- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation of Pure Mitochondria from Rat Kidneys and Western Blot of Mitochondrial Respiratory Chain Complexes


Published: Vol 9, Iss 19, Oct 5, 2019 DOI: 10.21769/BioProtoc.3379 Views: 11705
Reviewed by: Xiaoyi ZhengDhanya K NambiarMei Po Chan
Original research article
The authors used this protocol in:

Nov 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Cardiac, neuronal and renal tubular epithelial cells are the most metabolically active cells in the body. Their fate depends largely on their mitochondria as the primary energy generating system which participates in the control of apoptosis, cell cycle and metabolism. Thus, mitochondrial dysfunction is a hallmark of many chronic diseases including diabetic nephropathy. A drop in mitochondrial bioenergetics efficiency is often associated with altered expression of respiratory chain complexes. Moreover, recent studies demonstrate that cellular proteins can shuttle to mitochondria and modify their function directly. Here we illustrate two mitochondria isolation protocols; one is recommended if the purity of the mitochondrial fraction is a priority such as if the mitochondrial localization of a protein has to be validated, the other if a high yield of intact functional mitochondria is required for functional studies and quantitative Western blotting. Next, we provide a detailed protocol for Western blotting of isolated mitochondria and renal cortex either to prove the purity of isolated fractions or to quantify complexes of the mitochondrial respiratory chain. We used this approach to identify classically cell membrane bound angiotensin II receptors in mitochondria and to study the effect of these receptors on mitochondrial function in early stages of diabetic nephropathy.
Keywords: MitochondriaBackground
Mitochondria are the primary energy generating system in metabolically active cells with a high energy demand such as renal proximal tubular epithelial cells (PTEC), cardiac muscle and neuronal cells. Additionally, they participate in the regulation of calcium signaling, proliferation, cell death and metabolism (Antico Arciuch et al., 2012). Thus, it is not surprising that diabetic nephropathy (DN) correlates more closely to defective mitochondria and increased oxidative stress in the kidney than to hyperglycemia. Increased reactive oxygen species (ROS) generated by defective mitochondria may initiate a vicious cycle, aggravating oxidative stress and mitochondrial dysfunction (Arora and Singn, 2014; Higgins and Coughlan, 2014). Mitochondrial ATP is produced in the respiratory chain (ETC), an assembly of more than 20 discrete electron carriers that are mainly grouped into five multi-polypeptide complexes and localized to the inner mitochondrial membrane. Increased oxygen consumption for ATP production may contribute to ischemia. Increased ROS may alter a number of functional cellular processes. A major regulator of DN is angiotensin II (AngII) which primarily acts via its type 1 receptor (AT1R). A second AngII receptor type 2 (AT2R) often counteracts the AT1R mediated effects (Peters et al., 2012). We have previously shown that mitochondrial dysfunction already occurred in the very early stages of DN and was associated with an altered expression of the ETC complexes. Further we demonstrated that the AT2R, classically located at the cell membrane as a G-protein coupled receptor, can translocate to the mitochondria and protect against the diabetes induced drop in mitochondrial bioenergetics efficiency, the rise in mitochondrial superoxide production, metabolic reprogramming and increased PTEC proliferation. These effects were associated with altered expression of ETC complexes (Micakovic et al., 2018). Such translocation of cellular proteins to the mitochondria was recently reported for a number of proteins including renin (Peters et al., 1996; Wanka et al., 2009), ACE2 (Wilson et al., 2016), vitamin D receptor (Silvagno et al., 2013) and polycystin 1 (Lin et al., 2018). In order to prove their presence in mitochondria and to dissect their mitochondrial function from indirect effects, studies on isolated mitochondria are required. Isolation of intact, functional and pure mitochondria is crucial for in vitro approaches. Furthermore, we performed Western blotting to quantify the ETC complexes and to evaluate the mitochondrial fraction for possible contaminations with other cellular components like the endoplasmatic reticulum, cytosol and cell membrane. Mitochondrial isolation procedures still vary among research groups and inconsistent or contradictory results often found in the literature may be due to differences in the quality of mitochondria as a result of different isolation procedures (Gross et al., 2011).
In this paper, we illustrate two protocols with highly reproducible outcomes for the isolation of pure and structurally intact mitochondria from kidney cortex of experimental rats: 1. isolation using the QproteomeTM Mitochondria Isolation Kit and 2. isolation using the serial differential centrifugation method. Isolation of mitochondria using QproteomeTM Mitochondria Isolation Kit provides highly pure mitochondrial fraction, free of contamination by other cellular compartments. The main limitation of this method is that it uses only 60 mg of tissue and therefore results in a low mitochondrial yield and is very expensive. This method is performed according to the manufacturer’s protocol, with slight changes. During the isolation procedure, different subcellular fractions can be collected (cytosol, nuclei/cell debris, mitochondria, and plasma membrane/endoplasmatic reticulum (ER). This isolation method is recommended if the purity of the mitochondrial fraction is a priority, such as if the mitochondrial localization of a protein has to be validated or composition of the mitochondrial proteome has to be determined by mass spectroscopy (Figure 1A). Isolation of mitochondria using the differential centrifugation method provides a much higher yield of functionally active mitochondria. The mitochondrial fraction obtained has a slight reduction in purity compared to the QproteomeTM Mitochondria Isolation Kit, but still has very low levels of contamination (> 90% of purity). This isolation method can be used for mitochondrial functional studies which require a large quantity of structurally intact mitochondria. The homogenization procedure is a crucial step that can significantly impact the quality of resulting mitochondria. However, the isolation methods that result in high fractional yield typically require harsh homogenization that often damages the organelles. In contrast to other published mitochondria isolation protocols which harvested the 3,000-10,000 x g fraction (Graham, 2001), we used a very gentle homogenization in Dounce glass homogenizer with a manually driven glass pestle. We then harvested the 500-1,000 g fraction rich in pure mitochondria with preserved integrity after differential centrifugation at 100, 300, 500 and 1,000 x g (Peters et al., 1996). This approach minimizes both mitochondria damage and contamination with the microsomes/plasma membranes and nuclei (Figure 1B).
For validation of purity and structural integrity of isolated mitochondria, we describe a detailed protocol for Western blot performed on subcellular fractions or proteins isolated from the whole kidney cortex. To demonstrate the purity of the isolated mitochondrial fraction we used antibodies targeting a specific organelle marker: the presence of endoplasmic reticulum is detected by Anti-GRP78; cytosol by Anti-GAPDH; and nucleus by Anti-Histone H3. The presence of the endoplasmic reticulum is also detected by using Anti-KDEL monoclonal antibody. Anti-COX IV antibody is used as an effective loading control for mitochondria (Figures 1A and 1B). COX IV is polypeptide chains of cytochrome c oxidase, the terminal oxidase in mitochondrial electron transport, and it is generally expressed at a consistently high level. Mitochondrial integrity is an important consideration when assessing the function of isolated mitochondria since damaged mitochondrial membranes can become rate limiting factor for oxygen consumption and ATP synthesis. We detected representative subunits of each electron transport chain complex (ETC) (CI:NDUFB8, CII:SDHB, CIII:UQCRC2, CIV:MTC01, CV:ATPVA) in mitochondrial fraction using an OXPHOS cocktail of monoclonal antibodies. Anti VDAC-1/Porin monoclonal antibody is used to detect the mitochondrial outer membrane.
