- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In situ Hybridization of Plant-parasitic Nematode Globodera pallida Juveniles to Detect Gene Expression


Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3372 Views: 5565
Reviewed by: Demosthenis ChronisKrzysztof WieczorekAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In this study, we describe a standard whole mount in situ hybridization method which is used to determine the spatial-temporal expression pattern of genes from Globodera spp. Unlike more invasive radioactive labeling approaches, this technique is based on a safe, highly specific enzyme-linked immunoassay where a Digoxigenin (DIG)-tagged anti-sense probe hybridized to a target transcript is detected by anti-DIG antibodies conjugated with alkaline phosphatase enzyme (AP) (anti-DIG-AP). The hybrid molecules are visualized through an AP-catalyzed color reaction using as the substrate 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and nitro blue tetrazolium chloride (NBT). This method can be applied to both free-living pre-parasitic juveniles and early endoparasitic stages of cyst nematodes.
Keywords: In situ hybridizationBackground
Potato cyst nematodes (PCNs), Globodera pallida and G. rostochiensis, are a global threat to a potato production, causing in excess of 80% yield loss in infested fields (Brodie, 1989). PCNs are highly specialized sedentary endoparasites undergoing a complex life cycle with six developmental stages: egg, four juvenile stages, and male or female adult. The second stage juveniles (J2) hatched from eggs in soil are free-living mobile pre-parasitic nematodes that recognize and invade plant roots. After reaching cells in the root vascular cylinder, J2s become sedentary, feed and molt into third and fourth parasitic stage (J3 and J4, respectively). To complete their life cycle, nematodes undergo sexual differentiation into males and females. Following sexual reproduction, eggs are laid within the female body which eventually becomes a cyst. One way to understand this complex life cycle, is to use the in situ hybridization technique to monitor in vivo spatial gene expression at different nematode developmental stages to gain insight into the function of those genes. The in situ hybridization is a highly specific and sensitive assay based on the immunodetection of DIG-tagged probes hybridized to a target transcript. First, the polymerase chain reaction (PCR) is used to generate labeled probes through randomly incorporating DIG-coupled dUTP during enzymatic amplification of the cDNA template. Second, biological samples are fixed, mechanically cut, enzymatically permeabilized, and incubated with DIG-labeled probes for hybridization. Finally, hybridized probes are selectively detected by anti-DIG antibodies conjugated with alkaline phosphatase enzyme (anti-DIG-AP). AP catalyzes a color reaction using the substrate BCIP and NBT to visualize targeted hybrid molecules. Described here, the in situ hybridization protocol has been adapted from de Boer et al. (1998) and optimized for Globodera spp. Although this method has been routinely used to confirm esophageal gland expression of nematode effector genes, it can be applied to detect the expression pattern of any other nematode gene (Jones et al., 2002).
Materials and Reagents
- Laboratory gloves
- RNaseZap® RNase Decontamination Wipes (Thermo Fisher Scientific, catalog number: AM9786)
- 15 ml glass tubes
- 0.5 ml and 1.5 ml nonstick microcentrifuge tubes (e.g., VWR, catalog numbers: 20170-315 and 20170-650)
- 0.2 ml PCR tubes (e.g., Thermo Fisher Scientific, catalog number: E0030124707)
- Microscope slides and coverslips (e.g., Thermo Fisher Scientific, catalog numbers: 12-550-A3 and 10-016-24)
- Razor blades (e.g., Thermo Fisher Scientific, catalog number:12-640)
- Filtered DNase free tips (e.g., Mettler-Toledo, Rainin, catalog numbers: 17007957, 17002927, 17014361)
