can be stored at -20 °C)
0.25 M Acetate/KOH pH 5 - Acid invertase Assay Mix: for 1 reaction
10 µl Acid invertase assay buffer (see Recipe B4a)
35 µl water - Acid invertase Determination mix (protect from light): for 1 reaction
42 µl water
5 µl 1 M HEPES/KOH pH 7
1 µl Glucose oxidase (200 U ml-1 in 200 mM HEPES/KOH pH 7.0)
1 µl Horse Radish Peroxidase (0.2 U ml-1 in 200 mM HEPES/KOH pH 7.0)
1 µl 20 mM Amplex Red in DMSO
- Acid invertase assay Buffer (can be stored at -20 °C)
- Extraction buffers
Acknowledgments
We thank Deutsche Forschungsgemeinschaft (DFG TRR 175 The Green Hub – Central Coordinator of Acclimation in Plants; FA and ARF) for funding.
We also would like to thank the authors from Krueger et al. (2014), the previous work describing the NAF method, which we made the modification presented in the present protocol.
Competing interests
The authors declare no conflict of interest.
References
- Arrivault, S., Guenther, M., Florian, A., Encke, B., Feil, R., Vosloh, D., Lunn, J. E., Sulpice, R., Fernie, A. R., Stitt, M. and Schulze, W. X. (2014). Dissecting the subcellular compartmentation of proteins and metabolites in Arabidopsis leaves using non-aqueous fractionation. Mol Cell Proteomics 13(9): 2246-2259.
- Cross, J. M., von Korff, M., Altmann, T., Bartzetko, L., Sulpice, R., Gibon, Y., Palacios, N. and Stitt, M. (2006). Variation of enzyme activities and metabolite levels in 24 Arabidopsis accessions growing in carbon-limited conditions. Plant Physiol 142(4): 1574-1588.
- Elbers, R., Heldt, H. W., Schmucker, P., Soboll, S. and Wiese, H. (1974). Measurement of the ATP/ADP ratio in mitochondria and in the extramitochondrial compartment by fractionation of freeze-stopped liver tissue in non-aqueous media. Hoppe Seylers Z Physiol Chem 355(3): 378-393.
- Farre, E. M., Tiessen, A., Roessner, U., Geigenberger, P., Trethewey, R. N. and Willmitzer, L. (2001). Analysis of the compartmentation of glycolytic intermediates, nucleotides, sugars, organic acids, amino acids, and sugar alcohols in potato tubers using a nonaqueous fractionation method. Plant Physiol 127(2): 685-700.
- Fettke, J., Eckermann, N., Tiessen, A., Geigenberger, P. and Steup, M. (2005). Identification, subcellular localization and biochemical characterization of water-soluble heteroglycans (SHG) in leaves of Arabidopsis thaliana L.: distinct SHG reside in the cytosol and in the apoplast. Plant J 43(4): 568-585.
- Furtauer, L., Weckwerth, W. and Nagele, T. (2016). A benchtop fractionation procedure for subcellular analysis of the plant metabolome. Front Plant Sci 7: 1912.
- Gerhardt, R. and Heldt, H. W. (1984). Measurement of subcellular metabolite levels in leaves by fractionation of freeze-stopped material in nonaqueous media. Plant Physiol 75(3): 542-547.
- Gerhardt, R., Stitt, M. and Heldt, H. W. (1987). Subcellular metabolite levels in spinach leaves: regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiol 83(2): 399-407.
- Gibon, Y., et al. (2004). A Robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. The Plant Cell 16(12): 3304-3325.
- Klie, S., Krueger, S., Krall, L., Giavalisco, P., Flugge, U. I., Willmitzer, L. and Steinhauser, D. (2011). Analysis of the compartmentalized metabolome - a validation of the non-aqueous fractionation technique. Front Plant Sci 2: 55.
- Krueger, S., Giavalisco, P., Krall, L., Steinhauser, M. C., Bussis, D., Usadel, B., Flugge, U. I., Fernie, A. R., Willmitzer, L. and Steinhauser, D. (2011). A topological map of the compartmentalized Arabidopsis thaliana leaf metabolome. PLoS One 6(3): e17806.