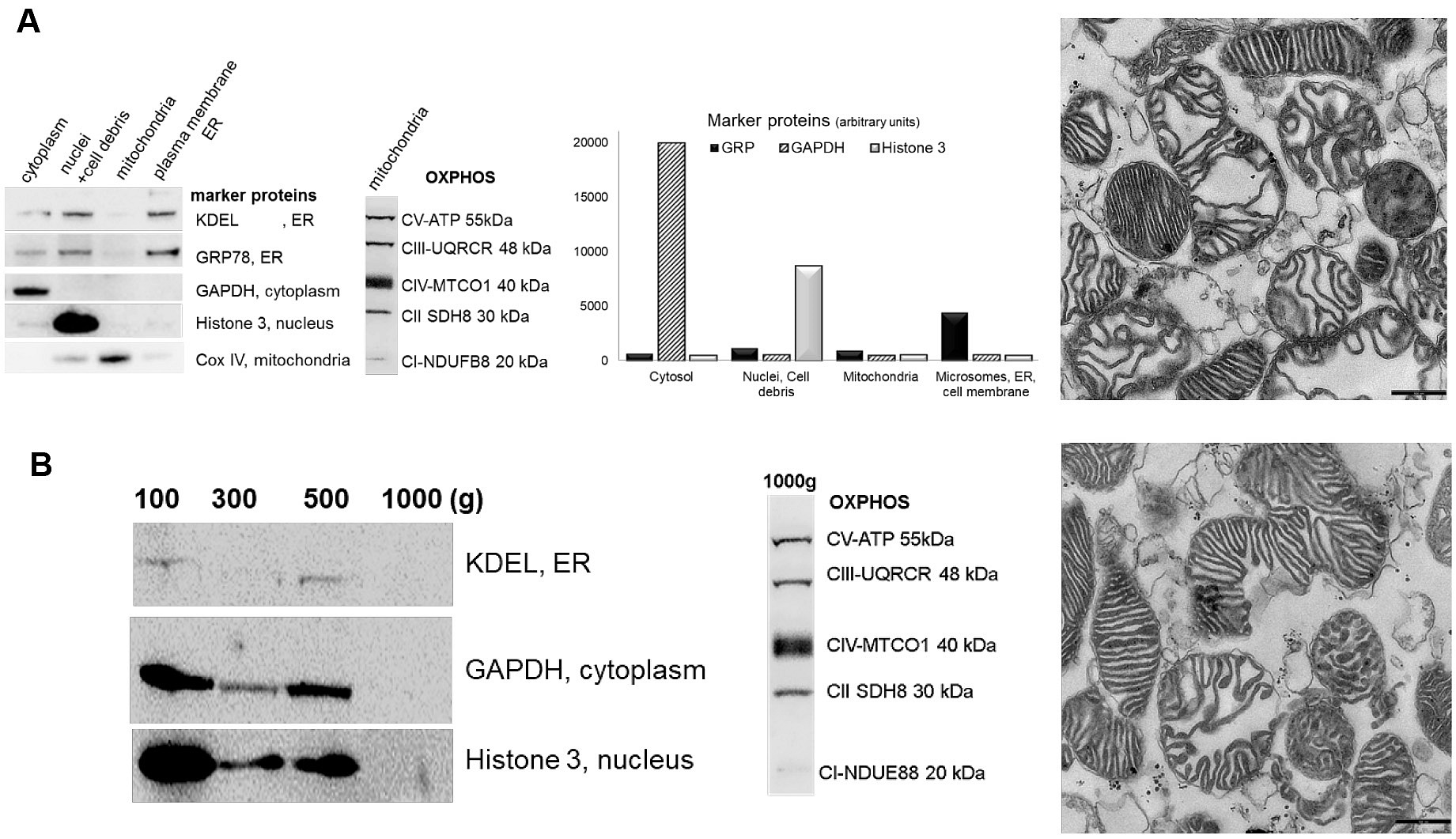
Figure 1. Western blot (left) and electron microscopy (right) demonstrating purity of mitochondrial fractions. A. QproteomeTM Mitochondria Isolation Kit. B. Differential centrifugation. Western blot of subcellular preparations probed with antibodies specific for organelle/cell compartment specific marker proteins: cytosol (GAPDH), nucleus (Histone 3), mitochondria (COXIV-ab16056, abcam) and microsome/ER/PM (KDEL-ab1223, abcam, GRP78) (GAPDH, GRP78, Histone 3 ab139415, abcam) fractions (left). OXPHOS antibody cocktail-ab110413, abcam Figure 2).
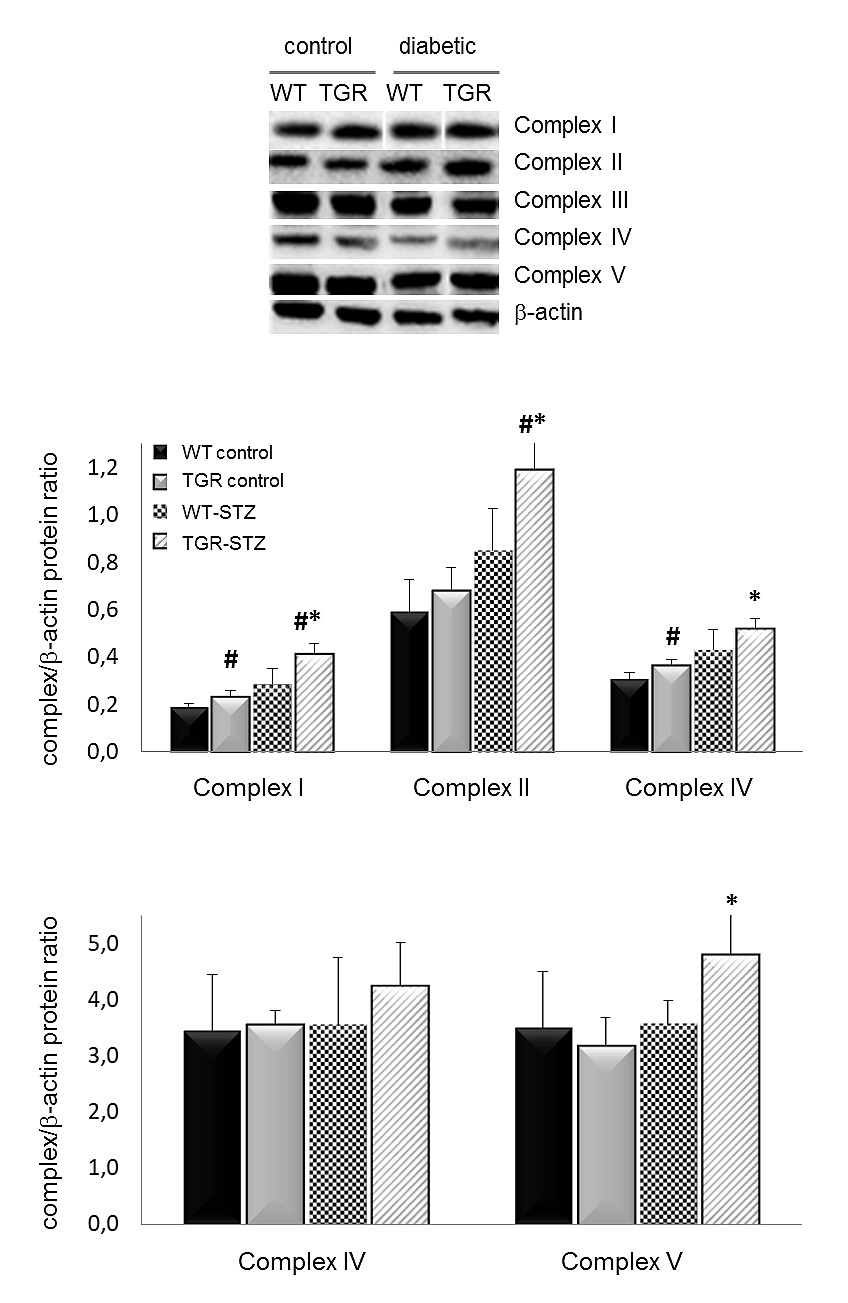
Figure 2. Western Blot of mitochondrial respiratory chain complexes. All complexes of the respiratory chain, except complex III, are increased in diabetic TGRs relative to their nondiabetic controls and diabetic WT-rats. The antibodies are against a subunit of each complex (CI:NDUFB8, CII:SDHB, CIII:UQCRC2, CIV:MTC01, CV:ATPVA) that is labile when its complex is not assembled. Results are expressed as means ± SD. *P ≤ 0.05 vs. control; #P ≤ 0.0 vs. wildtype, 3-4 rats/group. WT–wildtype rats, TGR–transgenic rats. STZ streptozotocin-diabetic.
