- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Yeast Single-cell RNA-seq, Cell by Cell and Step by Step
(*contributed equally to this work) Published: Vol 9, Iss 17, Sep 5, 2019 DOI: 10.21769/BioProtoc.3359 Views: 7713
Reviewed by: Alba BlesaHélène M. LégerShyam Solanki
Original research article
The authors used this protocol in:

Apr 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Single-cell RNA-seq (scRNA-seq) has become an established method for uncovering the intrinsic complexity within populations. Even within seemingly homogenous populations of isogenic yeast cells, there is a high degree of heterogeneity that originates from a compact and pervasively transcribed genome. Research with microorganisms such as yeast represents a major challenge for single-cell transcriptomics, due to their small size, rigid cell wall, and low RNA content per cell. Because of these technical challenges, yeast-specific scRNA-seq methodologies have recently started to appear, each one of them relying on different cell-isolation and library-preparation methods. Consequently, each approach harbors unique strengths and weaknesses that need to be considered. We have recently developed a yeast single-cell RNA-seq protocol (yscRNA-seq), which is inexpensive, high-throughput and easy-to-implement, tailored to the unique needs of yeast. yscRNA-seq provides a unique platform that combines single-cell phenotyping via index sorting with the incorporation of unique molecule identifiers on transcripts that allows to digitally count the number of molecules in a strand- and isoform-specific manner. Here, we provide a detailed, step-by-step description of the experimental and computational steps of yscRNA-seq protocol. This protocol will ease the implementation of yscRNA-seq in other laboratories and provide guidelines for the development of novel technologies.
Keywords: YeastBackground
The appearance of single-cell-omics has revolutionized our understanding of many biological processes and is a field that is rapidly evolving at both the experimental and computational level. Over the last decade, there has been a rapid increase in the number of features that can be measured within individual cells. Single-cell RNA-seq (scRNA-seq) has pioneered this endeavor, and it has become a routine experiment to perform in higher eukaryotes. While very close to the entirety of the repertoire of bulk experiments in higher eukaryotes now have single-cell counterparts, single-cell tools for microorganisms (beyond fluorescent reporter-based methods) are scarce. This technological gap is mainly due to technical limitations imposed by the intrinsic nature of yeast and other microorganisms.
Unlike the mammalian cells, in which scRNA-seq was developed, yeast cells are smaller in cell, genome and transcriptome size. The presence of a rigid cell wall, which needs to be removed prior to library preparation, has been one of the major technical challenges for single cell analysis. On top of that, the yeast genome is highly condensed and pervasively transcribed. Over 85% of their genome is expressed from both strands with extensive transcript isoform diversity per gene (David et al., 2006; Pelechano et al., 2013, 2014 and 2015), and this requires a sensitive, quantitative and strand-specific approach.
As was the case for higher eukaryotes, initial studies exploring scRNA-seq in yeast focused on the development and optimization of the method. Recently, three yeast single-cell RNA-seq approaches have been reported (Gasch et al., 2017; Nadal-Ribelles et al., 2019; Saint et al., 2019). Interestingly, each one of them uses a different cell-isolation strategy and library-preparation protocol. Cell isolation is one of the main differences across these studies that range from microfluidics (Gasch et al., 2017), micromanipulation (Saint et al., 2019) and index sorting (Nadal-Ribelles et al., 2019), the later described here. Microfluidic and micromanipulation-based approaches require specialized equipment and are labor intensive, but allow imaging of individual cells by microscopy as a way to measure phenotype. The protocol described here for yeast single-cell RNA-seq (yscRNA-seq), relies on index sorting as a strategy for recording phenotypic information such as cell morphology and any Fluorescent-Activated Cell-Sorting-compatible feature (fluorescent proteins, antibodies, etc.). Index sorting provides a seamless strategy for merging techniques for individual phenotyping and transcriptional profiling in cells.
A major tipping point for single-cell isolation was the appearance of droplet-based methodologies (Zhang et al., 2019). These technologies dramatically increase potential throughput by generating and isolating cDNA in barcoded gel emulsions, which encapsulate single cells on the order of thousands of cells per run, and thus the cost significantly decreases. Interestingly, a preliminary droplet-based study in yeast is underway (Gresham et al., 2019). While cell capturing boosts cell numbers to an unprecedented scale, the number of genes detected per cell tends to be much lower (Skinnider et al., 2019) and phenotypic information of individual cells is lost. In addition, customization of library preparation is complex and labor intensive in droplet based-approaches, which will most likely be bypassed as technologies are developed.