- Nuclease-free water (e.g., Thermo Fisher Scientific, catalog number: AM9937)
- Taq PCR polymerase (New England BioLabs, catalog number: M0273S)
- Deoxynucleotides (dNTPs) (Thermo Fisher Scientific, catalog number: 10297117)
- Forward and reverse primers for in situ hybridization probes (e.g., Sigma-Aldrich)
- PCR Purification Kit (e.g., ZYMO RESEARCH, catalog number: D4033)
- Digoxigenin (DIG) DNA Labeling Mix (Roche Diagnostics, catalog number: 11277065910)
- Proteinase K 20 mg/ml (Roche Diagnostics, catalog number: 03115887001)
- Boehringer blocking reagent (Roche Diagnostics, catalog number: 11096176001)
- Anti-Digoxigenin-AP-Fab fragments (Roche Diagnostics, catalog number:11093274910)
- 5-bromo-4-chloro-3-indolyl-phosphate, 4-toluidine salt BCIP (Roche Diagnostics, catalog number: 11383221001)
- 4-Nitro blue tetrazolium chloride NBT (Roche Diagnostics, catalog number: 11383213001)
- DNA sodium salt from salmon testes (Sigma-Aldrich, catalog number: D1626)
- tRNA from baker’s yeast (Sigma-Aldrich, catalog number: R8759, type X-SA)
- Denhardt’s solution 50x (Sigma-Aldrich, catalog number: D2532)
- Dry ice
- Sucrose (e.g., Sigma-Aldrich, catalog number: S0389)
- Agarose (e.g., VWR, catalog number: 0710)
- Potassium phosphate monobasic (KH2PO4) (e.g., Sigma-Aldrich, catalog number: P9791)
- Sodium phosphate dibasic (Na2HPO4) (e.g., Sigma-Aldrich, catalog number: S7907)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653)
- Sodium citrate (Na3C6H5O7) (Sigma-Aldrich, catalog number: 1613859)
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: 63138)
- 0.5 M EDTA pH 8 (Sigma-Aldrich, catalog number: 324504)
- Tris-base (Sigma-Aldrich, catalog number: T1503)
- Tween-20 (Sigma-Aldrich, catalog number: P1379)
- Acetone (Sigma-Aldrich, catalog number: 650501)
- Methanol (Sigma-Aldrich, catalog number: 34860)
- 37% formaldehyde solution (Sigma-Aldrich, catalog number: F15587)
- Maleic acid (Sigma-Aldrich, catalog number: M0375)
- SDS (Sigma-Aldrich, catalog number: 1614363)
- Formamide deionized (Sigma-Aldrich, catalog number: F9037)
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: H1758)
- Transparent nail polish (e.g., Pure Ice)
- Globodera pallida–pre-parasitic J2s (10,000) or parasitic stages (> 100)
- M9 buffer (see Recipes)
- Fixation Buffer (see Recipes)
- 20x SSC (see Recipes)
- Hybridization Buffer (see Recipes)
- Washing Buffer A (see Recipes)
- Washing Buffer B (see Recipes)
- Maleic Acid Buffer (see Recipes)
- Blocking Buffer (see Recipes)
- Detection Buffer (see Recipes)
Note: Recipes for in situ hybridization buffers can be found at the end of this protocol.
Equipment
- Pipets (e.g., Rainin, models: P2, P20, P200, P1000)
- Sieve of 2.8 mm/500 μm/250 μm/90 μm/25 μm/20 μm (e.g., Humboldt, catalog numbers: No. 7, 35, 60, 170, 500, 635)
- PCR thermocycler (e.g., Bio-Rad, model: T100)
- DNA electrophoresis system (e.g., Bio-Rad) and imaging system (e.g., Azure Biosystems, model: c300)
- NanoDrop (e.g., Thermo Fisher Scientific, model: 2000 Spectrophotometer)
- Centrifuge for 15 ml glass tubes (e.g., Eppendorf, model: 5804 R)
- Laboratory blender (e.g., Waring, model: WF2211214)
- Microcentrifuge for 0.5 ml and 1.5 ml tubes (e.g., Eppendorf, model: 5424)
- Tube rotator (e.g., Thermo Fisher Scientific, catalog number: 88881001)
- 4 °C Fridge
- -20 °C Freezer
- Hybridization oven (e.g., VWR, model: 230402V)
- Mini block heater (e.g., VWR, model: 10153-318)
- Light Inverted Microscope (Leica, model: DMi8)
Software
- Microscope imagining software–LAS V4.12 (Leica, https://www.leica-microsystems.com/products/microscope-software/p/leica-application-suite/)
Procedure
- Generate sense and anti-sense cDNA probes
- Identify a unique 200-250 bp region in the cDNA sequence of the gene of interest that will be detected by in situ probe and design standard PCR primers for that DNA fragment.