Materials and Reagents
- Materials
- Pipettes tips
- Glass bottle with in- and outtake socket
- Bulldog-clamp
- Plastic tray
- Adhesive tape
- Scissors
- Disposable Scalpel (Swann-Morton, Disposable Scalpel, catalog number: 0086)
- Razor blade (Häberle Labortechnik GmbH, Germany, catalog number: 6800353)
- Butterfly-19 gauge (OD-Ø 1.1 mm; ID-Ø 0.8 mm)
- Bulldog Clamp, Johns Hopkins bulldog clamp, straight, 3,8 cm (Fisher scientific, catalog number: 12-460-556)
- Einmal-Küvetten aus PS (neoLab Migge GmbH, ecoLab, catalog number: E-1642)
- Eppendorf Safe-Lock Tubes 2 ml, 1,5 ml (Eppendorf AG, Eppendorf Tubes®)
- Filter Paper (Merck KGaA, Whatman® cellulose chromatography papers, catalog number: WHA3030917)
- Homogeniser POTTER-ELVEHJEM with Glass pestle, 23 (17 ml) (Otto E. Kobe KG, catalog number: 9.651 677)
- Wheaton Mikro-Gewebe-Handhomogenisator Dounce 1 ml, with loose and tight glass pestle (NeoLab, catalog number: 9-0972)
- Homogeniser with glass pestle, 3 ml (NeoLab, catalog number: 9-0936)
- Injection needle (Becton, Dickinson, model: BD Microlance 27G REF300635)
- NuncTM 15 ml and 50 ml Conical Sterile Polypropylene Centrifuge Tubes (Thermo Fisher Scientific, NuncTM Centrifuge Tubes)
- NuPAGETM 4-12% Bis-Tris Protein Gels (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0321BOX)
- Petri dish 100 mm (Thermo Fisher Scientific, Nunc, catalog number: 4031)
- Petri dish 35 mm (Santa Cruz Biotechnology, catalog number: sc200284)
- Pipette tips (STARLAB INTERNATIONAL GmbH, TipOne®)
- PVDF-Membrane (Merck KGaA, Immobilon®, catalog number: IPVH00010)
- Razor blade (Häberle Labortechnik GmbH, Germany, catalog number: 6800353)
- Tuberkulin syringe Luer Lok, 1 ml Omnifix-F, oneway sterile (Fa. B. Brown, catalog number: 9161406V)
- XCell IITM Blot Module (Thermo Fisher Scientific, InvitrogenTM, catalog number: EI9051)
- XCell SureLock® Mini-Cell gel running tank (Thermo Fisher Scientific, InvitrogenTM, catalog number: EI0001)
- Reagents
- Antibodies
- Anti-COX IV Antibody, rabbit polyclonal to COX IV (Abcam, catalog number: ab16056), storage at -80 °C, dilution 1:1,000
- Anti-KDEL Antibody, mouse monoclonal [10C3] to KDEL (Abcam, catalog number: ab12223), storage at -80 °C, dilution 1:1,000
- Anti-VDAC1/Porin Antibody, mouse monoclonal [20B12AF2] to VDAC1/Porin (Abcam, catalog number: ab14734), storage at 4 °C, dilution 1:800
- Anti-β-Actin Antibody, mouse monoclonal to β-Actin (Sigma-Aldrich, catalog number: A2228), storage at -20 °C, dilution 1:2,000
- Endoplasmic Reticulum Fraction Western Blot Cocktail (GAPDH, GRP78, Histone 3) (Abcam, catalog number: ab139415), storage at -20 °C, dilution 1:500
Note: The cocktail already contains the HRP Conjugated Secondary Antibody Cocktail. - Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP (Thermo Fisher Scientific, catalog number: 32430), storage at 4 °C, dilution 1:2,000
- Goat anti-Rabbit IgG Secondary Antibody, HRP (Santa Cruz Biotechnology, catalog number: sc-2004), storage at 4 °C, dilution 1:2,000
- Total OXPHOS Rodent WB Antibody Cocktail (Abcam, catalog number: ab110413), storage at 4 °C, dilution 1:750
Note: The cocktail contains 5 mouse m-antibodies, one each against a subunit of each complex (CI:NDUFB8, CII:SDHB, CIII:UQCRC2, CIV:MTC01, CV:ATPVA), Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad Laboratories, catalog number: 500-0006), storage at 4 °C.
- Bovine Serum Albumine (Sigma-Aldrich, catalog number: A3311-50G)
- Mannitol, M = 182.17 g/mol (Sigma-Aldrich, catalog number: M9546)
- EDTA·2H2O, M = 372.24 g/mol (AppliChem, catalog number: A3553)
- EGTA, M = 380.4 g/mol (Sigma-Aldrich, catalog number: E3889)
- Heparin-sodium (Sigma-Aldrich, catalog number: H0200000)
- IGEPAL® CA-630 (Sigma-Aldrich, catalog number: 9036195)
- Magnesium Chloride, 6-Hydrate, Crystal, U.S.P. (MgCl2·6H2O), M = 203.3g/mol (J.T. Baker, catalog number: 7791-18-6)
- Morpholinopropane Sulfonic Acid (MOPS), M = 209.3 g/mol (Serva, catalog number: 29836)
- NuPAGETM LDS Sample Buffer (4x) (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0007), storage at 4 °C
- NuPAGETM MES SDS Running Buffer (20x) (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0002), storage at 4 °C
- NuPAGETM Sample Reducing Agent (10x) (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0004), storage at 4 °C
- NuPAGETM Transfer Buffer (20x) (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0006)
- Phenylmethanesulfonyl fluoride (PMSF), M = 174.19 g/mol (Gibco BRL, catalog number: 15521-016)
- Potassium Phosphate Dibasic Trihydrate (K2HPO4·3H2O), M = 228.22 g/mol (Sigma-Aldrich, catalog number: P966)
- cOmpleteTM Protease Inhibitor Cocktail (Sigma-Aldrich, catalog number: 11836145001), storage at 4 °C
- Qproteome Mitochondria Isolation Kit (Qiagen, catalog number: 37612)
- SeeBlue Plus2 Prestained Standard (Thermo Fisher Scientific, InvitrogenTM, catalog number: LC5925), storage at 4 °C
- Sodium Chloride (Sigma-Aldrich, catalog number: S3014)
- Sodium Deoxycholate (Sigma-Aldrich, catalog number: D6750)
- Sucrose, M = 342.3 g/mol (Sigma-Aldrich, catalog number: S0383)
- Trizma® base, M = 121.14 g/mol (Sigma-Aldrich, catalog number: T6066)
- Tween-20 (J.T. Baker, catalog number: 7374)
- Western Blotting Luminol Reagent (Santa-Cruz Biotechnology, catalog number: sc-2048), storage at 4 °C
- Milk powder (Carl Roth, catalog number: T145-1)
- EDTA disodium salt (J.T. Baker, catalog number: 6381-92-6)
- SDS (Sigma-Aldrich, catalog number: L4509)
- Methanol (Carl Roth, catalog number: 4627.4)
- HCl (Carl Roth, catalog number: 4625.1)
- Xylazin 20 mg/ml (Serumwerke Bernburg AG, catalog number: Ch.B: 01117)
- Ketamin 10% (WDT, Ch.B: J3218-03)
- Liquid N2
- Alcohol
- Acetone
- Homogenization Buffer (see Recipes)
- Isolation buffer (see Recipes)
- RIPA buffer (see Recipes)
- 1x TBS (see Recipes)
- TBST (see Recipes)
- Blocking Buffer (see Recipes)
- 1 M Sucrose (see Recipes)
- 1 M Tris-HCl (see Recipes)
- 0.5 M EDTA (see Recipes)
- 1 M Mannitol (see Recipes)
- 1 M MgCl2 (see Recipes)
- 0.5 M MOPS (see Recipes)
- 100 mM EGTA (see Recipes)
- 1 M K2HPO4 (see Recipes)
- 50 mM PMSF (see Recipes)
- Antibodies
Equipment
- Pipettes
- Dissection Instruments
- Centrifuge (Eppendorf AG, model: 5415R)
- Centrifuge Sorvall superspeed (Herolab GmbH Laborgeräte, model: Sorvall RC5CPlus)
- Chemiluminescence Imaging System (Vilber, model: Fusion Solo S)
- Multi-Sample Osmometer (Advanced Instruments, model: 2020)
- Nanodrop (Thermo Fisher Scientific, Life technologiesTM, model: 2000C)
- Overhead shaker GFL 3025 (Laborgeräte München)
- Mini-Blot-Schüttler, 3-D (VWR, Lab Services GmbH)
- Spectrophotometer (Harvard Bioscience, GeneQuant, catalog number: 80-2109-02)
- Jun Air compressor 87R-15B with manometer (-1 to +0.6bar) (JUN-AIR Gast Manufacturing, Inc, USA)
Software
- Excel (Microsoft Cooperation, Redmond, USA)
- Maschine Fusion SL, Vilber Lourmat Software: FusionCapt Advance SL4 16.08a
Procedure
- Tissue harvesting from rats
PREPARETION: Prepare all reagents and instruments ahead of time. You will need: dissection instruments, ice, small beakers, liquid N2, 1x air compressor with manometer, 1x glass bottle with in- and outtake socket + connecting tubes and per rat: 15 ml ice cold 0.9% NaCl, 300 ml perfusion solution (saline with 50 IU/ml Heparin, H 7.2-7.4, temperature 25-35 °C), xylazine/ketamine, 2 x 2 ml eppendorf tubes, 1 x 1 ml syringe, 1x injection needle 30 g, 1x Butterfly-19 gauge, 2x small bulldog-clamp. Prepare the perfusion system (Figure 3A): fill perfusion solution into the bottle, control pressure (220 mbar for rats > 200 g, 180 mbar for rats < 200 g body weight). Fix the Butterfly needle at the tubing.
