- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunofluorescence-based Determination of Centrosome Number in Tissue Samples

Published: Vol 9, Iss 20, Oct 20, 2019 DOI: 10.21769/BioProtoc.3396 Views: 4955
Reviewed by: Samantha E. R. DundonSander FrankManasa Chanduri
Original research article
The authors used this protocol in:

Mar 2019
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Centrosome numerical abnormalities have been reported in a variety of tumors. Centrosome numbers in cancer cells display both inter-tumor and intra-tumor heterogeneity. The over production of centrosomes (centrosome amplification) is unique in cancer cells and is a promising target for therapy. Thus, a method to quantify centrosome numbers on a single cell level is needed. Here, we describe a protocol to quantify centrosome numbers in formalin fixed paraffin embedded (FFPE) tissue samples by multiplexing antibodies to define bona fide centrosomes and cell borders. Centrosomes in single cells are identified using high resolution immunofluorescent microscopy with Z-sectioning. This protocol is easy to perform and has been used to quantify centrosome numbers on a single cell level in a variety of human tissue samples.
Keywords: CentrosomeBackground
The centrosome is the major microtubule-organizing center in cells and is crucial in defining mitotic spindle poles and forming cilia (Conduit et al., 2015). A centrosome consists of a pair of centrioles that serve as the duplicating elements of the centrosome and organize surrounding layers of pericentriolar material (PCM), a protein matrix with microtubule-nucleating activity (Bettencourt-Dias and Glover, 2007; Lawo et al., 2012). Centrosome numerical abnormalities, especially centrosome amplification, have been reported to be commonly present in a variety of human tumors and are correlated with advanced tumor grade, metastasis, recurrence, and poor survival (Bettencourt-Dias et al., 2011; Chan, 2011; Godinho and Pellman, 2014; Gonczy, 2015). Furthermore, centrosome numbers display both inter-tumor and intra-tumor heterogeneity in a broad range of tumors (Chan, 2011). Thus, a method to accurately quantify centrosome numbers on a single cell level in human tissue sample is useful to identify cancer cells with centrosome amplification, to study tumor heterogeneity, and to identify patients that might benefit from therapies that target cells with centrosome amplification.
Human centrioles are approximately 450-500 nm in length with a diameter of 200-250 nm (Gonczy, 2012; Greenan et al., 2018). Currently, centrosome quantification mainly relies on microscopy. Electron microscopy has been used as an accurate method to quantify centrosome numbers (Lingle et al., 1998). Super-resolution microscopy is another powerful way to study human centrosome structures (Sonnen et al., 2012). However, both electron microscopy and super-resolution microscopy require careful sample preparation and can be too laborious for quantifying centrosomes in a large number of cells and clinical tissue samples. Thus, most studies use high resolution microscopy like confocal and deconvolution microscopy. Further, the selection of centrosome markers is critical for accurate quantification. Most previous studies rely on PCM proteins like γ-Tubulin and pericentrin as centrosome markers, and centrosome numbers were quantified by counting PCM foci. However, PCM proteins are highly dynamic during centrosome assembly and can form assemblies or fragmentation without centrioles; they are not representative as bona fide centrosomes (Woodruff et al., 2014). Therefore, quantifying accentriolar PCM foci can lead to false positive results as centrosome amplification. On the other hand, cancer cells with centrosome amplification often undergo centrosome clustering during mitosis (Ganem et al., 2009; Silkworth et al., 2009). This leads multiple centrosomes in close proximity to each other and to appear as single PCM foci, which can result in false negative detection of centrosome numbers.
