- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Production of Quantum Dots-containing Influenza Virus Particles for Studying Viral Uncoating Processes

Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3366 Views: 6427
Reviewed by: George William CarnellWelsch Charles JeremyAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The genome of influenza A virus (IAV) comprises eight pinlike genomic segments called vRNPs enclosed in viral capsid. During infection, uncoating is the key step for viral replication and represents an antiviral therapeutic target, but it is difficult to observe the transient and dynamic event in detail. Here, we report a protocol for production of quantum dots-containing influenza virus particles by encapsulating quantum dot-conjugated vRNPs during viral assembly. These labeled virions can be used for monitoring viral trafficking in real time and studying viral uncoating processes.
Keywords: Influenza A virusBackground
Influenza A virus (IAV) is one of the most important human pathogens. Viral entry before virus-endosome fusion has been widely studied by various methods, including single-particle tracking. However, the viral uncoating process following endocytosis remains elusive. Robust live imaging strategies that offer high temporal and spatial resolution are essential to study this dynamic and transient event. However, the influenza RNA genome is intolerant to insertion of large genetic materials, and rescue of viruses using fluorescent protein fused to viral genomic core proteins have been limited (Lakdawala et al., 2014). This has delayed progress in the field of influenza-uncoating and vRNPs-dynamics live-imaging studies. Although purified vRNPs have been dye-labeled and microinjected into cells to track their intracellular transportation and nuclear import process in living cells in real time (Babcock et al., 2004), this method cannot capture the real infection process of incoming virions or determine vRNP behaviors during uncoating. Fluorescence in situ hybridization (FISH) and colocalization analyses with single-molecule resolution have also been used to study the behavior of vRNPs in infected cells (Chou et al., 2013), but the fixation procedures are not compatible with imaging vRNP dynamics in living cells. Semiconductor quantum dots (QDs) have unique optical properties, such as remarkable brightness and superior photostability, and are well-suited to single-particle tracking of viral infection. Single-particle tracking of virions with QDs-labeled genetic material provides an opportunity for tracking viral uncoating and post-uncoating genetic behaviors in real time. As a subunit of polymerase complex, PA protein participates in the composition of vRNP-genetic core of IAV. In PNAS, Qin et al. (2019) developed a nanotechnology that labels IAV viral ribonucleoprotein complexes (vRNPs) with QDs. Briefly, the biotin acceptor peptide (AP tag) was genetically fused to the PA protein to construct a recombinant PR8 strain. The rescued rPR8-PA-AP virus was then cultured in BirA-expressing MDCK cells in the presence of biotin. Streptavidin-coated QDs (SA-QDs) were introduced into these cells by lipofection to allow noncovalent binding of the QDs to biotinylated PA. QDs-containing influenza virus particles were thus generated. This approach will advance the mechanistic understanding of influenza virus uncoating using live fluorescence microscopy.
Materials and Reagents
- Materials
- 1.5 ml graduated microcentrifuge tubes (QSP, catalog number: 509-GRD-Q)
- 2 ml graduated microcentrifuge tubes (QSP, catalog number: 508-GRD-Q)
- SW32 Ultra-Clear Tubes (Beckman Coulter, catalog number: 344058)
- SW41 Ultra-Clear Tubes (Beckman Coulter, catalog number: 344059)
- 0.1-10 µl pipette tips (QSP, catalog number: T104RS-Q)
- 10-200 µl pipette tips (QSP, catalog number: TTW110RS-Q)
- 100-1250 µl pipette tips (QSP, catalog number: T112NXLRS-Q)
- 12-well plates (Corning Costar, catalog number: 3513)
- 6-well plates (Thermo Scientific, catalog number: 140675)
- V-bottom 96-well plates (Solarbio, catalog number: YA0590)
- Scalpel Blade (Hong Yue, catalog number: GC-HY00388)
(link: https://www.casmart.com.cn/product-details/page/103/11044449) - 100 mm Nunc EasYDishes (Thermo Scientific, catalog number: 150464)
- 75 cm2 Nunc EasYFlasks (Thermo Scientific, catalog number: 156472)
- 25 cm2 Nunc EasYFlasks (Thermo Scientific, catalog number: 156340)
- 0.45 μm filter (Millex-HV, catalog number: SLHU033RB)