Note: Remove air from all tubings. Air bubbles in the vessels of animals can damage the tissue!- Anesthesia
Anaesthetize rats with xylazine/ketamine mixture (5 mg/kg/100 mg/kg) via intraperitoneal injection using the 1 ml syringe with a 30 gauge needle. Test the depth of anesthesia with pedal withdrawal reflex before beginning with the operation!! Administer additional anesthetics as necessary before beginning with the operation to maintain a surgical plane of anesthesia. - Perfusion
- Fix anesthetized rat with adhesive tape on a cork table and put the cork in a plastic tray to prevent perfusion fluid from leaking onto the bench and floor.
- Swab with a little 70% alcohol to wet the fur, as otherwise fur will get into the operating site.
- The abdominal skin and abdominal wall is then incised by a longitudinal cut (laparatomy) and two cuts to both sides giving free access to the abdomen; put the gut to the left side.
- Free the Aorta abdominalis and Vena cava from connective tissue and fat (from the iliac artery and vein up to the Aa. renales) with the help of cotton buds. Be sure that the vessels are completely free of connective tissue otherwise you can’t place the needle correctly (Figure 3B).
- Insert the needle into the Aorta–caudal third, approximately 1 cm cranially of the beginning of the Aa. iliacae in the rat. Fix the needle in the vessel with a small bulldog-clamp and watch the needle carefully later on (Figure 3C)!!
Note: The opening of the needle should point downwards, you otherwise may push the needle through both walls of the vessel. - Open the Vena cava over a length of 1.5 cm (rats) with small and very sharp scissors! The opening should be big enough to allow a rapid drain of the perfusion solutions. Remove the clamp near the Aa. renales if there is one (Figure 3D)!!
- Perfuse the rat 3 min with 220 or 180 mbar depending on body weight with perfusion solution (saline with 50IU/ml Heparin, pH 7.2-7.4, temperature 25-35 °C). The kidneys should have changed their appearance to a pale red color after perfusion, this means they are free of erythrocytes. If they appear patchy, the perfusion is not complete.
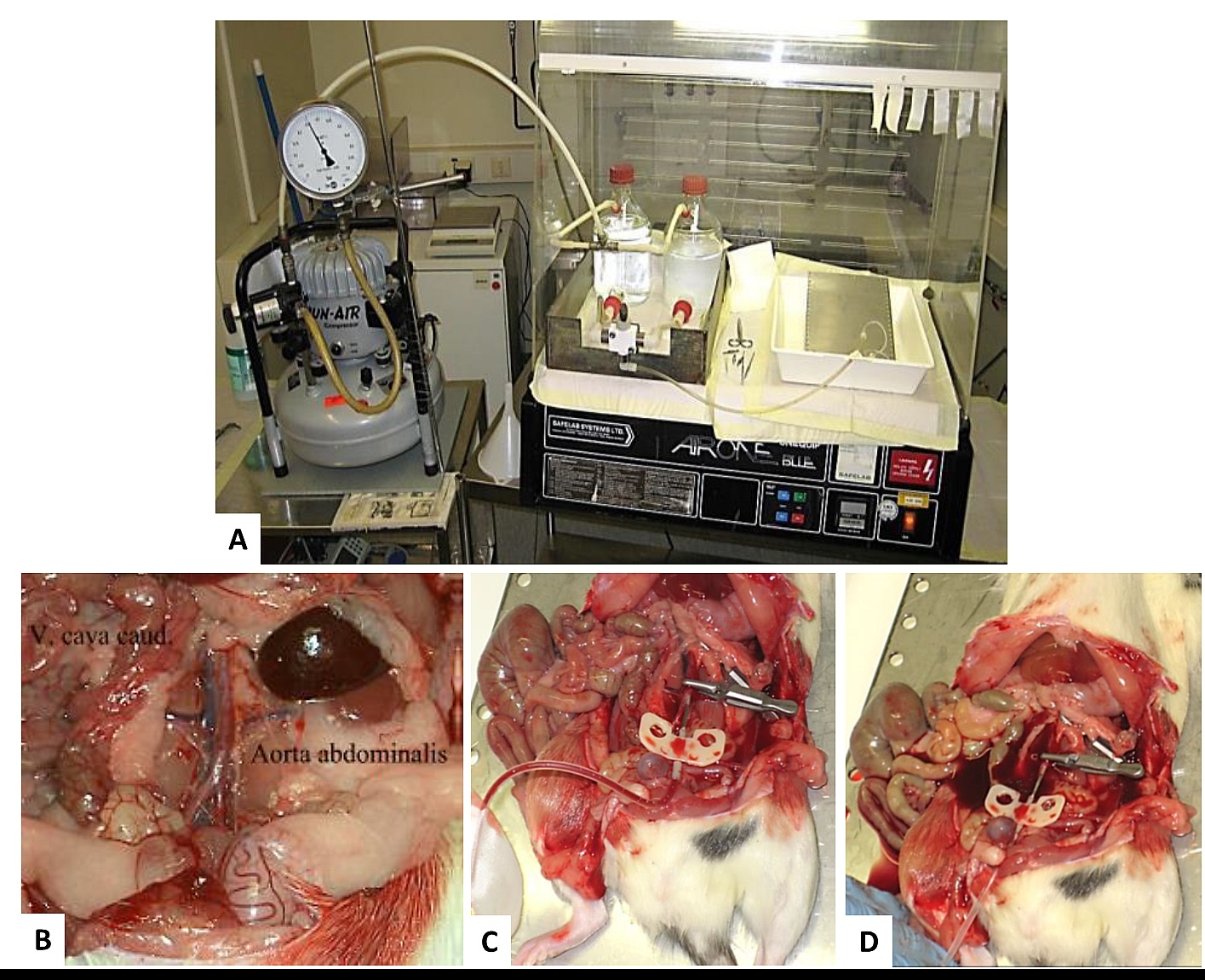
Figure 3. Perfusion system. A. assemble of the perfusion system with Jun air compressor, B-D. Scheme for needle setting in the rat.
- Harvest kidney cortex
- Harvest kidneys and immediately remove the renal capsule by hand (Figure 4A). Place the kidneys in a small beaker containing 5 ml ice cold 0.9% NaCl. Rinse 3 times in fresh 0.9% NaCl.
- After rinsing, bisect kidneys along the coronal axis (Figures 4B-4D), remove medulla with fine scissor and discard it (Figures 4E-4F).
Note: For Western blotting cut the renal cortex into small pieces, place it in a 2 ml Eppendorf tube, freeze down directly in liquid N2 and store it in -80 °C; for mitochondria isolation proceed immediately with the fresh tissue.
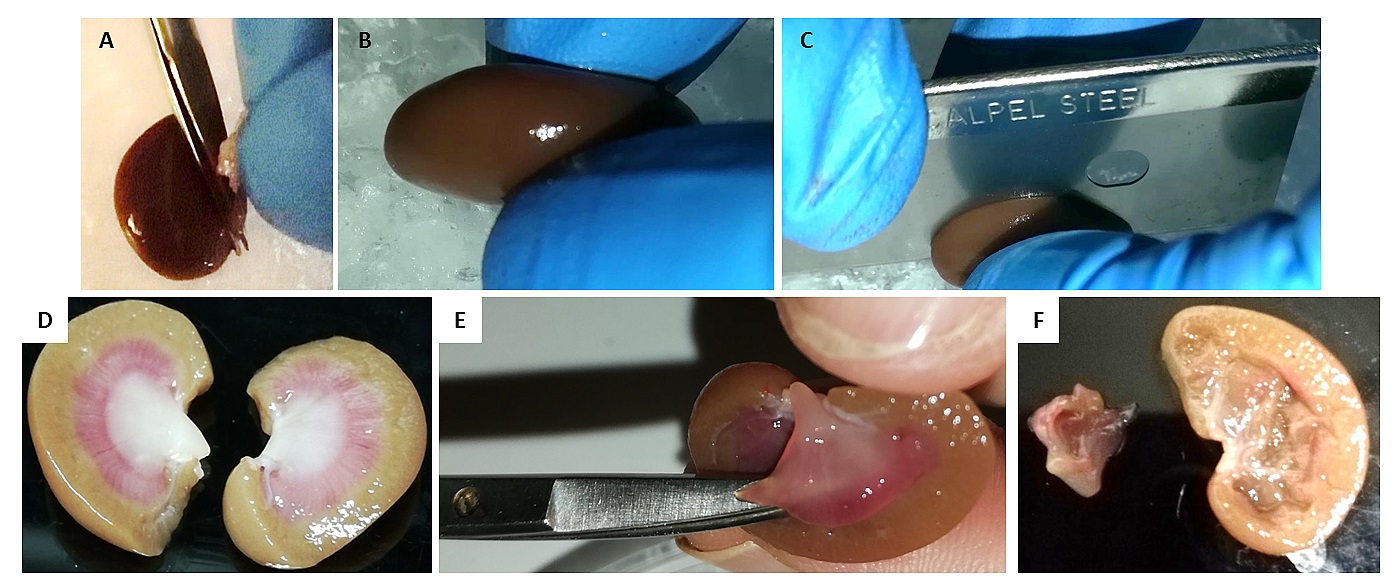
Figure 4. Harvesting of kidney cortex. A. Removal of renal capsule. B-C. Bisect kidneys along the coronal axis. D. Kidney in half, red line mark the cutting position. E-F. Removal of medulla.