- Obtain nematode RNA and synthesize cDNA as described (Casavant et al., 2017).
- Follow the standard PCR protocol for the DNA polymerase (e.g., Taq polymerase) to amplify desired gene fragment from nematode cDNA. Verify a single PCR product by running a small amount on a 1% DNA agarose gel and purify it using commercially available PCR purification kit.
- Set up two independent asymmetric PCR reactions with DIG DNA Labeling Mix (1 mM dATP, 1 mM dCTP, 1 mM dGTP, 0.65 mM dTTP, 0.35 mM DIG-dUTP) to separately synthesize sense (negative control) and anti-sense cDNA probes as follows:
X µl PCR template from A3 (20 ng)
4 µl 5x Taq Buffer
2 µl DIG DNA Labeling Mix
4 µl 10 µM Forward primer (sense probe) OR Reverse primer (anti-sense probe)
1 µl Taq polymerase
X µl ddH2O
Total volume 20 µl
Run the following program in a PCR cycler as:- 95 °C – 30 s
- 95 °C – 15 s
- 60 °C – 30 s (temperature might be adjusted depending on the primers)
- 68 °C – 90 s (repeat b-c 35 times)
- 68 °C – 5 min
- Check your DIG-labeling PCR step, by running a small amount of labeled probes on the 1% DNA agarose gel site-by-site with the original PCR template. The labeled probes should show an increase in molecular mass compare with unlabeled DNA due to the incorporated DIG (Figure 1).
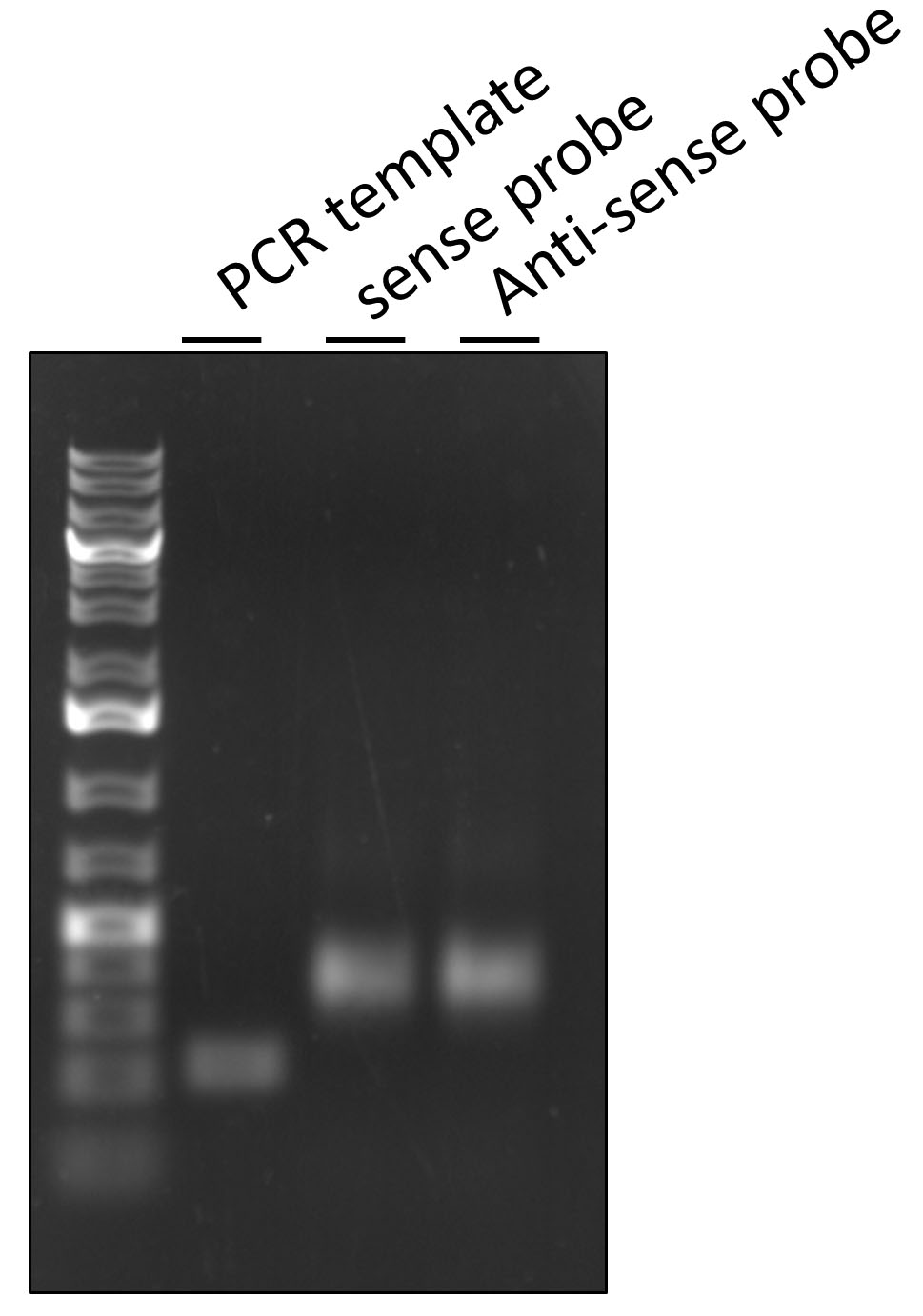
Figure 1. DNA 1% agarose gel to check DIG-labeling PCR step. Lane 1–PCR template, Lane 2–DIG-labelled sense negative probe synthesized with Forward primer, and Lane 3–DIG-labeled anti-sense probe synthesized with Reverse primer. Both DIG-labeled probes have increased molecular mass due to incorporation of DIG. - Purify DIG-labeled probes with commercially available PCR purification kit and store them at -20 °C until needed.
- Fixation–Day 1
- Collect nematode juveniles
- For pre-parasitic juveniles
- Hatched J2s nematodes as described previously (Casavant et al., 2017).
- Spin down freshly hatched J2s nematodes by centrifugation in 15 ml glass tubes (150 x g for 5 min, 4 °C) and remove supernatant.
- For parasitic juveniles