- Filter paper (Biorad, catalog number: 1703932)
- 3 ml Pasteur pipettes (Nest, catalog number: 318314)
- 1 ml syringes (Gemtier, catalog number: JT-001)
- 10 ml syringes (Gemtier, catalog number: JT-010)
- 15 ml centrifuge tubes (Nest, catalog number: 601052)
- 50 ml centrifuge tubes (Nest, catalog number: 602052)
- Cell lines
- 293T cells (Boster, catalog number: CX0008; ATCC, catalog number: ACS-4500)
- MDCK cells (Boster, catalog number: CX0206; ATCC, catalog number: CRL-2936)
- Plasmids
Note: All plasmids are available from our lab. BirA is an enzyme that catalyzes ligation of Biotin and AP tag.- Eight plasmids (pHW2000) each containing individual cDNA segment of the influenza virus strain A/Puerto Rico/8/34 (H1N1) (Hoffmann et al., 2000)
- pcDNA3.1(+)-BirA
- Primers
Linearized Vector-F: GTGCTACTATTTGCTATCCATACTGTCC
Linearized Vector-R: ACATCTTCTCCAATCTCATCGAGCTCTATC
Insert-F1: GAAAATCGAATGGCACGAATAGTCTAGAAAACTGCTTCTTATCGTTCAGG
Insert-R1: TGGACAGTATGGATAGCAAATAGTAGCAC
Insert-F2: GATATCTTCGAAGCTCAGAAAATCGAATGGCACGAATAGTCT
Insert-R2: TGGACAGTATGGATAGCAAATAGTAGCAC
Insert-F3: GAGTGGTTCAGGAGGCGGTCTGAACGATATCTTCGAAGCTCAGAAAATCGAAT
Insert-R3: TGGACAGTATGGATAGCAAATAGTAGCAC
Insert-F4: GATAGAGCTCGATGAGATTGGAGAAGATGT
Insert-R4: GTTCAGACCGCCTCCTGAACCACTCAATGCATGTGTAAGGAAGGAGTTG
Insert-F5: GATAGAGCTCGATGAGATTGGAGAAGATGT
Insert-R5: TGGACAGTATGGATAGCAAATAGTAGCAC
PA-F: AGCGAAAGCAGGTACTGATCCAAAATG
PA-R: AGTAGAAACAAGGTACTTTTTTGGACAGTATG
Note: All primers were synthesized by Sangon Biotech. - Reagents
- SPF embryonated chicken eggs (Merial-vital)
- Competent cells DH5α (TIANGEN, catalog number: CB101-01)
(Link: http://www.tiangen.com/?productShow/t1/6/id/160.html) - 70% ethanol (Amresco, catalog number: E505)
- 2 kb Plus DNA ladder (TransGen, catalog number: BM121)
- Ampicillin (MDBio, catalog number: 69-52-3)
- SA-QD625 (Invitrogen, catalog number: A10196)
- KOD FX DNA polymerase enzyme (TOYOBO, catalog number: KFX-101)
- T4 DNA polymerase (NEB, catalog number: M0203S)
- 50x TAE buffer (Sangon Biotech, catalog number: B548101-0500)
- Agarose G-10 (BIOWESTE, catalog number: 111860)
- Omega Cycle-Pure Kit (OMEGA, catalog number: D6492-01)
- Omega Viral RNA Kit (OMEGA, catalog number: R6874-02)
- Omega gel extraction kit (OMEGA, catalog number: D2500-02)
- Omega Plasmid Mini kit (OMEGA, catalog number: D6943-02)
- One Step SYBR PrimeScript PLUS RT-PCR Kit (TaKaRa, catalog number: RR096A)
- DMEM (Gibco, catalog number: 11965118)
- MEM (Genom, catalog number: GNM41500)
- DMEM Powder (Gibco, catalog number: 12100061)
- Low gelling temperature Agarose (Sigma-Aldrich, catalog number: A9045)
- FBS (Gibco, catalog number: 1787602)
- 7.5% BSA (Solarbio, catalog number: H1130)
- TPCK-trypsin (Sigma-Aldrich, catalog number: T1426)
- 1x PBS (Gibco, catalog number: 10010049)
- Lipofectamine 2000 (Invitrogen, catalog number: 11668019)
- Biotin (Sigma-Aldrich, catalog number: B4639)
- Sucrose (HUSHI, catalog number: 10021418)
(Link: https://www.reagent.com.cn/goodsDetail/94d364146ee0495d955089dbd33dade3) - NaCl (HUSHI, catalog number: 10019318)
(Link: https://www.reagent.com.cn/goodsDetail/d8517dae2fd14870aeeaddbb8b0a13f4) - Tryptone (OXOID, catalog number: LP0042)
- Yeast extract (OXOID, catalog number: LP0021)
- LB medium with 100 μg/ml ampicillin (see Recipes)
- LB agar plate with 100 μg/ml ampicillin (see Recipes)
- Cell culture medium (see Recipes)
- Infection medium (see Recipes)
- 0.5% Chicken red blood cell (RBC) (see Recipes)
- 2x plaque medium (see Recipes)
- 1.6% agar (see Recipes)
- Sucrose solution (see Recipes)
Equipment
- Eppendorf Pipettes (0.1-10 µl, 10-200 µl, 100-1250 µl)
- 6 W UV transilluminator (UVP, model: UVLM-26)
- Rotating incubator (FUMA, model: QYC-200)
- Centrifuge (Eppendorf, model: 5810R)
- Ultracentrifuge (Beckman, model: OPTIMA XE-100)
- Water bath (YIHENG17, model: DK-8D)
- Beckman SW32 rotor
- Beckman SW41 rotor
- PCR machine (GnenAmp, PCR System 9700)
- 100 kV Transmission electron microscope (Hitachi, model: H-7000 FA)
Procedure
- Construction of pHW2000-PA-AP tag
The PA-AP segment was constructed by inserting a sequence encoding the short linker GSGG (Nucleotide sequence: GGTTCAGGAGGC), a 15-amino acid biotin AP tag (Nucleotide sequence: GGTCTGAACGATATCTTCGAGCTCAGAAAATCGAATGGCACGAA), and a duplication of 165 base pairs of the 3ʹ end of the PA-coding region after the PA ORF (stop codon deleted) using one-step sequence and ligation-independent cloning method (Jeong et al., 2012). Schematic of pHW2000 PA-AP tag is shown in Figure 1.