- Anesthesia
- Isolation of pure mitochondrial fraction
Two protocols with highly reproducible outcomes were established for the isolation of pure and structurally intact mitochondria from the kidney cortex of experimental rats. Both protocols provide high quality intact mitochondria. Protocol 1 describes the isolation of mitochondria using QproteomeTM Mitochondria Isolation Kit. It provides highly pure mitochondrial fraction, completely free of contamination by other cellular compartments, but at the expense of a lower yield and higher costs. Protocol 2 describes the isolation of mitochondria using the differential centrifugation method which results in a much higher yield of mitochondria but at the expense of a small reduction in purity (Purity > 90%).- Isolation of pure mitochondrial fraction using QproteomeTM Mitochondria Isolation Kit (Figure 5)
Note: Lysis Buffer, Disruption Buffer, Protease Inhibitor Solution (100x), Mitochondria Storage Buffer and Mitochondria Purification Buffer are provided within of QproteomeTM Mitochondria Isolation Kit.
PREPARETION: Pre-cool all buffers, centrifuges and other equipment to 4 °C and homogenizers to -20 °C (overnight) beforehand. You will need ice, Petri dish, razor blade, 1.5 ml and 2 ml tubes, 1 ml and 2 ml glass homogenizers with glass pestle, 80% and 100% acetone pre-cooled to 20 °C. Immediately before use, supplement Lysis Buffer and Disruption Buffer with 1:100 volume of Protease Inhibitor Solution (100x) (i.e., if using 2 ml of Lysis Buffer, add 20 μl of Protease Inhibitor Solution [100x]).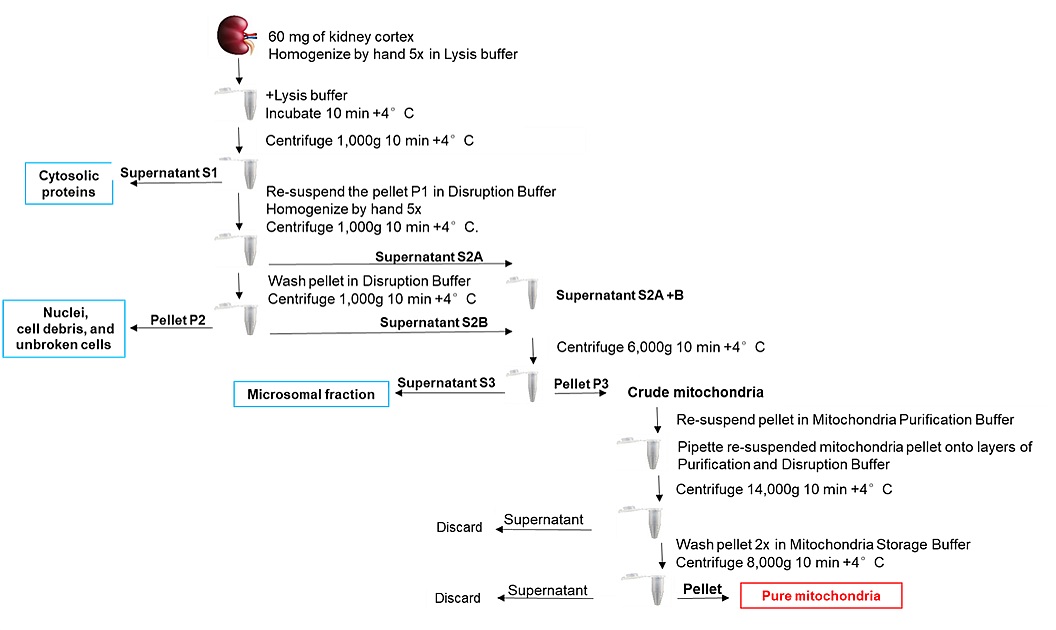
Figure 5. Scheme illustrates the work flow of mitochondria isolation using QproteomeTM Mitochondria Isolation Kit
Note: All steps should be performed at 4 °C or on ice. Separated protein fractions should be snap-frozen in liquid nitrogen and stored at -80 °C.- Immediately after harvesting, weigh out 60 mg of kidney cortex and, place it into a 30 mm Petri dish on ice, cover it with 2 ml ice cold storage buffer and mince it into very small pieces (mush) using the razor blade.
Note: Have no more than 5 min between tissue harvesting and cutting. - Place the mush into a 15 ml tube and centrifuge at 200 x g, 4 °C, for 1 min.
- Discard the supernatant and suspend the minced kidney pellet in 0.5 ml of ice-cold Lysis Buffer supplemented with Protease Inhibitor Solution.
- Transfer the suspension to the pre-cooled 1 ml glass-glass Dounce homogenizer and homogenize the tissue by hand on ice. Gently operate the glass pestle in an up and down manner by hand, rotating it during the movement. Repeat up and down movement 5-10 times until you get a homogenous solution, 30 s rest on ice, add 1.5 ml ice-cold Lysis Buffer supplemented with Protease Inhibitor Solution, then 5 strokes again.
Note: Avoid creating air bubbles during the homogenization. - Transfer the homogenate into a 2 ml tube and incubate on an over-head shaker for 10 min at 4 °C.
- Centrifuge the homogenate at 1,000 x g for 10 min at 4 °C.
- Carefully remove the supernatant (S1). This fraction primarily contains cytosolic proteins. Keep it on ice (see Steps B1bb-B1ee).
Note: Remove very carefully all the remaining fluid from the pellet and tube to avoid contamination of the pellet with cytosolic proteins.
Disrupt the cells to get S2 with mitochondria and P2 with nuclei, cell debris and unbroken cells- Resuspend the pellet P1 in 1.5 ml ice-cold Disruption Buffer supplemented with Protease Inhibitor Solution by pipetting up and down using a 1 ml pipette tip.
Note: The pellet is dense. In order to resuspend it more easily, cut the end of a 1 ml pipette tip and energetically pipette. Then take a new tip and continue pipetting up and down until the pellet is completely re-suspended. - Transfer the suspension into a 2 ml glass-glass homogenizer and homogenize it by hand as described in Step B1d.
- Transfer the suspension into a 2 ml tube and centrifuge it at 1,000 x g for 10 min at 4 °C.
- Carefully transfer the supernatant (S2A) containing mitochondria into a new 2 ml tube and keep it on ice until Step B1I.
Note: To aspirate the supernatant direct the pipette tip to the tube side wall and leave some remaining fluid over the pellet in order to avoid any contamination with the pellet containing nuclei, cell debris, unbroken cells! Keep on ice. - Wash the pellet P2 in 0.5 ml ice-cold Disruption Buffer supplemented with Protease Inhibitor Solution. Resuspend it as described in Step B1h and centrifuge the suspension at 1,000 x g for 10 min at 4 °C.
- Carefully take the supernatant S2B and add it to S2A. Keep it on ice until the next step. The pellet P2 contains nuclei, cell debris, and unbroken cells. Resuspend it in 150 μl of Mitochondria Storage Buffer and aliquot in two vial, keep 10 µl for protein measurements, freeze immediately down in liquid N2 and store it at -80 °C until further analysis.
Note: Remove all of remaining fluid very carefully to avoid contaminations.
- Centrifuge combined supernatant S2A+B from Steps k and m at 6,000 x g for 10 min at 4 °C.
- The resulting supernatant from the above step (S3) constitutes the microsomal fraction. Carefully remove it and keep it on ice until Step ff.
- The pellet P3 contains a crude mitochondrial fraction.
Note: Remove all remaining fluid from the pellet and tube to avoid contaminations!
- Resuspend pellet P3 in 750 μl Mitochondria Purification Buffer by carefully pipetting up and down using a 1 ml pipette tip.