- Infect potato plants with G. pallida cysts (~ 10 cysts/6-inch pot) as described previously (Dandurand and Knudsen, 2016).
- At the desired infection stage, gently pull the infected root system from the soil and rinse it with water to remove larger soil particles.
- Cut roots into 2 cm long sections, place in a blender, cover with water, and blend 5 times for 2 s at the low setting.
- To separate nematode juveniles from root debris, pour the blended root-nematode suspension through a series of sieves (2.8 mm, 500 μm, 250 μm, 90 μm, 20 μm). Nematodes will be collected on the last 20 μm sieve.
Note: Re-blend larger roots and repeat this step if needed. - To remove small soil and root residue, purify the nematodes by sucrose gradient centrifugation.1)Prepare 50% w/v sucrose solution and cool at 4 °C.
Note: Sucrose solution can be made a day earlier and stored overnight at 4 °C.2)In 15 ml glass tubes add 5 ml of cold 50% sucrose, then gently overlay it with 5 ml of the collected nematode solution.3)Spin down tubes for 10 min at 1,250 x g at 4 °C (acceleration and break set to “zero”). Nematodes will accumulate at the water-sucrose solution border, whereas, small soil particles and root residues will pellet on the bottom of the tube (Figure 2).4)Collect the nematode layer with P1000 pipette and wash nematodes 5 times with water on the 25 μm sieve to remove sucrose.
Figure 2. Purification of parasitic nematodes through sucrose gradient centrifugation
- For pre-parasitic juveniles
- Transfer nematodes (10,000 pre-parasitic J2s or > 100 parasitic worms) to a 1.5 ml nonstick microcentrifuge tube.
- Wash twice by resuspending nematodes in 1 ml of water and then centrifuge them at 6,000 x g for 2 min.
- Remove as much water as possible and resuspend nematodes in 1 ml of Fixation Buffer.
- Fixate nematodes by lying tubes flat for 18 h at 4 °C, then move samples to room temperature and incubate for additional 4 h.
- Collect nematode juveniles
- Cutting of nematodes, permeabilization and hybridization–Day 2
- Wipe everything down with RNase wipes to protect RNA.