Figure 1. Schematic of the constructed plasmid pHW2000 PA-AP- PCR amplification and gel purification of linearized vector fragment
- The linearized vector fragment (pHW2000: 2,980 bp, partial PA sequence: 1,368 bp) is amplified using the KOD FX DNA polymerase enzyme in a PCR machine, according to the manufacturer’s protocol. Reaction system (Table 1) and parameters (Table 2) are shown as follows:
Table 1. The PCR reaction mixture used for the linearized vector fragment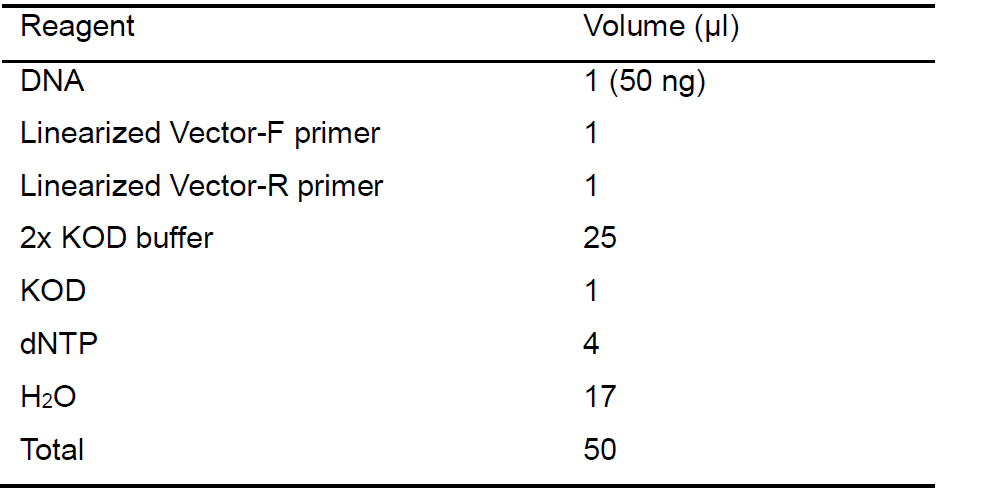
Table 2. The PCR reaction parameters used for the linearized vector fragment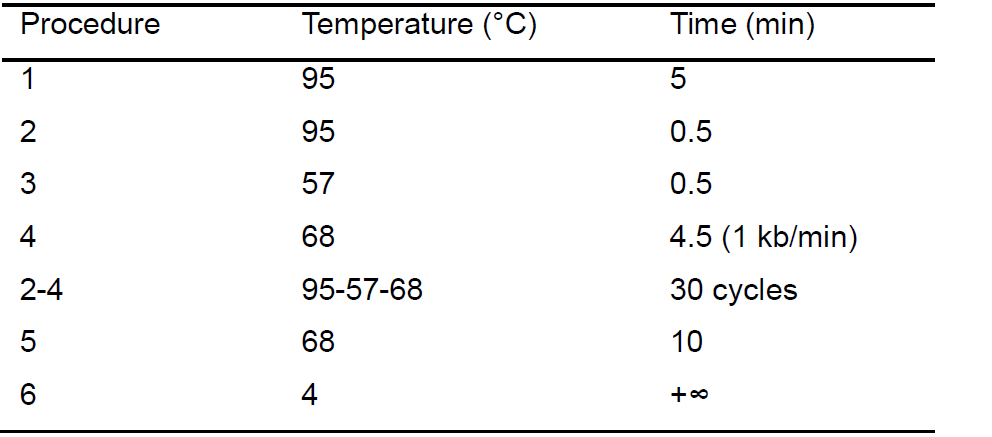
- Prepare a 1% agarose-TAE gel (Add 3 μl of 1 mg/ml EB into 30 ml solution for a gel).
- Add 1/10 volume of 10x DNA loading dye to each PCR sample and load 50 μl reaction onto the gel. In addition, load the 2 kb Plus DNA ladder for size verification.
- Run the gel in 1x TAE until the bands of the loading dye run to approximately three quarters of the way down the gel.
- Visualize the PCR product on a UV transilluminator. Cut the ~4,300 bp band using a clean blade. Place the gel slice into a clean 1.5 ml microcentrifuge tube.
- Purify the PCR product using the Omega gel extraction kit as per the manufacturer's protocol. Elute the linearized vector fragment PCR product with 50 μl elution buffer.
- The linearized vector fragment (pHW2000: 2,980 bp, partial PA sequence: 1,368 bp) is amplified using the KOD FX DNA polymerase enzyme in a PCR machine, according to the manufacturer’s protocol. Reaction system (Table 1) and parameters (Table 2) are shown as follows:
- PCR amplification and gel purification of insert fragment
- Acquire the insert fragment (partial PA sequence: 895 bp, linker-AP tag: 45 bp, and duplicated packaging sequence of PA gene: 205 bp) through a 5-steps PCR procedure. For a flow chart of these steps see Figure 2.
- Firstly, amplify duplicated packaging fragment from PA gene with Insert-F1 and Insert-R1 primers.
- Then duplicated packaging fragment is used as the template to amplify AP tag-duplicated packaging fragment with Insert-F2 and Insert-R2 primers.
- The same linker-AP tag-duplicated packaging fragment can be acquired in the third step.
- Partial PA fragment is acquired from PA gene using Insert-F4 and Insert-R4 primers in the fourth step.
- Finally, the linker-AP tag-duplicated packaging fragment and partial PA fragment are fused together and amplified using Insert-F5 and Insert-R5 primers by overlap extension PCR.