- Make the gradient (Figure 6):
- Pipette 750 µl Mitochondria Purification Buffer into a 2 ml tube.
- Slowly pipet 500 µl Disruption Buffer under the Mitochondria Purification Buffer by placing the pipette tip onto the bottom of the tube. Due to their different viscosities, buffers will not readily mix and 2 layers will be created (Figure 6A).
- Carefully pipet the 750 µl mitochondrial suspension (from step q) on top of the Mitochondria Purification Buffer layer, by directing the pipette tip to the side wall of the tube, so that the drop slowly runs down at the wall (Figure 6B).
- Centrifuge at 14,000 x g for 15 min at 4 °C.
- A soft pellet containing pure mitochondria will be formed at the bottom. Carefully remove 1.5 ml of the supernatant S4 without dislodging the mitochondrial pellet from the bottom of the tube (Figure 6C).
- Then, carefully aspirate the remaining 500 µl solution containing the pellet from the bottom of the tube and transfer into a new 2 ml tube using a top cut 1 ml pipette tip (Figure 6D).
- Add 1.5 ml Mitochondria Storage Buffer and resuspend the mitochondrial pellet using a 1 ml pipette tip.
- Centrifuge at 8,000 x g for 10 min at 4 °C.
- Wash mitochondrial pellet by repeating Steps B1w and B1x two times.
- The pellet should have a brownish color.
- Discard the supernatant, and resuspend pellet in 50 μl Mitochondria Storage Buffer, shock freeze in N2 and store it at -80 °C until proceeding further analysis.
- To extract the cytosolic proteins from the fraction obtained in Step B1g, add 4 volumes of ice-cold acetone to the supernatant and incubate for 15 min on ice.
- Centrifuge at 12,000 x g for 10 min at 4 °C.
- Discard the supernatant and wash the pellet with 1 ml 80% acetone.
- Air dry the pellet and resuspend it in 50 μl Mitochondria Storage Buffer. Store it at -80 °C until proceeding with further analysis.
- To extract the proteins from the microsomal fraction containing the cell membrane obtained in Step B1o, centrifuge supernatant S3 at 45,000 x g for 30 min at 4 °C in a Sorvall centrifuge.

Figure 6. Scheme illustrates the workflow of preparing the gradient to isolate pure mitochondria using QproteomeTM Mitochondria Isolation Kit
- Immediately after harvesting, weigh out 60 mg of kidney cortex and, place it into a 30 mm Petri dish on ice, cover it with 2 ml ice cold storage buffer and mince it into very small pieces (mush) using the razor blade.
- Isolation of mitochondria using differential centrifugation (Figure 7)
PREPARATION: Prepare Homogenization Buffer 10 ml/rat kidney and Isolation Buffer (500 µl/rat kidney) according to the recipes. Immediately before use, supplement Isolation Buffer with 0.1 mM phenylmethanesulfonyl fluoride (PMSF) (i.e., if using 50 ml Homogenization Buffer, add 100 µl of 50 mM PMSF). Filter through a 0.2 µm Filter. Additionally, you will need ice and per rat: 1x 10 mm Petri dishes, 1x razor blade, 1 x 1.5 ml and 4 x 15 ml tubes and 1 x 15 ml glass-glass Dounce homogenizers.
Note: All steps should be performed at 4 °C. Pre-cool all buffers, centrifuges, and other equipment before starting the procedure. Place the homogenizers overnight into -20 °C freezer.- Immediately after harvesting, weight out 1 g of kidney cortex, place it onto a 10 mm Petri dishes cooled by ice and mince into very small pieces (mush) using a razor blade.
- Transfer the kidney mush to the 17 ml glass POTTER-homogenizator and add 10 ml of Homogenization Buffer (ratio: kidney weight/Homogenization Buffer = 1 g/10 ml).
Note: Always remember to supplement Homogenization Buffer with 0.1 mM PMSF before use. - Homogenize the tissue by hand at 4 °C using a loose glass pestle. Slowly operate a pestle in an up-and-down manner, rotating it during the movement. Repeat up-and-down movement 5-10 times. Repeat the procedure using a tight glass pestle.
Note: Avoid creating air bubbles during the homogenization. - Leave homogenate for 2 min on ice.
- Transfer the homogenate into a 15 ml blue cup tube and centrifuge it at 100 x g for 10 min at 4 °C.
- Transfer the supernatant into a new 15 ml tube and centrifuge it at 300 x g for 10 min at 4 °C.
- Transfer the supernatant into a new 15 ml tube and centrifuge it at 500 x g for 10 min at 4 °C.
- Transfer the supernatant into a new 15 ml tube and centrifuge it at 1,000 x g for 10 min at 4 °C.
- Discard the supernatant and wash the pellet in 5 ml ice cold Isolation Buffer. Centrifuge it at 1,000 x g for 10 min at 4 °C.
- Discard the supernatant. The brownish pellet containing pure mitochondria fraction should be dissolved in 200-400 µl of ice-cold Isolation Buffer, transferred to a 1.5 ml Eppendorf tube, shock freezed in liquid N2 and stored at -80 °C until proceeding further analysis.

Figure 7. Scheme illustrates work flow of mitochondria isolation using differential centrifugation
- Isolation of pure mitochondrial fraction using QproteomeTM Mitochondria Isolation Kit (Figure 5)
- Protein isolation from renal cortex
PREPARATION: Prepare RIPA buffer according to the recipe. Immediately before use, supplement RIPA buffer with 1 tablet of Protease Inhibitor/50 ml buffer. Additionally, you will need ice, glass homogenizer with glass pestle and 1.5 ml and 0.5 ml tubes.
Notes:- Work on ice. Pre-cool all buffers, centrifuges and other equipment to 4 °C and homogenizers overnight in -20 °C before starting the procedure.
- As soon as lysis occurs during homogenization proteolysis, dephosphorylation and denaturation begin. These events can be slowed down significantly if samples are kept on ice or at 4 °C at all times and appropriate inhibitors are added fresh to the lysis buffer.
- Take around 100 mg of tissue, mince to fine mush with razor blade and place in 1 ml hand homogenizer.
- Add 500 µl of RIPA buffer to the homogenizer.
- Homogenize the tissue using simultaneous twisting and up/down movements. Work on ice.
- Transfer the homogenate to a new 1.5 ml tube.
- Wash the homogenizer with 500 µl RIPA buffer and transfer the buffer to the same tube and vortex.
Note: Wash the homogenizer 3 times with tap water and 3 times with double-distilled water (ddH2O) between the samples. - Allow the samples to stand 5 min on ice.
- Centrifuge the homogenate at 12,000 x g for 20 min at 4 °C. This separates the total protein (supernatant) from the cellular debris (pellet).
- Gently remove the tubes from the centrifuge and place it on ice, Aspirate the supernatant carefully directing the tip to the tube side wall.
- Transfer the supernatant to a new 1.5 ml tube kept on ice and shock-freeze the samples in liquid N2 and store at -80 °C if not needed immediately.
Note: Aliquot around 200 µl in an additional 0.5 ml tube for measuring the protein concentration.
- Measuring protein concentration
PREPARATION: Prepare beforehand 1 ml of 1 mg/ml Bovine Serum Albumin (BSA) as a standard. Dilute 1 part Bio-Rad Protein Assay Dye Reagent Concentrate with 4 parts deionized ddH2O. Filter through Whatman #1 filter (or equivalent) to remove particulates. Additionally, you will need ice, several cuvettes and parafilm.
Note: Pre-cool all buffers to 4 °C before starting the procedure.- Prepare 14 cuvettes for the standard curve and 3 cuvettes for each sample (measure always triplicates) add 1 ml of diluted Bio-Rad Protein Assay Dye Reagent to each.
- To perform a standard curve, you will need 1 cuvette with 0 µg BSA (reference) and 2 cuvettes per 5, 10, 20, 30, 50 µg BSA each (standard curve sample).
Note: i.e., to prepare a cuvette with 5 µg BSA, add 5 µl of 1 mg/ml BSA to 1 ml reagent. - Cover the top with parafilm and mix carefully by inverting. Incubate for 10 min at room temperature.
- Measure the protein absorption at 595 nm and transfer the data to excel file. Create a curve and trend line with R2 and equation.
Note: Proceed only, if you are satisfied with standard curve and R2. Otherwise repeat the Steps D1-D4. - If the standard curve is good proceed with your samples. Prepare for each sample three cuvettes each with 1 ml of Bio-Rad Protein Assay Dye Reagent, adding 1 µl of sample successively to the cuvette, trying to attain the color in linear range of your standard curve samples. (Triplicate the measurement for better accuracy.)