- Centrifuge nematodes (6,000 x g for 2 min), remove supernatant and resuspend nematode pellet with 200 μl of 10-fold diluted (in M9 buffer) Fixation Buffer.
- Spread ~70 μl of the nematode suspension onto a glass slide and cut them using a flat razor blade until ~80% of nematodes are cut (inspect under microscope) (Figure 3).
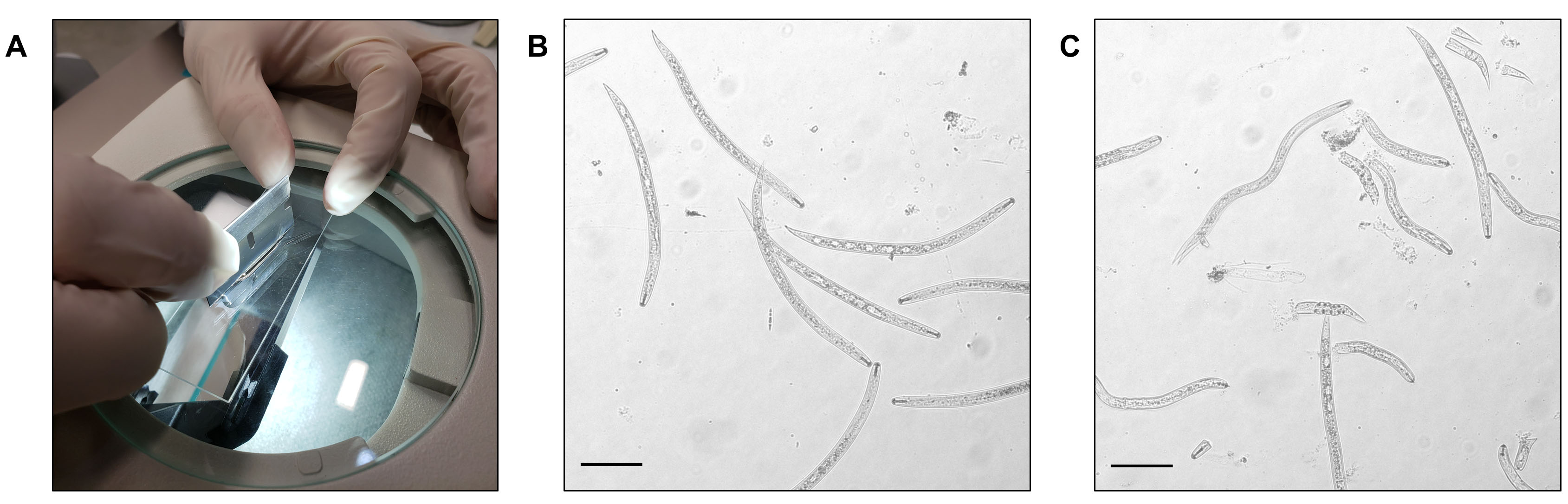
Figure 3. Cutting fixed nematodes. A. Cutting station. B. Nematodes before cutting. C. Nematodes after cutting. Scale bars = 100 μm. - Collect chopped nematodes into a 1.5 ml nonstick microcentrifuge tube by gently rinsing a glass slide with 10-fold diluted Fixation Buffer, and repeat Step C3 until all nematodes are cut.
- Spin down nematodes (6,000 x g for 2 min) and remove supernatant.
- Wash nematodes twice by resuspending in 500 μl of M9 buffer and then centrifuging at 6,000 x g for 2 min.
- Add 500 μl of proteinase K solution (0.5 mg/ml in M9 buffer) and carry out permeabilization on a rotator for 30 min at room temperature. For nematodes of parasitic stages, permeabilization time should be increased to 45-60 min.
- Spin down the sample and wash the nematode pellet twice with 500 μl of M9 buffer (6,000 x g for 2 min). After the second wash, remove as much M9 buffer as possible.
- Freeze the nematode pellet on dry ice for 15 min.
- Re-suspend nematodes in 1 ml of cold methanol (stored at -20 °C) and incubate nematode pellet on dry ice for an additional 30 s.