- Krueger, S., Niehl, A., Lopez Martin, M. C., Steinhauser, D., Donath, A., Hildebrandt, T., Romero, L. C., Hoefgen, R., Gotor, C. and Hesse, H. (2009). Analysis of cytosolic and plastidic serine acetyltransferase mutants and subcellular metabolite distributions suggests interplay of the cellular compartments for cysteine biosynthesis in Arabidopsis. Plant Cell Environ 32(4): 349-367.
- Krueger, S., Steinhauser, D., Lisec, J. and Giavalisco, P. (2014). Analysis of subcellular metabolite distributions within Arabidopsis thaliana leaf tissue: a primer for subcellular metabolomics. Methods Mol Biol 1062: 575-596.
- Lunn, J. E. (2007). Compartmentation in plant metabolism. J Exp Bot 58(1): 35-47.
- Shapiguzov, A., Vainonen, J. P., Hunter, K., Tossavainen, H., Tiwari, A., Jarvi, S., Hellman, M., Aarabi, F., Alseekh, S., Wybouw, B., Van Der Kelen, K., Nikkanen, L., Krasensky-Wrzaczek, J., Sipari, N., Keinanen, M., Tyystjarvi, E., Rintamaki, E., De Rybel, B., Salojarvi, J., Van Breusegem, F., Fernie, A. R., Brosche, M., Permi, P., Aro, E. M., Wrzaczek, M. and Kangasjarvi, J. (2019). Arabidopsis RCD1 coordinates chloroplast and mitochondrial functions through interaction with ANAC transcription factors. Elife 8. pii:e43284.
- Sharkey, T. D., and Vanderveer, P. J. (1989). Stromal phosphate concentration is low during feedback limited photosynthesis. Plant Physiol. 91: 679-684.
- Sweetlove, L. J. and Fernie, A. R. (2013). The spatial organization of metabolism within the plant cell. Annu Rev Plant Biol 64: 723-746.
- Weiner, H., and Heldt, H. W. (1992). Intercellular and intracellular-distribution of amino acids and other metabolites in maize (Zea-Mays L) leaves. Planta 187(2),:242-246.
- Winter, H., Robinson, D. G.and Heldt, H. W. (1993). Subcellular volumes and metabolite concentrations in barley leaves. Planta 191(2): 180-190.
- Yamada, K., Norikoshi, R., Suzuki, K., Imanishi, H. and Ichimura, K. (2009). Determination of subcellular concentrations of soluble carbohydrates in rose petals during opening by nonaqueous fractionation method combined with infiltration-centrifugation method. Planta 230(6): 1115-1127.
Article Information
Copyright
![]() Medeiros et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Medeiros et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Medeiros, D. B., Arrivault, S., Alpers, J., Fernie, A. R. and Arabi, F. (2019). Non-aqueous Fractionation (NAF) for Metabolite Analysis in Subcellular Compartments of Arabidopsis Leaf Tissues. Bio-protocol 9(20): e3399. DOI: 10.21769/BioProtoc.3399.
- Shapiguzov, A., Vainonen, J. P., Hunter, K., Tossavainen, H., Tiwari, A., Jarvi, S., Hellman, M., Aarabi, F., Alseekh, S., Wybouw, B., Van Der Kelen, K., Nikkanen, L., Krasensky-Wrzaczek, J., Sipari, N., Keinanen, M., Tyystjarvi, E., Rintamaki, E., De Rybel, B., Salojarvi, J., Van Breusegem, F., Fernie, A. R., Brosche, M., Permi, P., Aro, E. M., Wrzaczek, M. and Kangasjarvi, J. (2019). Arabidopsis RCD1 coordinates chloroplast and mitochondrial functions through interaction with ANAC transcription factors. Elife 8. pii:e43284.
Category
Plant Science > Plant biochemistry > Metabolite
Systems Biology > Metabolomics > Leaf
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.