Note: Take note of the total volume added and always add the same volume of each sample. - Cover the top with parafilm and mix carefully by inverting. Incubate for 10 min at room temperature.
- Measure the protein absorption at 595 nm and transfer the data to an excel file.
- Calculate the average absorption for each sample and use the standard curve equation for calculating the protein concentration in each sample. Your standard curve will have the following form:
y=m∙x+b
where, y = absorption, m = slope, x = protein concentration and b = y-intercept.
To calculate the protein concentration, which depends on the added sample volume, change the formula to: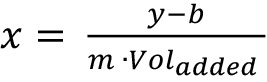 .
.
- Western Blotting
To validate the purity and structural integrity of isolated mitochondria, we describe below a detailed protocol for Western blot performed on subcellular fractions or proteins isolated from the whole kidney cortex. We used antibodies listed in Table 1 to identify specific marker proteins for the individual fractions.
Table 1. Antibodies, targeting specific organelle marker
- Sample Preparation
PREPARATION: Pre-heat your heating block to 97 °C or 50 °C depending on the antibody used. You will further need LDS sample buffer, NuPAGETM Sample Reducing Agent (10x), 1x MES SDS running buffer, NuPAGETM Transfer Buffer , XCell SureLock® Mini-Cell gel running tank and NuPAGETM Novex® 4-12% Bis-Tris Protein Gel, SeeBlue Plus 2 Marker.
Note: Work with 0.5 ml (or smaller) tubes.- Take 20 µg of sample protein and dilute as appropriate in ddH2O to obtain the total volume of 13 µl.
- Add 5 µl of NuPAGETM LDS Sample Buffer (4x) and 2 µl of NuPAGETM Sample Reducing Agent (10x).
- Denature the proteins by heating the samples at 97 °C or 50 °C for 10 min, depending on the antibodies used.
Notes:- Antibodies typically recognize a small epitope of the protein of interest. To enable access of the antibody to this portion it is necessary to unfold the protein, i.e., denature it.
- Exceptional treatment when OXPHOS cocktail is used. The antibody CIV:MTC01 is a highly hydrophobic protein, which is very sensitive to heating. That’s why proteins for the Western blotting using this antibody cocktail are only heated at 50 °C!!!
- Gel electrophoresis
- Set up the XCell SureLock® Mini-Cell gel running tank and NuPAGETM Novex® 4-12% Bis-Tris Protein Gel according to the manufacturer's instruction. Fill the chamber with ice-cold 1x MES SDS Running Buffer.
Note: Wash the wells 3 times with 1x MES SDS Running Buffer beforehand to remove gel residues. - Load 8 µl of SeeBlue Plus 2 Marker in one well.
- Load 20 µl of sample into the remaining wells.
- Run electrophoresis at 190 V for 45 min, or until the dye reaches the end of the gel.
- Set up the XCell SureLock® Mini-Cell gel running tank and NuPAGETM Novex® 4-12% Bis-Tris Protein Gel according to the manufacturer's instruction. Fill the chamber with ice-cold 1x MES SDS Running Buffer.
- Protein transfer
PREPARATION: Cut Whatman- filter paper and PVDF membrane to the appropriate size for the transfer chamber. Soak pads and filter paper in NuPAGETM Transfer Buffer until saturated.
Note: Use the gel immediately following the run. Do not soak the gel in transfer buffer.- During the run of electrophoresis activate the PVDF membrane by placing it in methanol for 30 s. Then briefly rinse in ddH2O and place the membrane in a shallow dish and wash with ice-cold NuPAGETM Transfer Buffer for at least one minute.
- Immediately after electrophoresis, assemble the XCell IITM Blot Module according to the manufacturer's instruction. The construct should be built in the chamber for electroblotting as presented in Figure 8.
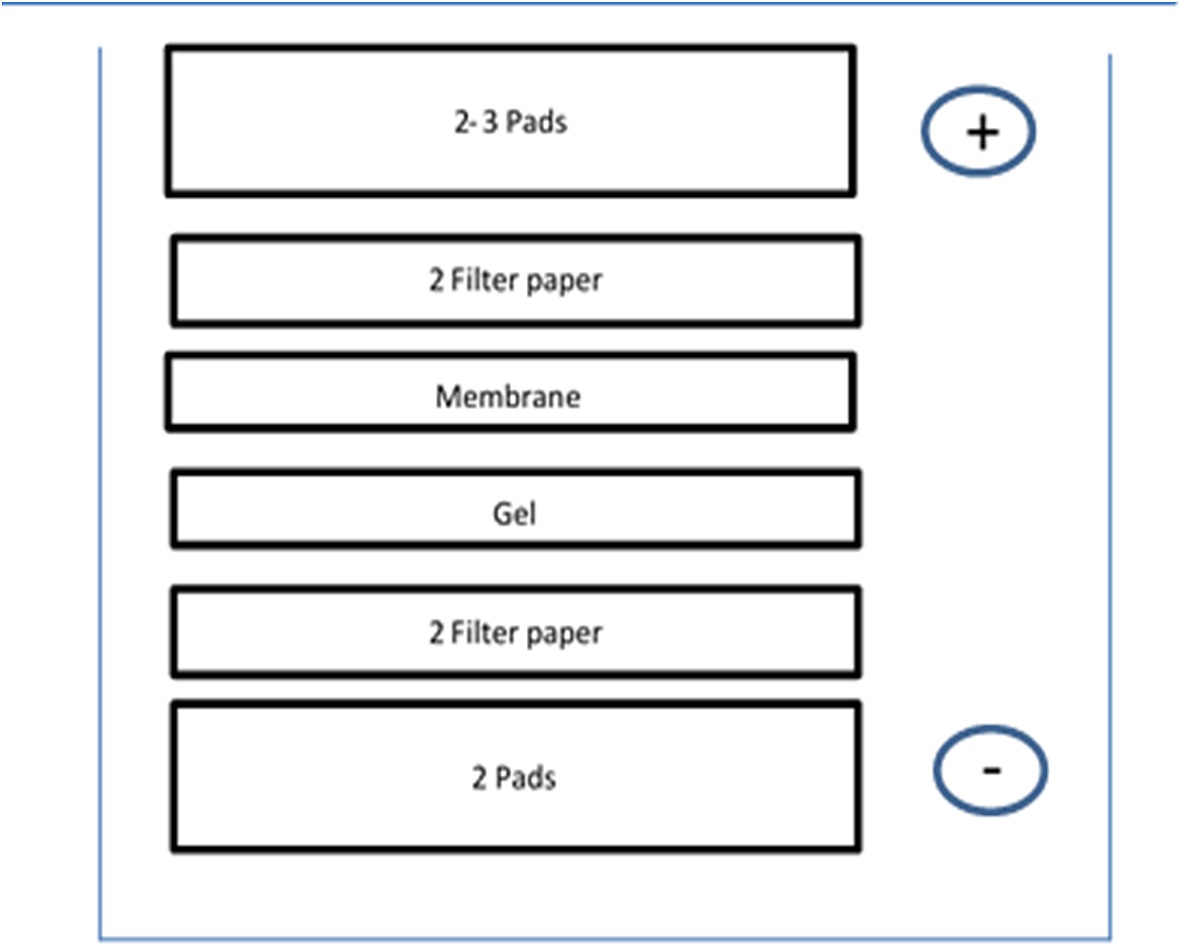
Figure 8. Electroblotting construct - Place the construct in the chamber and fill the electroblotting construct with ice-cold NuPAGETM Transfer Buffer. Fill the chamber with ddH2O.
- Carry out the blotting procedure at 30 V for 90 min.
- Immunodetection
PREPARATION: Prepare TBST Buffer and Blocking Buffer beforehand. Use only plastic forceps to handle membrane.- To prevent unspecific binding of antibodies, block the membrane with Blocking Buffer. Incubate for 90 min at room temperature with gentle agitation on a rocket shaker.
Note: You can also block overnight at 4 °C with agitation. - Dilute primary antibodies in Blocking Buffer to the recommended dilution (see Materials and Reagents)
- Incubate the membrane with the appropriate dilutions of primary antibody in Blocking Buffer overnight at 4 °C with agitation.
- Wash the membrane in TBST Buffer three times for 5 min each at room temperature.
- Incubate the membrane with the recommended dilution of appropriate horseradish peroxidase (HRP)-conjugated secondary antibody in blocking buffer at room temperature for 1 h with agitation (see Materials and Reagents).
- Wash the membrane in TBST Buffer three times for 5 min each at room temperature.