- Spin down the nematodes at 21,000 x g for 30 s (room temperature) and remove the methanol.
- Re-suspend the nematodes in 1 ml of cold acetone (stored at -20 °C) and incubate the nematode pellet on dry ice for 1 min.
- Spin down the nematodes at 21,000 x g for 30 s and discard all but 100 μl of the acetone.
- Slowly, drop-by-drop, rehydrate the sample by adding 100 μl of H2O.
- Pre-heat the Hybridization Buffer at 50 °C.
- Spin down the sample and wash the nematode pellet with 500 μl of the Hybridization Buffer (6,000 x g for 2 min at room temperature). Discard the supernatant.
- Add 300 μl of the pre-heated Hybridization Buffer and transfer 150 μl aliquots to two 0.5 ml nonstick microcentrifuge tubes (for sense and anti-sense sample).
- Pre-hybridize samples for 15 min at 50 °C in the hybridization oven.
- MEANWHILE: denature probes for 10 min at 100 °C and place them on ice immediately to prevent re-annealing.
- Add separately sense and anti-sense probes to two nematode samples.
- Hybridize by rotating in a hybridization oven at 50 °C overnight.
Note: Hybridization temperature might be adjusted depending on length and probe specificity.
- Washing and staining–Day 3
- Spin down the nematodes (6,000 x g for 2 min at room temperature) and remove the supernatant.
- Wash the samples 3 times each with 100 μl of Wash Buffer 1 by rotating 15 min in hybridization oven at 50 °C followed by centrifugation at 6,000 x g for 2 min.
- Wash the samples 3 times each with 100 μl of Wash Buffer 2 by rotating 20 min in a hybridization oven at 50 °C followed by centrifugation at 6,000 x g for 2 min (room temperature).
- Wash samples by resuspending them in 100 μl of Maleic Acid Buffer and then centrifuging at 6,000 x g for 2 min.
- Add 100 μl of Blocking Buffer and incubate for 30 min by rotating.
- Spin down samples (6,000 x g for 2 min), resuspend nematode pellet in 100 μl of Blocking Buffer containing the anti-DIG-AP (1 μl anti-DIG-AP/1,000 μl Blocking Buffer), and incubate while rotating for 2 h at room temperate.
- Wash the samples 3 times with 100 μl of Maleic Acid Buffer each by rotating for 15 min followed by centrifugation at 6,000 x g for 2 min.
- Wash the samples briefly with 100 μl Detection Buffer (6,000 x g for 2 min).
- Add 150 μl of freshly made AP Substrate Solution (3.5 μl BCIP and 3.4 μl NBT in 1,000 μl Detection Buffer) and incubate overnight without agitation at 4 °C.
- Imaging–Day 4
- Wash the nematodes twice with 100 μl 0.01% Tween-20 for 1 min (6,000 x g for 2 min) and remove the supernatant.
- Resuspend the nematode pellet with 50 μl 0.01% Tween-20 and apply 10 μl/slide.
- Cover with coverslip and seal edges with nail polish.
- Examine stained nematode sections under microscope.