We have established a method using both centriolar protein CEP135 and PCM protein γ-Tubulin together as centrosome markers. We multiplexed centrosome markers with cell-cell junction markers to quantify centrosomes on a single cell level by high resolution microscopy (Wang et al., 2019a). We have tested different centriolar and cell-cell junction markers. Cell-cell junction proteins like E-Cadherin and α6 integrin are used to distinguish single cells from the neighbor cells and to identify the whole cell volume (Figure 1A). Foci with both centriolar proteins and PCM proteins are considered as centrosomes. Using this method, we have successfully performed centrosome quantification on a variety of normal and cancer tissues (Wang et al., 2019a; Wang et al., 2019b). Further, this method can be used to detect cells with centrosome amplification in both interphase and mitotic cells (Figure 1B). Here, we describe a detailed protocol to perform this method.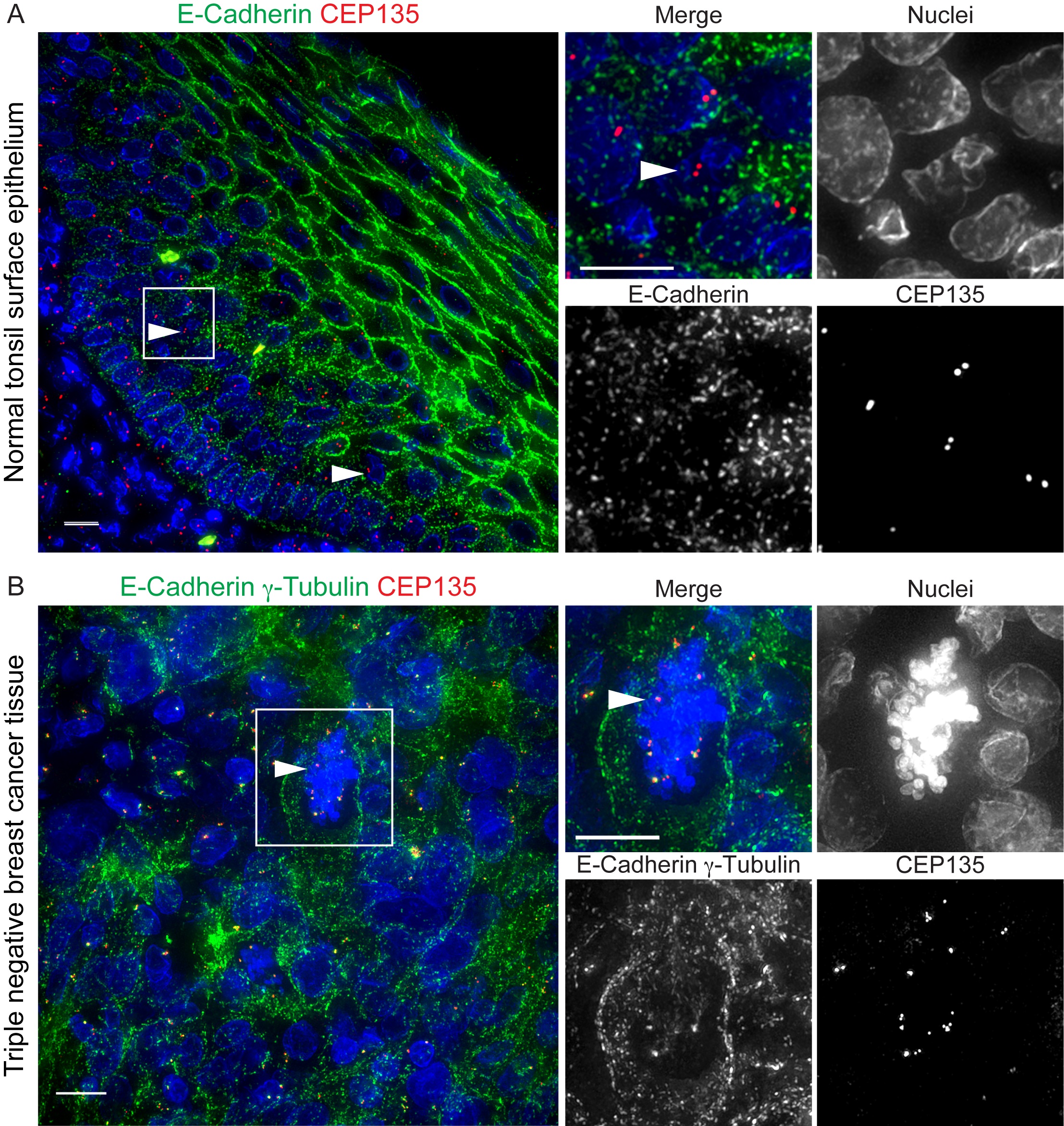
Figure 1. Centrosomes in human normal and cancer tissue. A. A human normal tonsil FFPE tissue sample stained with CEP135 (red) and E-Cadherin (green). Nuclei in blue. Scale bars: 10 μm. B. A human triple-negative breast cancer FFPE tissue sample stained with CEP135 (red), γ-Tubulin (green) and E-Cadherin (green). Nuclei in blue. Scale bars: 10 μm. Arrowheads indicate centrosomes.