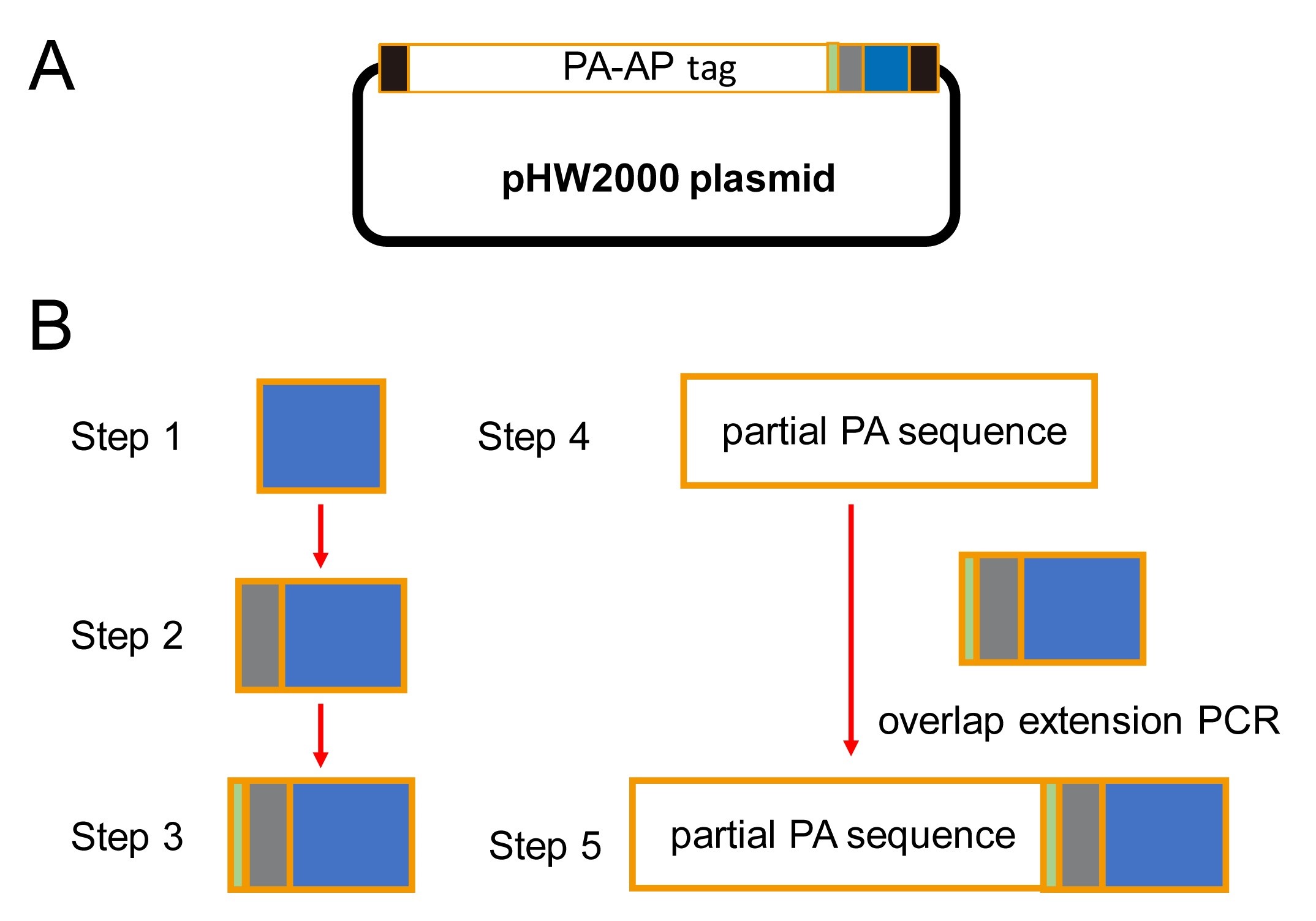
Figure 2. Flow chart diagram of the insert fragment acquisition. A. Schematic diagram of pHW2000 PA-AP. B. Flow chart diagram of the 5-steps PCR procedure. - PCR amplification and gel purification are performed as described above with reference to linearized vector fragments. Elute the Purified PCR product (~1,200 bp) with 50 μl elution buffer.
- Acquire the insert fragment (partial PA sequence: 895 bp, linker-AP tag: 45 bp, and duplicated packaging sequence of PA gene: 205 bp) through a 5-steps PCR procedure. For a flow chart of these steps see Figure 2.
- One-step sequence- and ligation-independent cloning
- Mix the linearized vector fragment and insert fragment at a molar ratio of 1:2 in a 1.5 ml tube. Reaction system is shown in Table 3.
Table 3. Reaction system of One-Step sequence- and ligation-independent cloning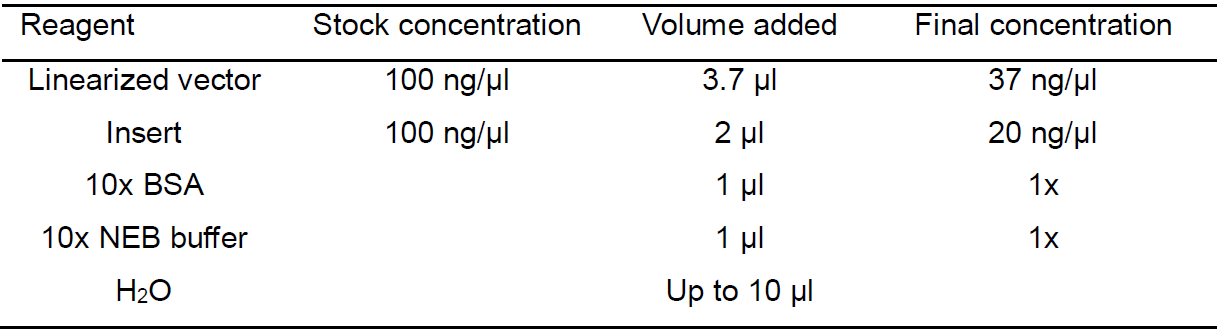
- Add 0.5 μl of T4 DNA polymerase (3 U/μl, NEB) to the mixture and incubate at room temperature for 2.5 min. Then, put the reaction mixture on ice immediately and incubate for 10 min. At the same time, thaw chemically competent cells DH5α (200 μl) on ice for ~10 min.