While cell-isolation strategies determine the number of cells and phenotypic information that can be recorded, the choice of library-preparation approach defines the kind of transcriptional profiling. Due to the intertwined nature of the yeast transcriptome, and the diversity of isoforms per gene, library preparation requires both gene- and strand-isoform-specific resolutions. YscRNA-seq is based on STRT-seq (Islam et al., 2014), and its library design bypasses the technical limitations of working with yeast, fulfilling the requirements for a high-resolution transcriptome profiling method. First, unique molecular identifiers (UMI) are incorporated during cDNA synthesis via a biotinylated template-switching oligo (TSO) at the 5′-end of the molecule to digitally count the absolute abundance of a gene, along with its transcription start site (TSS) position. Second, the use of a homemade Tn5 enzyme to incorporate cell-specific adaptors provides a cost-effective strategy that greatly reduces experimental costs, compared to commercial Tn5 (Hennig et al., 2018). Finally, biotinylated primers allow selectively recovering 5′ ends. Moreover, the high affinity between streptavidin and biotin leads to a sequestration of the biotinylated strand after a brief denaturing step, which releases the non-biotinylated strand into the supernatant for sequencing. These features make yscRNA-seq one of the most sensitive methods available, and the most sensitive method for yeast reported to date (Nadal-Ribelles et al., 2019).
We anticipate that the development of novel methodologies with different aims will soon start to emerge, and yscRNA-seq will be extended to any yeast species or microorganism. The protocol described here, along with future optimizations, could serve as a starting point for the development of new methods. There are many unanswered biological questions that influence our understanding of human health, such as the emergence of drug-resistant microbial phenotypes. Generation of solid frameworks to profile microorganisms at single-cell resolution promises to expand our understanding, and perhaps answer some of these questions once and for all.
Materials and Reagents
Note: Reagents can be from different suppliers as far as they are only used for single-cell RNA-seq protocols and nuclease free. The ones listed here were used to develop the protocol. The regular molecular biology reagents are assumed to be already present in each laboratory (e.g., water, Tris, or NaCl). For plastic labware (filter tips and tubes), we have used several brands, with identical results as far as the material is certified to be nuclease-free and low binding.
- Eppendorf LoBind 1.5 ml (Eppendorf, catalog number: 30108051)
- Break-away plates (Thomas Scientific, catalog number: EK-75118)
- 96-well plates (Thomas Scientific, catalog number: EK-75012)
- Filter Tips (Mettler Toledo, catalog numbers: 17007954, 17014973, 17014973)
- qPCR plates (Applied Biosystems, catalog number: 4309849)
- Universal PCR plate seal (Sigma-Aldrich, catalog number: Z742420-100EA)
- UMI_Oligo dT_T31 (100 μM) (Integrated DNA Technologies)
- UMI_TSO6 (Integrated DNA Technologies)
- STRT-adaptors 96 different oligos (96 well plate Integrated DNA Technologies)
- UMI PCR (96 well plate Integrated DNA Technologies
- dNTP 25 mM (Thermo Fisher, catalog number: R0181)
- 50x Advantage 2 Polymerase (Takara, catalog number: 639202)
- PvuI (Cutsmart 10x provided) (New England Biolabs, catalog number: R3150L)
- DynabeadsTM MyOneTM Streptavidin C1 (Thermo Fisher, catalog number: 65001)
- Zymolyase 100T (100 mg/ml) (US Bio, catalog number:37340-57-1)
- RNase Zap (Thermo Fisher, catalog number: AM9780)
- RNase Inhibitor (40 U/ml) (Takara, catalog number: 2313A)
- RNase-nuclease free water (Thermo Fisher, catalog number: 10977035)
- ERCC RNA spike ins (Thermo Fisher, catalog number: 4456740)