Data analysis
The stained nematode sections were analyzed under Leica Microscope and images were captured using LAS V4.12 imaging software. The representative results are described in the example of dorsal glad specific expression of Globodera pallida effector RHA1B (Kud et al., 2019) (Figure 4). The positive in situ hybridization signal in a parasitic-J2 is visualized as dark violet/brown coloring of dorsal glands (Figure 4A). In contrast, no signal is detected for the same gene in a pre-parasitic J2 (Figure 4B), showing that parasitic/developmental stage-dependent gene expression directly impacts in situ hybridization results. Positive results are typically accompanied by a negative control, sense probe-incubated sample with no signal detected (Figures 4C and 4D), to confirm signal specificity. The staining efficiency may vary from experiment to experiment; therefore, this assay should be repeated several times with similar results. At least 20 stained nematodes should be checked for each assay to ensure consistency.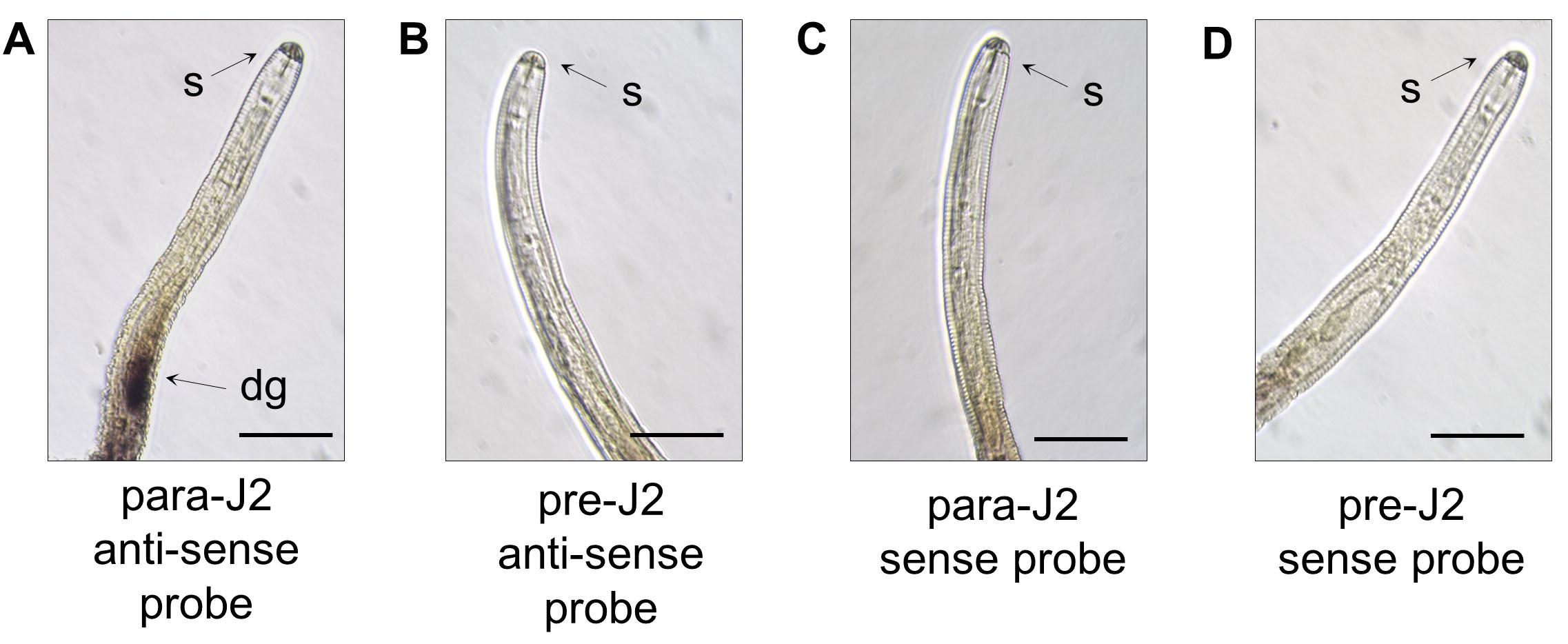
Figure 4. Representative results showing in situ hybridization results for a dorsal gland expression of G. pallida RHA1B effector. A. Sample with RHA1B anti-sense probe in a para-J2–strong signal confirming glad specific expression at this parasitic stage. B. Sample with RHA1B anti-sense probe in a pre-J2s–no signal detected due to very low expression or RHA1B at this stage. C. Negative control with RHA1B sense probe in a parasitic J2 (para-J2). D. Negative control with RHA1B sense probe in a pre-parasitic J2. Scale bars = 50 μm. s–stylet and dg–dorsal glands. This figure has been modified from Kud et al. (2019).
- Because even a small amount of RNase can compromise RNA integrity, it is recommended to wear gloves, use only RNase-free plastic- and glassware, and frequently wipe lab surfaces with commercially available decontamination reagents, such as RNaseZap wipes, to ensure RNase and DNase-free environment.
- Most of hybridization buffers can be purchased as premade stock solutions.
- Make sure to always use fresh deionized formamide for Fixation Buffer.
- The probe length is a tradeoff between sensitivity and penetrability, where a shorter probe gets into nematode tissue, but a longer probe results in better detection/hybridization to the tested transcript.