- Gently mix the cells with 10 μl of the reaction mixture and incubate the cells on ice for 30 min. Heat shock the cells at 42 °C for 90 s. Incubate the cells on ice for 2 min. Add 800 μl of LB medium to the cells and transfer the cells to 15 ml glass tube. Incubate the cells at 37 °C for 1 h. Then plate the cells on agar plates containing ampicillin antibiotics. Normally, we spread 200 μl of culture onto an agar plate. Incubate the plates upside down at 37 °C for 16 h.
- Pick six colonies and inoculate each into LB medium supplemented with ampicillin in 15 ml glass tubes. Grow overnight in a 37 °C rotating incubator. Extract plasmid DNA from the above cultures using the Omega Plasmid Mini kit as per the manufacturer’s protocol.
- Sequence clones with PA-F and PA-R sequencing primers to confirm the proper insertion of linker-AP tag-duplicated packaging sequence into the pHW2000-PA backbone.
- The resulting plasmid is herein referred to as pHW2000-PA-AP tag.
- Mix the linearized vector fragment and insert fragment at a molar ratio of 1:2 in a 1.5 ml tube. Reaction system is shown in Table 3.
- PCR amplification and gel purification of linearized vector fragment
- Rescue of infectious recombinant rPR8-PA-AP
- Transfect the cells with rPR8-PA-AP rescue plasmids.
- Transfect 293T cells in Opti-MEM as per the manufacturer’s protocol for 6-well plate, and the following per well:
2 x 106 293T cells
1 μg pHW2000-PB2
1 μg pHW2000-PB1
1 μg pHW2000-HA
1 μg pHW2000-NP
1 μg pHW2000-NA
1 μg pHW2000-M
1 μg pHW2000-NS
1 μg pHW2000-PA-AP tag
16 μl Lipofectamine 2000
1.5 ml Opti-MEM - Incubate the transfected cells for 6 h at 37 °C, 5% CO2. Carefully aspirate the Opti-MEM/lipofectamine mix from the transfected cells, and replace with 1.5 ml of fresh Opti-MEM. After 48 h, transfer the supernatant from the transfected cells into a 2.0 ml microcentrifuge tube. Centrifuge the supernatant for 1 min, 19,419 x g to remove cell debris.
- Transfect 293T cells in Opti-MEM as per the manufacturer’s protocol for 6-well plate, and the following per well:
- Rescue rPR8-PA-AP from transfection supernatants
Infection of 10-day-old chicken embryonated eggs- Candle the 10-day-old eggs using a light egg candler in a darkroom to mark the interface between the air sac and the allantoic cavity. Make a hole with a 10 ml syringe needle in the eggshell. Inoculate each egg with 200 μl of the tissue culture supernatants using a 1 ml syringe. Cover the hole in the eggshell with melted wax, then incubate the infected eggs at 37 °C for 3 days.
- Prior to harvesting the allantoic fluid, incubate the chicken eggs overnight at 4 °C to kill the embryo and coagulate the blood.
- Clean the eggshells with 70% ethanol and open the embryo over the air cavity by tapping with a spoon. Remove the broken eggshell and allantoic membrane with forceps. Stabilize the chicken embryo and collect as much allantoic fluid as possible into a 15 ml centrifuge tube on ice by a 1 ml pipette. Centrifuge for 5 min, 19,419 x g at 4 °C and transfer the allantoic fluid to fresh 15 ml centrifuge tubes. Store the allantoic fluid at 4 °C until they are checked for the presence of rescued virus with a hemagglutination (HA) assay.
HA assays- HA assays are carried out in V-bottom 96-well plates. Add 50 μl of 1x PBS into each well of the V-bottom 96-well plate.
- Add 50 μl of allantoic fluid to the first well and make 2-fold serial dilutions for the following wells. Discard the extra 50 μl from the last well.
- Add 50 μl of 1.0% chicken red blood cells (prepared in 1x PBS) to each well.
- Incubate the V-bottom 96-well plate for 30 min (until a red dot is visible in the bottom) at RT. A positive viral rescue can be confirmed by HA assay (Figure 3).
Plaque purification of rescued rPR8-PA-AP- Seed 2x 105 MDCK cells/well to 90% confluency in 12-well plates using 1.5 ml growth medium per well.
- Dilute HA positive allantoic fluid and perform eleven 1:10 dilutions using infection medium.
- Heat 1.2% agar until dissolved and place it in a 56 °C water bath. Add TPCK-trypsin to the 2x plaque assay medium at concentration of 2 μg/ml and place it in a 37 °C water bath.
- Aspirate the growth medium from the MDCK cells, and wash thrice in 1x PBS.
- Add the diluted samples (200 μl) to each 12-well plate, then incubate the well plate for 1 h at 37 °C, 5% CO2 and shake the plate every 15 min.
- Carefully aspirate the inoculums from the MDCK cells, and wash the cells once in 1x PBS.
- Mix 2x plaque assay medium plus TPCK-trypsin (7 ml) with 1.2% agar (7 ml) with a ratio of 1:1 and immediately overlay each well with 1 ml mixture, and solidify at room temperature for 30 min.
- Invert the plate and incubate for 48-72 h at 37 °C, 5% CO2 until the plaques are visible (Figure 4A).
- Outline the individual plaques and pick about 10 plaques. Using a 200 μl pipet tip, stab the plaque and scrape the cells forming the plaque. Inoculate each plaque into 500 μl of infection medium.
- Detect and sequence PA-AP gene from the plaques by RT-PCR using the OMEGA Viral RNA kit and TaKaRa one step SYBR PrimeScript PLUS RT-PCR Kit with PA-F and PA-R primers following the manufacturer’s protocol. The correct clonal stocks are amplified in chicken eggs for further use.
- Transfect the cells with rPR8-PA-AP rescue plasmids.