Materials and Reagents
- Glass Coverslips 24 x 50 mm, No.1.5 (Globe scientific inc., catalog number: 1415-15)
- Forceps
- Humidified chamber (Biolegend, catalog number: SIG-31031)
- Coplin Jar
- Slide Staining Dish (IHC WORLD, catalog number: IW-2511)
- Slide Staining Rack (IHC WORLD, catalog number: IW-2512)
- Xylene (Fisher Scientific, catalog numbers: HC7001GAL, 22-110-676 and X16-4)
- Isopropanol (Fisher Scientific, catalog number: A416-4)
- EnVision FLEX Target Retrieval Solution, High pH (50x) (Dako Omnis, Code No: GV804)
- Tris-HCl (Sigma-Aldrich, catalog number: T5941)
- NaCl (Sigma-Aldrich, catalog number: S9888)
- Tween 20 (Sigma-Aldrich, catalog number: BP337)
- Triton X-100 (Sigma-Aldrich, catalog number: X100)
- NaN3 (Fisher Scientific, catalog number: S2271-25)
- Normal Goat Serum (Sigma-Aldrich, catalog number: G9023)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A7030)
- Tris Base (Fisher Scientific, catalog number: BP152-5)
- Anti- γ-tubulin (1:100, Sigma-Aldrich, catalog number: T5326)
- Anti-Pericentrin (1:200, Abcam, catalog number: 28144)
- Anti-CEP135 (1:50, Abcam, catalog number: ab75005)
- Anti-Centrin-3 (1:100, Abnova, catalog number: H00001070-M01)
- Anti-CP110 (1:100, proteintechTM, catalog number: 12780-I-AP)
- Anti-E-Cadherin (1:100, Abcam, catalog number: ab76055 M168)
- Secondary antibodies, purchased from Jackson Immunoresearch and used at 1:200
- Hoechst 33342 (1:1,000; Invitrogen, catalog number: H3570)
- ProLong® Diamond Antifade Mountant (Thermo Fisher Scientific, catalog number: P36970)
- EDTA (Fisher Scientific, catalog number: BP120-500)
- Washing buffer for FFPE tissue (see Recipes)
- Blocking buffer for FFPE tissue (see Recipes)
- Antigen retrieval buffer (see Recipes)
- Tris-EDTA Buffer for Antigen Retrieval (pH 9.0) (see Recipes)
Equipment
- Hot air oven (Labnet ProBlotTM 6 Hybridization Oven)
- Fume hood
- Decloaking chamber or pressure cooker (Biocare medical, DC0228)
- High-resolution fluorescence microscope (DeltaVision Core system (GE Healthcare Bio-Sciences, Pittsburgh, United States of America) equipped with Olympus IX71 Camera (CoolSNAP HQ2; Photometrics)
Note: Because centrioles are small (0.5 μm in length) and can localize at close proximity depending on cell cycle, high resolution images at a resolution around 200 nm are required to accurately separate each centrosome. High resolution microscopy like confocal and deconvolution microscopy can be used. We have successfully detected individual centrosomes using Deconvolution microscopy (DeltaVision) with a 60x, NA 1.42 objective. Because the positions of centrosomes vary depending on the tissue type, Z-section images through the whole cell volumes are required to detect and measure most centrioles in different z-sections within individual cells.