- If a strong background signal is observed, a few troubleshooting steps can be applied. 1) Designing a probe that spans two exons will reduce its affinity to DNA. 2) In our hands, hybridization temperature of 50 °C is a good starting point. Depending on length and probe specificity, the perfect hybridization temperature might be optimized experimentally (range of 45 °C to 55 °C). 3) Although generally the wash temperature should match the hybridization temperature, slightly increasing that temperature and/or SSC salt concentration can reduce background signal.
- To additionally validate in situ hybridization results, the second in situ probe aligning with a different region of transcript of interest can be used.
Recipes
- M9 buffer (pH 7)
22 mM KH2PO4
42 mM Na2HPO4
86 mM NaCl
Sterilize by autoclaving - Fixation Buffer
2% formaldehyde
M9 buffer - 20x SSC (pH 7)
3 M NaCl
0.3 M sodium citrate
Sterilize by autoclaving - Hybridization Buffer
50% Deionized formamide
4x SSC
1% Blocking reagent
2% SDS
1x Denhardt’s solution
1 mM EDTA, pH 8
200 μg/ml DNA sodium salt from salmon testes
3.125 U/ml Yeast tRNA - Washing Buffer A
4x SSC - Washing Buffer B
0.1x SSC
0.1% SDS - Maleic Acid Buffer (pH 7.5)
0.1 M maleic acid
0.15 M NaCl
Sterilize by autoclaving - Blocking Buffer
1% Boehringer blocking reagent
Maleic Acid Buffer
Sterilize by autoclaving - Detection Buffer (pH 9.5)
0.1 M Tris-HCl
0.1 M NaCl
50 mM MgCl2·6H2O
Acknowledgments
This work was supported by the Agriculture and Food Research Initiative competitive grant (2015-69004-23634, 2017-67014-26197; 2017-67014-26591) of the USDA National Institute of Food and Agriculture, USDA-NIFA Farm Bill, Northwest Potato Consortium, and ISDA Specialty Crop.
Described here in situ hybridization protocol has been adapted from de Boer et al. (1998). We would like to thank Dr. John Jones’ and Dr. Vivian Block’s lab at The James Hutton Institute for providing useful tips to optimize this method.
Competing interests
The authors declare no financial and non-financial competing interests.
References
- Brodie, B. B. (1989). Control of the golden nematode in the United States. Annu. Rev. Phytopathol 27: 443-461.
- Casavant, N. C., Kuhl, J. C., Xiao, F., Caplan, A. B. and Dandurand, L. M. (2017). Assessment of Globodera pallida RNA extracted from solanum roots. J Nematol 49(1): 12-20.
- Dandurand, L.-M. and Knudsen, G. R. (2016). Effect of the trap crop Solanum sisymbriifolium and two biocontrol fungi on reproduction of the potato cyst nematode, Globodera pallida. Annals of Applied Biology 169(2): 180-189.
- de Boer, J. M., Yan, Y., Smant, G., Davis, E. L. and Baum, T. J. (1998). In-situ hybridization to messenger RNA in Heterodera glycines. J Nematol 30(3): 309-312.
- Jones, J., Blok, V. and Smant, G. (2002). SXP/RAL-2 proteins of the potato cyst nematode Globodera rostochiensis: secreted proteins of the hypodermis and amphids. Nematology 2(8): 887-893.
- Kud, J., Wang, W., Gross, R., Fan, Y., Huang, L., Yuan, Y., Gray, A., Duarte, A., Kuhl, J. C., Caplan, A., Goverse, A., Liu, Y., Dandurand, L. M. and Xiao, F. (2019). The potato cyst nematode effector RHA1B is a ubiquitin ligase and uses two distinct mechanisms to suppress plant immune signaling. PLoS Pathog 15(4): e1007720.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kud, J., Solo, N., Caplan, A., Kuhl, J. C., Dandurand, L. and Xiao, F. (2019). In situ Hybridization of Plant-parasitic Nematode Globodera pallida Juveniles to Detect Gene Expression. Bio-protocol 9(18): e3372. DOI: 10.21769/BioProtoc.3372.
Category
Molecular Biology > DNA > DNA labeling
Cell Biology > Cell imaging > Fixed-tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.