- Production of IAV-QDs
- Seed 5x 106 MDCK cells/well to reach 80% confluency in 100 mm dishes using 10 ml growth medium per dish.
- Transfect MDCK cells in Opti-MEM as per the manufacturer’s protocol.
15 µg pcDNA3.1(+)-BirA
37.5 µl Lipofectamine 2000
6 ml Opti-MEM containing 50 µM biotin - Incubate the transfected cells for 6 h at 37 °C, 5% CO2. Aspirate the Opti MEM/ lipofectamine mix from the transfected cells and wash twice in 1x PBS.
- Incubate with rPR8 PA-AP viruses at a multiplicity of infection (MOI) of 10 about 1 ml for 1 h at 37 °C, 5% CO2. Rock the dishes every 15 min to keep the inoculums evenly distributed.
- Remove the inoculums from the MDCK cells, wash twice in 1x PBS and add 10 ml infection medium containing 50 µM biotin.
- After a 2-h incubation, deliver SA-QD625 into MDCK cells by liposomal transfection:
4 μl SA-QD625
10 μl Lipofectamine 2000
1 ml Opti-MEM - Harvest cell culture supernatants every 12 h after virus infection and supplement with fresh infection medium containing 50 µM biotin until 3 days post-infection or ~80% CPE.
Note: The purpose of adding biotin during the 6 h incubation after the transfection of the BirA is to internalize biotin into cells before PA protein expression. Biotinylation of PA protein is dependent on BirA catalyzing the ligation of biotin to AP tag. Quantum dots introduced into cells are coupled with biotinylated PA proteins during virus infection. - Centrifuge the collected supernatants at 4 °C, 19,419 x g for 10 min and filter through a 0.45 μm filter to remove cell debris.
- Centrifuge at 4 °C, 153,125 x g for 2.5 h with Beckman SW32 rotor on 5 ml of a 30% (w/v) sucrose cushion in PBS to concentrate the supernatants. Then layer the pellet onto a 20-60% sucrose density gradient and centrifuge at 4 °C, 245,853 x g for 3.5 h with SW41 rotor.
- Collect fractions containing QD625 under UV excitation (Figure 5), and centrifuge at 4 °C, 161,560 x g for 1.5 h to remove sucrose.
- Resuspend purified virus in PBS (200-500 µl) to acquire quantum dots-containing influenza virus particles (IAV-QDs).
- Transmission electron microscopy of IAV-QDs
- Load IAV-QDs sample onto carbon-coated copper grids, adsorb for 5 to 10 min, and remove the excess liquid using filter papers.