- After washing, remove excess fluid gently via capillary action by touching filter to the edge of the block, place the membrane on a plastic foil and add 1 ml Western Blotting Luminol Reagent Solution gel (prepare the working solution just before use) according to the manufacturer's instruction), make sure that the membrane is adequately covered with reagent and incubate the membrane for 1 min.
Note: Horseradish peroxidase (HRP)-conjugated to secondary antibody catalyzes the oxidation of luminol to 3-aminophthalate in the presence of oxygen peroxide and the reaction is accompanied by light emission at 428 nm. - Detect signal using an appropriate imaging system.
Note: Try three exposures; 30 s, 2 min, and 5 min.
- To prevent unspecific binding of antibodies, block the membrane with Blocking Buffer. Incubate for 90 min at room temperature with gentle agitation on a rocket shaker.
- Sample Preparation
Recipes
- Homogenization Buffer, pH 7.4 (100 ml)
Note: Buffer should be stored at 4 °C for a maximum of one week and used ice cold.
Notes:Final con. Volume of stock Stock buffer 250 mM sucrose 25 ml 1 M Sucrose 10 mM Tris-HCl 1 ml 1 M Tris-HCl pH 7.4 2 mM EDTA 0.4 ml 0.5 M EDTA pH 8.0 H2O Add up to 100 ml 0.1 mM PMSF* - Adjust the pH to 7.4 with 37% HCl before adding PMSF.
- *Add PMSF just before use (stable in water solution for 30 min). For 50 ml of Homogenization Buffer, add 100 µl 50 mM PMSF.
- Isolation buffer, pH 7.4 (100 ml)
Note: Buffer should be stored at 4 °C for a maximum of one week and used ice cold.
Adjust the pH to 7.4 with 10 M KOHFinal con. Volume of stock Stock buffer 210 mM mannitol 21 ml 1 M Mannitol 70 mM Sucrose 7 ml 1 M Sucrose 10 mM MgCl2 1 ml 1 M MgCl2 5 mM K2HPO4 0.5 ml 1 M K2HPO4 10 mM MOPS 2 ml 0.5 M MOPS 1 mM EGTA 1 ml 100 mM EGTA H2O Add up to 100 ml - RIPA buffer, pH 7.2 (100 ml)
Note: Aliquot in 50 ml tubes and store at -20 °C. Shortly before using, add 1 tablet of Protease Inhibitor/50 ml.
Adjust the pH to 7.2 with 1 M HClFinal con. Volume of stock Stock buffer 50 mM Tris-HCl 5 ml 1 M Tris-HCl pH 7.4 2.5 mM EDTA 0.5 ml 0.5 M EDTA pH 8.0 2% IGEPAL CA-630 2 ml IGEPAL CA-630 0.1% SDS 1 ml 10% SDS 250 mM NaCl 5 ml 5 M NaCl 0.5% Sodium Deoxycholate 5 ml 10% Sodium Deoxycholate H2O Add up to 100 ml - 1x TBS, pH 7.4 (1,000 ml)
Adjust the pH to 7.4 with 1 M HClFinal con. Volume of stock Stock buffer 20 mM Tris-HCl 20 ml 1 M Tris-HCl pH 7.4 150 mM NaCl 30 ml 5 M NaCl H2O Add up to 1000 ml - TBST (1,000 ml)
0.1% Tween-20/TBST 1 ml Tween-20/1,000 ml TBS - Blocking Buffer (100 ml)
5% Milk powder/TBST 5 g Milk powder/100 ml TBSTStock solutions for buffers
- 1 M Sucrose
Sucrose 171.5 g in 500 µl ddH2O, filtrate - 1 M Tris-HCl
Trizma base 12.114 g in 100 ml ddH2O
Adjust the pH to 7.4 with HCl, autoclave - 0.5 M EDTA
EDTA·2H2O 18.612 g in 100 ml ddH2O
Adjust the pH to 8, autoclave - 1 M Mannitol
Mannitol 91.085 g in 500 µl ddH2O, filtrate - 1 M MgCl2
MgCl2·6H2O 20.33 g in 100 ml ddH2O - 0.5 M MOPS
MOPS 10.465 g in 100 ml ddH2O - 100 mM EGTA
EGTA 3.804 g in 100 ml ddH2O - 1 M K2HPO4
K2HPO4·3H2O 22.822 g in 100 ml ddH2O - 50 mM PMSF
PMSF 0.087 g in 10 ml abs. EtOH, store at 4 °C, light protected
Acknowledgments
The research was funded by grants from Deutsche Forschungsgemeinschaft (SFB 1118) and by the grant of the NIH (5RO1DK100482).
Competing interests
The authors have declared that no conflict of financial and non-financial competing interests exists.
Ethics
All animal studies were conducted in accordance with federal and local laws and were approved by the Regierungspräsidium Karlsruhe, Land Baden-Württemberg (G21/13, I-15/09, I-15/10).
References
- Antico Arciuch, V. G., Elguero, M. E., Poderoso, J. J. and Carreras, M. C. (2012). Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 16(10): 1150-1180.
- Arora, M. K. and Singh, U. K. (2014). Oxidative stress: meeting multiple targets in pathogenesis of diabetic nephropathy. Curr Drug Targets 15(5): 531-538.
- Graham, J. M. (2001). Isolation of mitochondria from tissues and cells by differential centrifugation. Curr Protoc Cell Biol Chapter 3: Unit 3.3.
- Gross, V. S., Greenberg, H. K., Baranov, S. V., Carlson, G. M., Stavrovskaya, I. G., Lazarev, A. V. and Kristal, B. S. (2011). Isolation of functional mitochondria from rat kidney and skeletal muscle without manual homogenization. Anal Biochem 418(2): 213-223.
- Higgins, G. C. and Coughlan, M. T. (2014). Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol 171(8): 1917-1942.
- Lin, C. C., Kurashige, M., Liu, Y., Terabayashi, T., Ishimoto, Y., Wang, T., Choudhary, V., Hobbs, R., Liu, L. K., Lee, P. H., Outeda, P., Zhou, F., Restifo, N. P., Watnick, T., Kawano, H., Horie, S., Prinz, W., Xu, H., Menezes, L. F. and Germino, G. G. (2018). A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci Rep 8(1): 2743.
- Micakovic, T., Papagiannarou, S., Clark, E., Kuzay, Y., Abramovic, K., Peters, J., Sticht, C., Volk, N., Fleming, T., Nawroth, P., Hammes, H. P., Alenina, N., Grone, H. J. and Hoffmann, S. C. (2018). The angiotensin II type 2 receptors protect renal tubule mitochondria in early stages of diabetes mellitus. Kidney Int 94(5): 937-950.
- Peters, J., Kranzlin, B., Schaeffer, S., Zimmer, J., Resch, S., Bachmann, S., Gretz, N. and Hackenthal, E. (1996). Presence of renin within intramitochondrial dense bodies of the rat adrenal cortex. Am J Physiol 271(3 Pt 1): E439-450.
- Peters, B., Podlich, D., Ritter, M., Muller, A., Wanka, H., Maser-Gluth, C., Seitz, C., de Boni, L., Maier, E., Gretz, N., Peters, J. and Hoffmann, S. C. (2012). A new transgenic rat model overexpressing the angiotensin II type 2 receptor provides evidence for inhibition of cell proliferation in the outer adrenal cortex. Am J Physiol Endocrinol Metab 302(9): E1044-1054.
- Silvagno, F., Consiglio, M., Foglizzo, V., Destefanis, M. and Pescarmona, G. (2013). Mitochondrial translocation of vitamin D receptor is mediated by the permeability transition pore in human keratinocyte cell line. PLoS One 8(1): e54716.
- Wanka, H., Kessler, N., Ellmer, J., Endlich, N., Peters, B. S., Clausmeyer, S. and Peters, J. (2009). Cytosolic renin is targeted to mitochondria and induces apoptosis in H9c2 rat cardiomyoblasts. J Cell Mol Med 13(9A): 2926-2937.
- Wilson, B. A., Nautiyal, M., Gwathmey, T. M., Rose, J. C. and Chappell, M. C. (2016). Evidence for a mitochondrial angiotensin-(1-7) system in the kidney. Am J Physiol Renal Physiol 310(7): F637-F645.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Micakovic, T., Banczyk, W. Z., Clark, E., Kränzlin, B., Peters, J. and Hoffmann, S. C. (2019). Isolation of Pure Mitochondria from Rat Kidneys and Western Blot of Mitochondrial Respiratory Chain Complexes. Bio-protocol 9(19): e3379. DOI: 10.21769/BioProtoc.3379.
Category
Cell Biology > Organelle isolation > Mitochondria
Molecular Biology > Protein > Intercellular translocation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.