Software
- Image acquisition software (softWoRx v1.2 software [Applied Science])
- Adobe photoshop (https://www.adobe.com/products/photoshopfamily.html)
- ImageJ or Fiji (Schindelin et al., 2012; Schneider et al., 2012)
Procedure
- Staining FFPE tissue samples
Notes:- Tissue sections should be sliced at 5 to 10 μm thickness. Sectioning the tissue too thin can result in loss of cell volume and false negative detection of centrosomes. The optimal thickness should be determined according to the specific tissue samples used.
- For each section, we recommend the use of an H&E stained section as a guide slide to identify regions of interest.
- Tissue preparation. Timing: ~1 h.
Bake tissues 1 h or overnight at 65 °C in a hot air oven.
Note: This can vary according to the experiments based on tissue thickness. - Deparaffinization. Timing: ~25 min.
- Immerse tissue slides in a Coplin jar containing 100% fresh Xylene for 7 min at room temperature in a fume hood.
- Repeat Step 2a twice, 3 times total.
- Rehydration. Timing: ~45 min.
- Incubate slides in a Coplin jar containing 100% Isopropanol for 7 min at room temperature in a fume hood twice.
- Incubate slides in a Coplin jar containing 70% Isopropanol for 7 min at room temperature in a fume hood.
- Incubate slides in a Coplin jar containing 50% Isopropanol for 7 min at room temperature in a fume hood.
- Incubate slides in a Coplin jar containing ddH2O for 7 min at room temperature twice.
- Antigen retrieval. Timing: ~1 h.
Note: This can vary depending on the device used.- Add 500 ml deionized or filtered water into a decloaking chamber or a pressure cooker.
- Add 200 ml freshly prepared antigen retrieval buffer into a slide staining dish and immerse the slides into antigen retrieval buffer.
- Balance the decloaking chamber using another slide staining dish with 200 ml ddH2O in it.
- Close the lid of the decloaking chamber and rotate it to make sure the decloaking chamber is appropriately sealed. Turn on the decloaking chamber to incubate the slides for 20 min at its full pressure. For Decloaking chamber (Biocare Medical, DC0228), it is set to approximately 103 kPa/15 psi, and it will achieve a temperature of approximately 120 °C at full pressure.
- Open the decloaking chamber after the pressure is completely released and the decloaking chamber is cooled down.
- Transfer slides to a humidified chamber and incubate slides in washing buffer by placing a droplet of washing buffer on top of each slide to cover the tissue for 5 min at room temperature.
Notes:- Declocking chamber at full pressure is dangerous. The pressure must be completely released before opening.
- From this step on, it is very important that the slides do not dry out. Make sure to perform all the following steps in a humidified chamber.
- Blocking. Timing: ~30 min
- Gently tap the slides by holding the slides horizontally with label facing the investigator and tap on the bottom edge to remove excess liquid.
- Cover the tissue section with 100 μl of blocking buffer.
- Gently place a piece of coverslip on top of the tissue to evenly spread the liquid and incubate slides for 30 min at room temperature.
Note: The slides can be kept in blocking buffer at 4 °C overnight before proceeding.
- Primary antibody incubation. Timing: Overnight.
Note: This can vary according to the experiment.- Remove coverslips and gently tap the slides to remove excess liquid.
- Cover the tissue section with 100 μl of diluted antibodies in blocking buffer.
- Gently place a piece of coverslip on top of the tissue to evenly spread the liquid and incubate slides overnight at 4 °C. Alternatively, tissue slides can be incubated in primary antibodies for 1 h at room temperature.