- Wash the grids once with PBS and negatively stain with 10 μl of 1% phosphotungstate (PTA) for 20 s at room temperature.
- Remove the excess phosphotungstate using filter papers and let the copper grids dry naturally.
- Examine the prepared copper grids under a transmission electron microscope.
Data analysis
- For virus rescue, read the results as indicated in Figure 3 Virus rescue causes hemagglutination of RBC, while no rescue of virus causes the formation of a red pellet of RBC in the bottom of the well.

Figure 3. Hemagglutinin assay. A representative result from an HA assay is shown with no detectable levels of virus (top) or presence of virus (bottom). - For plaque purification, we picked 10 plaques, as shown in Figure 4A, 5 small plaques (white arrow) and 5 large plaque (black arrow). A single band of the expected size was detected from the purified rPR8-PA-AP by RT-PCR, which is larger than the wild-type PA gene due to insertion of the foreign sequence (Figure 4B).
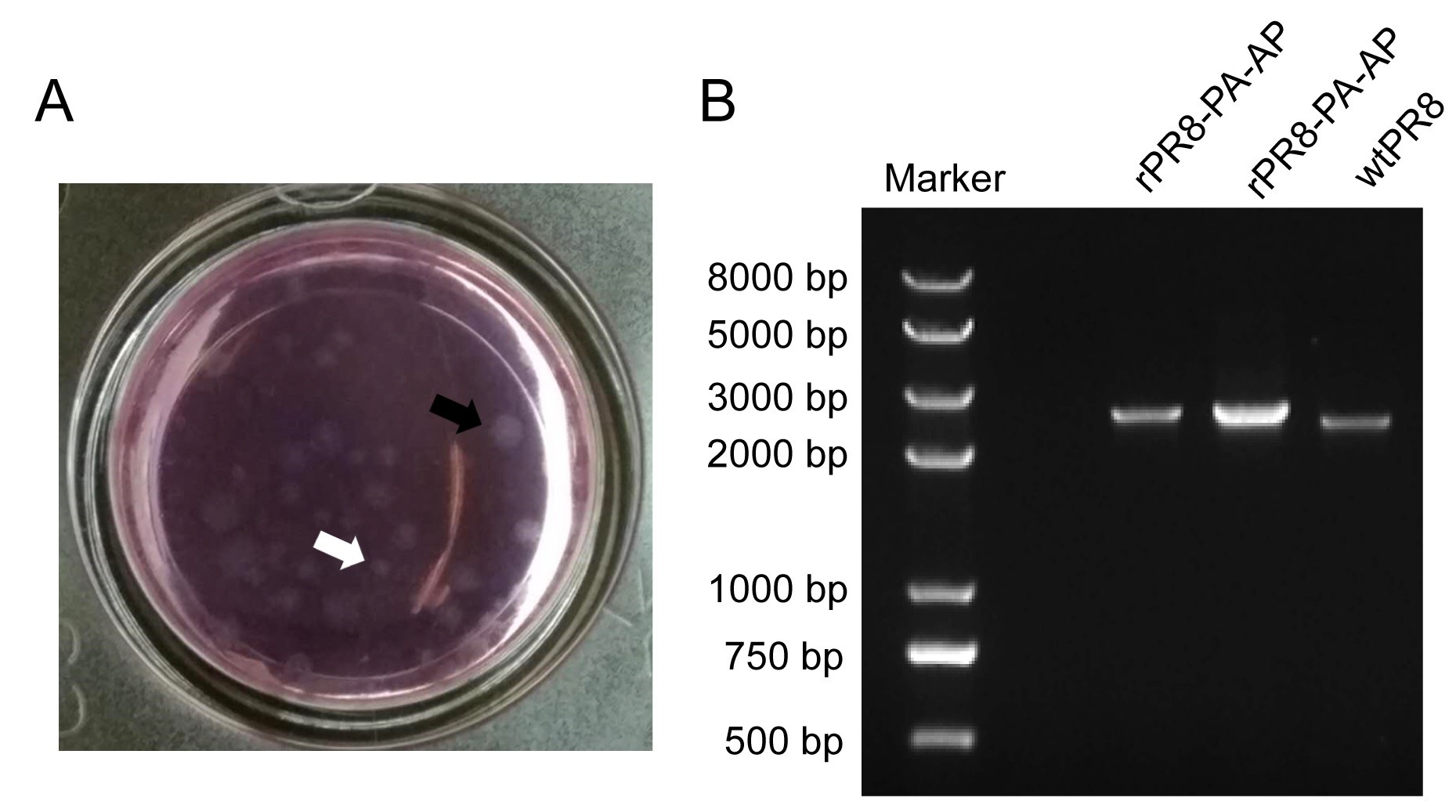
Figure 4. Virus purification. A. Viral plaques appear as white round spots. White arrow indicates small plaque, black arrow indicates large plaque. B. RT-PCR detection of PA-AP gene of purified rPR8-PA-AP. - For IAV-QDs production, after sucrose density gradient centrifugation, the labeled virus band shows red color under UV excitation and is located in the 50% sucrose layer, as shown in Figure 5.
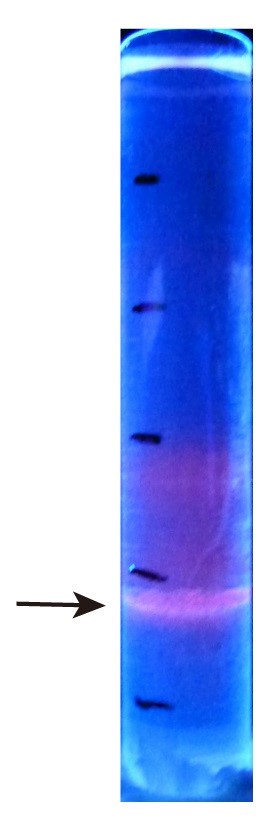
Figure 5. Sucrose density gradient ultracentrifugation. Arrow indicates IAV-QD625 band under UV excitation. - For TEM imaging of IAV-QDs, we tested different times and concentration gradients to determine the optimal conditions for IAV-QDs staining. We set staining times from 5 s to 3 min and PTA concentration from 0.25% to 2%. Under the condition of staining with 1% PTA for 20 s, we get the highest-resolution images, as shown in Figure 6. TEM images show the structure of virus as well as dark quantum dots clearly.
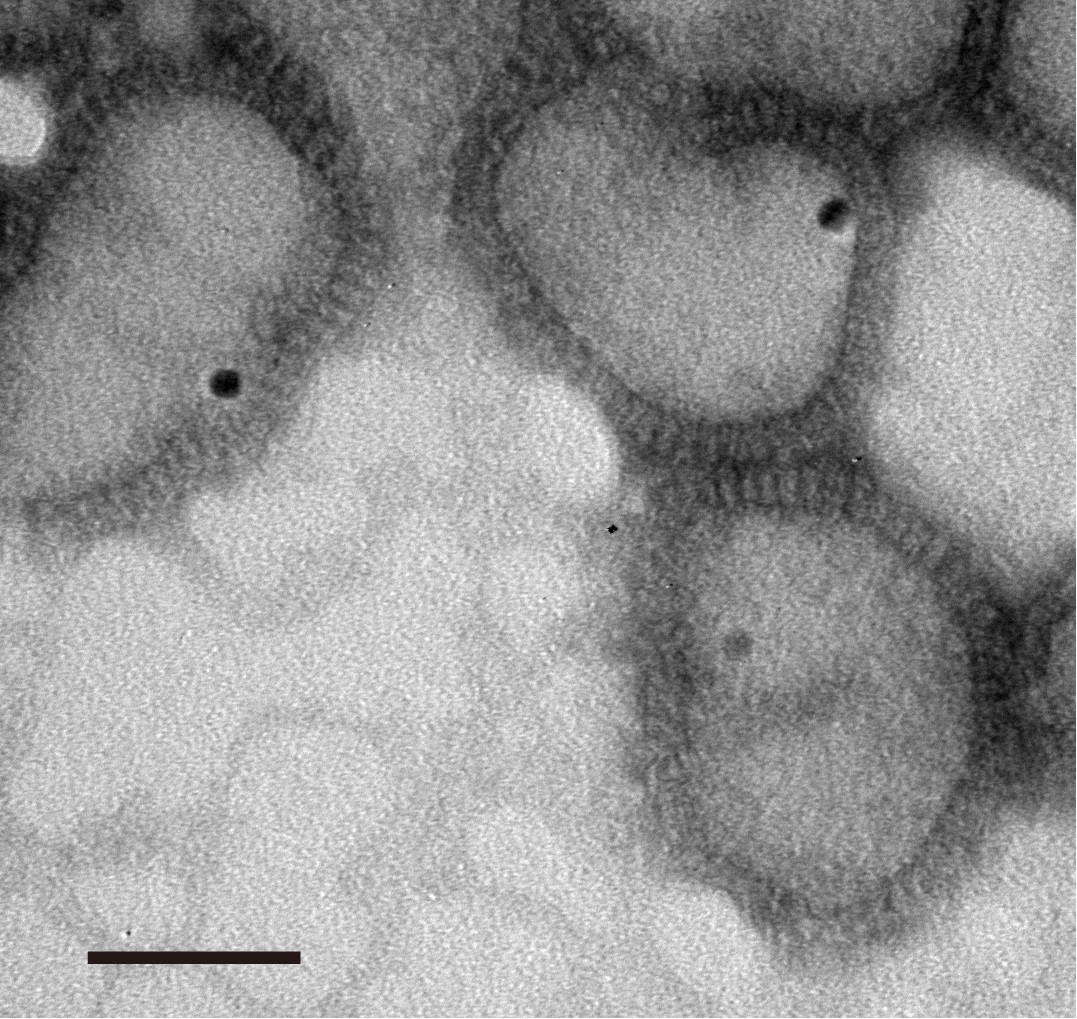
Figure 6. TEM images of IAV-QD625. The dark round dots indicate QD625s, Scale bar: 50 nm.