- TAPS (Sigma-Aldrich, catalog number: T5316)
- DMF (Sigma-Aldrich, catalog number: 74438)
- 5x Superscript First strand buffer (Thermo Fisher, catalog number: 18064014)
- MgCl2 (1 M) (Thermo Fisher, catalog number: AM9530G)
- Betaine (5 M) (Sigma-Aldrich, catalog number: 61962)
- Superscript II (Thermo Fisher, catalog number: 18064014)
- 10x Advantage 2 PCR Buffer (Takara, catalog number: 639202)
- Tris (Sigma-Aldrich, catalog number: T2319)
- Tween-20 (Sigma-Aldrich, P9416-50ML)
- Glycerol (Sigma-Aldrich, catalog number: G5516)
- KAPA library quantification (Kapa Biosystems, catalog number: KR0405)
- Ampure beads (Beckman Coulter, catalog number: A63881)
- TE 10x (Sigma-Aldrich, catalog number: T9285)
- NaCl (Sigma-Aldrich, catalog number: S5150)
- EDTA (Thermo Fisher, catalog number: AM9261)
- Elution Buffer (EB) (Qiagen, catalog number: 19086)
- PB Buffer (PB) (Qiagen, catalog number: 19066)
- Sybergreen 2x mastermix (Invitrogen, catalog number: 4309155)
- LNA primers (Exiqon)
- Primers for qPCR
- SOMN17 Fw_TDH3_probe: TCGTCAAGTTGGTCTCCTGG
- SOMN18 Rv_TDH3_probe: GGCAACGTGTTCAACCAAGT
- SOMN21 Fw_ADH1_probe: TGGTGCCAAGTGTTGTTCTG
- SOMN22 Rv_ADH1_probe: GGCGAAGAAGTCCAAAGCTT
- SOMN310 Fw_5_ERCC_00130: CGGAAAAGTACTGACCAGCG
- SOMN311 Rv_5_ERCC_00130: TGCCAATGACTTCAGCTGAC样式
- Optical lids for qPCR (Applied Biosystems, catalog number: 4311971)
- DNA High Sensitivity CHIP (Agilent, catalog number: 5067-4626)
- 1% Triton X-100 (Sigma-Aldrich, catalog number: X100-1L)
- 100 mM DTT (Thermo Fisher, catalog number: 18064014)
- Propidium Iodide (PI) (Sigma Aldrich, P4170-10MG)
- Cell capturing and lysis solution (see Recipes)
- 2x BWT Buffer (see Recipes)
- TNT Buffer (see Recipes)
Equipment
- 96-well plate magnet (Thermo Fisher, catalog number: 12331D)
- 1.5-2 ml tube magnet (Thermo Fisher, catalog number: 12303D)
- Bioanalyzer (Agilent)
- HiSeq 2000 (Illumina)
- FACS (BD Influx, or Aria II, nozzle 70 and 100 microns)
- qPCR (Applied Biosystems)
- Qubit (Thermo Fisher, catalog number: Q32854)
- Thermocycler (any vendor)
- Thermomixer (any vendor)
- Multichannel pipettes (any vendor)
- Plate centrifuge (any vendor)
Software
- Novocraft (Novocraft Technologies Sdn Bhd, http://www.novocraft.com/)
- Samtools v1.3.1 (Li et al., 2009, http://samtools.sourceforge.net/)
- R programming language v.3.5.0 (R Core Team, 2019, https://www.r-project.org/)
- Genomic Alignments R package v.1.18.1 (Lawrence et al., 2013)
Procedure
Note: If possible, generate a single-cell library preparation space in the laboratory. If not, wash the surface of the bench and all the material with RNase Zap at the beginning and end of each library. All material and reagents are handled with gloves and the standard precautions for RNA work should be taken (RNase free material and filter tips).
- Cell growth
On the day before sorting, grow the desired pre-inoculum of your desired yeast strain in their corresponding media overnight (O/N). To profile exponentially growing cells, we recommend the initial culture not to grow over optical density OD660 = 1. - Cell sorting
- The next morning (sorting day), dilute cells to OD660 = 0.05 in the corresponding media and allow for at least 2 cell divisions (3 h approximately for wild type strains) prior to cell isolation.
- Prepare 96- or 384-well plates containing 5 µl absolute ethanol in each well to fix cells immediately for sorting.
Notes:- During protocol optimization, we recommend using break-away plates. These plates allow breaking 96 well plates by rows, and thus several tests can be done using the same plate. Check with your facility the compatibility of the plates.
- We have obtained the same results sorting cells directly into 5 µl of “Cell capturing and lysis solution” (see below). If doing so, prepare plates right before sorting and keep them on 4 °C ice.