Note: The dilutions for primary antibodies can vary depending on the targets. Optimal antibody concentrations should be determined on a case-by-case basis. We have successfully stained different centrosomal markers. The primary antibodies and dilutions can be found in Materials and Reagents. To quantify centrosome numbers per cell, investigators should choose centrosomal markers and cell border markers accordingly. To simultaneously stain different targets, investigators should choose the antibodies that are raised in different species. For example, we have successfully co-stained CEP135 (rabbit) to mark centrioles and E-Cadherin (mouse) to mark epithelial cell borders in different tissue types (Figure 1A).
- Secondary antibody incubation. Timing: 45 min to 1 h.
- Remove coverslips and wash slides three times in washing buffer by placing a droplet of washing buffer on top of each slide to cover the tissue, 5 min each at room temperature.
- Dilute secondary antibodies in the blocking buffer. For each tissue slide, 100 μl blocking buffer is recommended.
- Remove coverslips and gently tap the slides to remove excess liquid.
- Cover the tissue section with 100 μl of diluted antibodies in blocking buffer.
- Gently place a piece of coverslip on top of the tissue to evenly spread the liquid and incubate for 30 min to1 h at room temperature.
Note: For co-staining different markers, investigators should choose the secondary antibodies conjugated to different fluorophores according to the primary antibodies used and the filter sets on the microscope available. For example, image E-Cadherin (mouse), CEP135 (rabbit) and DNA together using a microscope equipped with FITC, TRITC, and DAPI filter sets. We used Alexa-488 Goat anti-mouse for E-Cadherin (mouse), and Alexa-568 Goat anti-rabbit for CEP135 (rabbit). The dilutions for secondary antibodies can vary. We have successfully used AlexaTM conjugated, highly cross-absorbed secondary antibodies from Invitrogen used at a 1:200 dilution. Samples stained with secondary antibodies only can be used as negative controls to increase the robustness of the analysis.
- Counterstain. Timing: ~30 min.
- Remove coverslip and wash slides three times in washing buffer, 5 min each at room temperature.
- Counterstain with Hoechst (1:1,000; 10 µg/ml) in PBS for 20 min at room temperature.
- Wash slides twice in washing buffer by placing a droplet of washing buffer on top of each slide to cover the tissue, 5 min each at room temperature.
- Mount. Timing: 24 h.
- Mount tissue sections using ProLong® Diamond Antifade Mountant and store in the dark at room temperature overnight for mountant to cure.
Note: Avoid bubbles in the tissue area. You can add a drop of mountant on coverslips and invert the coverslip to cover the tissue section. - Seal with nail polish after cure.
Note: Image the sample next day after the mountant has cured, or store at -20 °C for future analysis. Protect the samples from light. The samples can be stored at -20 °C, protected from light for more than a year.
- Mount tissue sections using ProLong® Diamond Antifade Mountant and store in the dark at room temperature overnight for mountant to cure.
- Imaging
- Use low magnification objective (e.g., 10x, N.A 0.7) to find the region of interest.
- Use high magnification objective (e.g., 60x, N.A 1.42 oil) to image the region of interest defined from Step B1 by Z-sections. Use a cell-cell junction marker (e.g., E-Cadherin) to find the whole cell volume and set the Z-section thick enough to cover the whole tissue section. We have successfully separated centrosomes at different focal planes using Z-sectioning with a 0.2 μm interval on DeltaVision Core system with a 60x objective, NA 1.42. Export images to .tiff format before performing proceeding to dara analysis.
Note: High resolution microscope systems like confocal or deconvolution microscopy can be used to image the region of interest. Note that a confocal microscope uses a pinhole to block out-of-focus light in the z-plane while deconvolution microscope uses computational methods to remove the out-of -focus light after capturing the image. Since each deconvolution method should be generated specifically for each objective on the imaging system, we only used deconvolution files generated on the DeltaVision core system using softWoRx.
Representative graphs using the protocol to display the quantification of centrosome numbers in tissue was recently published (Wang et al., 2019a, Wang et al., 2019b). They can be found in figure 2 in our published paper (Wang et al., 2019b).
Data analysis
Use the Z-sectioning images taken from high-resolution microscope for quantifying centrosome number per cell.
- Use ImageJ or Fiji to open Z-section images using “stacks” under “image”.