Recipes
- LB medium with 100 μg/ml ampicillin
10 g NaCl
10 g Tryptone
5 g yeast extract
1 L ddH2O
Autoclave at 121 °C, 15 min and let cool to RT
1 ml of 100 mg/ml ampicillin - LB agar plate with 100 μg/ml ampicillin
2 g NaCl
2 g Tryptone
1 g yeast extract
3 g agar
200 ml ddH2O
Autoclave at 121 °C, 15 min and let cool to 55 °C
200 μl of 100 mg/ml ampicillin - Cell culture medium
445 ml DMEM
50 ml FBS
5 ml penicillin/streptomycin - Infection medium
429 ml MEM
66 ml 7.5% BSA
5 ml penicillin/streptomycin
0.5 μg/ml TPCK-trypsin (final concentration) - 0.5% Chicken red blood cell (RBC)
0.5 ml RBC
100 ml physiological saline solution - 2x plaque medium
100 ml 2x DMEM
1 μg/ml TPCK-trypsin - 1.6% agar
1.6 g low gelling temperature agarose
100 ml ddH2O
Autoclave at 121 °C, 15 min - Sucrose solution
20% (w/v) sucrose: 20 g sucrose, 100 ml 1x PBS
30% (w/v) sucrose: 30 g sucrose, 100 ml 1x PBS
40% (w/v) sucrose: 40 g sucrose, 100 ml 1x PBS
50% (w/v) sucrose: 50 g sucrose, 100 ml 1x PBS
60% (w/v) sucrose: 60 g sucrose, 100 ml 1x PBS
Acknowledgments
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDB29050201), the National Key Research and Development Program of China (Grant 2018ZX10301405), the National Natural Science Foundation of China (Grants 31470269, 21727816, 91743108, and 31470837), and the Youth Innovation Promotion Association of the Chinese Academy of Sciences. This protocol is mainly derived from the experimental procedures used in our previous work (Qin et al., 2019).
Competing interests
The authors declare that they do not have any conflicts of interests or competing interests.
References
- Babcock, H. P., Chen, C. and Zhuang, X. (2004). Using single-particle tracking to study nuclear trafficking of viral genes. Biophys J 87(4): 2749-2758.
- Chou, Y. Y., Heaton, N. S., Gao, Q., Palese, P., Singer, R. H. and Lionnet, T. (2013). Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog 9(5): e1003358.
- Curry, A., Appleton, H. and Dowsett, B. (2006). Application of transmission electron microscopy to the clinical study of viral and bacterial infections: present and future. Micron 37(2): 91-106.
- Hoffmann, E., Neumann, G., Kawaoka, Y., Hobom, G. and Webster, R. G. (2000). A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97(11): 6108-6113.
- Hutchinson E.C. and Stegmann M. (2018). Purification and proteomics of influenza virions: methods and protocols. Humana Press, New York, NY.
- Jeong, J. Y., Yim, H. S., Ryu, J. Y., Lee, H. S., Lee, J. H., Seen, D. S. and Kang, S. G. (2012). One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl Environ Microbiol 78(15): 5440-5443.
- Lakdawala, S. S., Wu, Y., Wawrzusin, P., Kabat, J., Broadbent, A. J., Lamirande, E. W., Fodor, E., Altan-Bonnet, N., Shroff, H. and Subbarao, K. (2014). Influenza a virus assembly intermediates fuse in the cytoplasm. PLoS Pathog 10(3): e1003971.
- Qin, C., Li, W., Li, Q., Yin, W., Zhang, X., Zhang, Z., Zhang, X. E. and Cui, Z. (2019). Real-time dissection of dynamic uncoating of individual influenza viruses. Proc Natl Acad Sci U S A 116(7): 2577-2582.
- Martinez-Sobrido, L. and Garcia-Sastre, A. (2010). Generation of recombinant influenza virus from plasmid DNA. J Vis Exp(42): e2057.
- Tse, L. V., Zhang, Y., Whittaker, G. R. (2013). Immunoplaque assay (Influenza virus). Bio-protocol 3(21): e959.
- WHO global influenza surveillance network. (2011). Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Qin, C., Zhang, X. and Cui, Z. (2019). Production of Quantum Dots-containing Influenza Virus Particles for Studying Viral Uncoating Processes. Bio-protocol 9(18): e3366. DOI: 10.21769/BioProtoc.3366.
Category
Microbiology > Microbe-host interactions > Virus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.