- If imported as multichannel RGB images, separate each channel using “image”→“color”→ “split channels”.
- Adjust contrast and brightness for each channel to reduce the background and increase the signal intensity. Merge all the channels after adjusting each channel.
Note: Adjustment of brightness and contracts should be linear, and the changes should be cautiously used without losing or oversaturating signals. - Generate a maximum intensity projection using command “image”→ “stacks”→ “Z project”. Use this maximum intensity projection image to find regions with centrosomes and check if cells overlap in the Z dimension. If there are no cells or centrosomes overlapping in the image, investigators can use maximum projection to quantify centrosomes per cell. If overlap occurs, go to step 5.
- Go through Z-section to quantify centrosome numbers per cell manually using the centrosome markers (e.g., CEP135 foci) and cell border marker (e.g., E-Cadherin).
Note: Depending on the tissue type and sample sectioning, cells in tissue can overlap on different focal planes. If the cells overlapping with each other at different focal planes, maximum projection images from Z-section images should not be used to quantify centrosomes for individual cells.
Notes
- Tissue fixation is critical for the staining results. Do not use over-fixed tissue, or re-fix tissue samples that previously were frozen.
- To practice the procedure and to test specific antibodies, we recommend using human tonsil tissue because it contains many different normal cell types.
- The thickness of tissue sections is critical for accurately quantifying centrosome numbers per cell accurately. Sectioning tissue too thin could result in loss of cell volume and false negative results. Too thick of the tissue samples results in cells overlapping. Depending on the tissue quality and tissue type, we recommend carefully testing the appropriate tissue section thickness to avoid loss of tissue or the complication of overlapping cells. A thickness of 5 or 10 μm is a good starting point.
- Keep all the reagents sterile.
Recipes
- Washing buffer for FFPE tissue
0.1 M Tris-HCl
0.3 M NaCl
0.1% Tween 20
7.7 mM NaN3
Adjust pH to 7.6 at 25 °C.
To make a 50 ml stock solution:- Add 5 ml of 1 M Tris-HCl, 5 ml of 5 M NaCl, 50 ul of Tween 20 and 0.025 g of NaN3 into a beaker, and ddH2O
- Adjust pH to 7.6 at room temperature
Note: Tween 20 is viscous. When adding Tween 20, pipette slowly to avoid air bubbles entering the pipette tip. One can also cut the end of the pipette tip to facilitate the procedure. Washing buffer can be stored at room temperature indefinitely.
- Blocking buffer for FFPE tissue
5% (v/v) Normal Goat Serum (NGS)
0.1 M Tris-HCl
0.15 M NaCl
7.7 mM NaN3
Adjust pH to 7.6 at 25 °C
To make a 50 ml stock solution:- Add 5 ml of 1 M Tris-HCl, 5 ml of 5 M NaCl, 2.5 ml of NGS and 0.025 g of NaN3 into beaker, and ddH2O.
- Adjust pH to 7.6 at room temperature. Blocking buffer can be stored at room temperature indefinitely. Warm up to room temperature before use
Note: NGS can be substituted with 5% (w/v) bovine serum albumin (BSA) depending on the primary antibody one is using. If the primary antibody was generated in goat, one needs to change the blocking reagent to BSA.
- Antigen retrieval buffer
Dilute 50x EnVision FLEX Target Retrieval Solution into ddH2O
Note: For staining less than 24 slices (the number of slides can vary depending on the slide rack one uses), one can add 4 ml 50x EnVision FLEX Target Retrieval Solution into 200 ml of ddH2O in a Slide Canister. Unused diluted solution may be stored at 2-8 °C for one month. - Tris-EDTA Buffer Antigen Retrieval (pH 9.0) (1x)
Note: This buffer is an alternative antigen retrieval buffer for this protocol.
10 mM Tris Base
1 mM EDTA Solution
0.05% Tween 20
Adjust pH to 9.0 at 25 °C
To make 100 ml 10x stock solution:- Add 1.21 g Tris Base, 0.37 g EDTA, and 0.5 ml Tween 2, and 100 ml ddH2O into a beaker
- Mix well and adjust pH to 9.0 at room temperature. Store this solution at 4 °C
Acknowledgments
We are grateful to National Cancer Institute (NCI) P30CA23074, which provided the research support services of the Tissue Acquisition and Molecular Analysis Service. This work was also supported by National Institutes of Health (NIH) Grants R01GM110166 and R01GM126035 to G.C.R., NIH-NCI RO1CA159406 to A.E.C., and a Tim and Diane Bowden Cancer Biology Research Fellowship to M.W. The protocol expands the details of the procedure from our previous work (PMID: 30699045) on a method of quantifying centrosomes at the single-cell level in human normal and cancer tissue.
Competing interests
The authors declare no conflict of interest.
Ethics
We accessed the tissue through the tissue acquisition and molecular analysis service research core (TACMASR) that was provided by the UA Cancer Center. The UA Cancer Center is an NCI designated comprehensive center and supported by P30 CA 23074 that utilizes best practices for management of the core services in accordance with NCI policy. The specimens/data are unidentifiable and obtained from a provider that is prohibited from releasing identifiers by established regulations or policies with oversight and approval from the Institutional Review Board.
References
- Bettencourt-Dias, M. and Glover, D. M. (2007). Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol 8(6): 451-463.
- Bettencourt-Dias, M., Hildebrandt, F., Pellman, D., Woods, G. and Godinho, S. A. (2011). Centrosomes and cilia in human disease. Trends Genet 27(8): 307-315.
- Chan, J. Y. (2011). A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7(8): 1122-1144.
- Conduit, P. T., Wainman, A. and Raff, J. W. (2015). Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol 16(10): 611-624.
- Ganem, N. J., Godinho, S. A. and Pellman, D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460(7252): 278-282.
- Gonczy, P. (2015). Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer 15(11): 639-652.
- Godinho, S. A. and Pellman, D. (2014). Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci 369(1650).
- Gonczy, P. (2012). Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol 13(7): 425-435.
- Greenan, G. A., Keszthelyi, B., Vale, R. D. and Agard D. A. (2018). Insights into centriole geometry revealed by cryotomography of doublet and triplet centrioles. Elife 7. doi:10.7554/elife.36851.
- Lawo, S., Hasegan, M., Gupta, G. D. and Pelletier, L. (2012). Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat Cell Biol 14(11): 1148-1158.
- Lingle, W. L., Lutz, W. H., Ingle, J. N., Maihle, N. J. and Salisbury, J. L. (1998). Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A 95(6): 2950-2955.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
- Silkworth, W. T., Nardi, I. K., Scholl, L. M.and Cimini, D. (2009). Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 4(8): e6564.
- Sonnen, K. F., Schermelleh, L, Leonhardt H.and Nigg , E. A. (2012). 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open. 1(10): 965-976.
- Wang, M., Knudsen, B. S., Nagle, R. B., Rogers, G. C. and Cress, A. E. (2019a). A method of quantifying centrosomes at the single-cell level in human normal and cancer tissue. Mol Biol Cell 30(7): 811-819.
- Wang, M., Nagle, R. B., Knudsen, B. S., Cress, A. E. and Rogers, G. C. (2019b). Centrosome loss results in an unstable genome and malignant prostate tumors. Oncogene. doi: 10.1038/s41388-019-0995-z.
- Woodruff, J. B., Wueseke, O. and Hyman, A. A. (2014). Pericentriolar material structure and dynamics. Philos Trans R Soc Lond B Biol Sci 369(1650).
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, M., Rogers, G. C. and Cress, A. E. (2019). Immunofluorescence-based Determination of Centrosome Number in Tissue Samples. Bio-protocol 9(20): e3396. DOI: 10.21769/BioProtoc.3396.
Category
Cancer Biology > General technique > Cell biology assays
Cell Biology > Cell imaging > Fixed-tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.