- Dilute cells prior to sorting to OD = 0.05 in 3 ml of growth media and vortex vigorously to separate cell clumps.
Note: At this step, propidium iodide (PI) (4 µg/ml) can be added to check for cell viability. Adjust the culture volume to your needs. In our experience, 3 ml is enough to sort at least 10 plates. - At the FACS facility, filter cells with Cell Strainer Tubes (check with your facility which tubes they prefer) and put cells in the appropriate sorting tube for live single-cell sorting.
- Check the alignment of the plate with the sorter. For example, this can be done by sorting a drop into a covered plate, and ensuring that the droplet would fall into the center of each well.
- Sort live single yeast (propidium iodide (PI) negative) into each well of the plates, being sure to leave one well as a negative control. We index-sorted from the population using the forward and side scatter (FSC and SSC respectively).
Note: To include a positive control, sort 100 cells into one well. - Cover plates with Universal PCR plate seal.
- Quick spin plates to collect cells at the bottom of each well (short spin to collect all liquid to the bottom of the wells).
- Let the ethanol evaporate in a sterile environment (i.e., sterile hood) for no more than 45 min.
- Once the ethanol is completely evaporated, add 5 µl yeast “Cell capturing and lysis solution”. Spin down and freeze immediately.
Note: Regardless of whether the cells are sorted into ethanol or directly into “Cell capturing and lysis solution” (see Recipes), frozen plates can be stored at -80 °C for at least 6 months. ERCCs are spike in RNAs that provide an accurate measure of technical noise, while we recommend using them, they can be excluded. - Perform the following lysis cycle from freshly-sorted or frozen plates (Table 1).
Table 1. Temperature conditions for cell wall digestion and cell lysis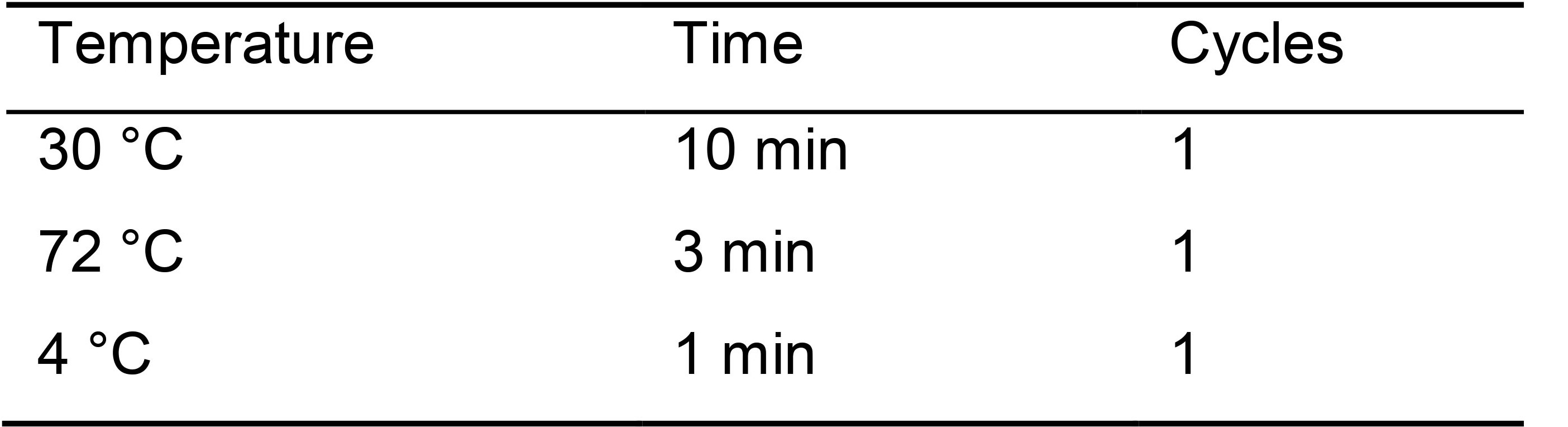
Note: Digestion can be extended up to 30 min. - Immediately proceed to add the RT reaction for first strand cDNA synthesis. Add 5 µl Reverse Transcription mix (RT mix) (Table 2).
Table 2. Master mix reagents for first strand cDNA synthesis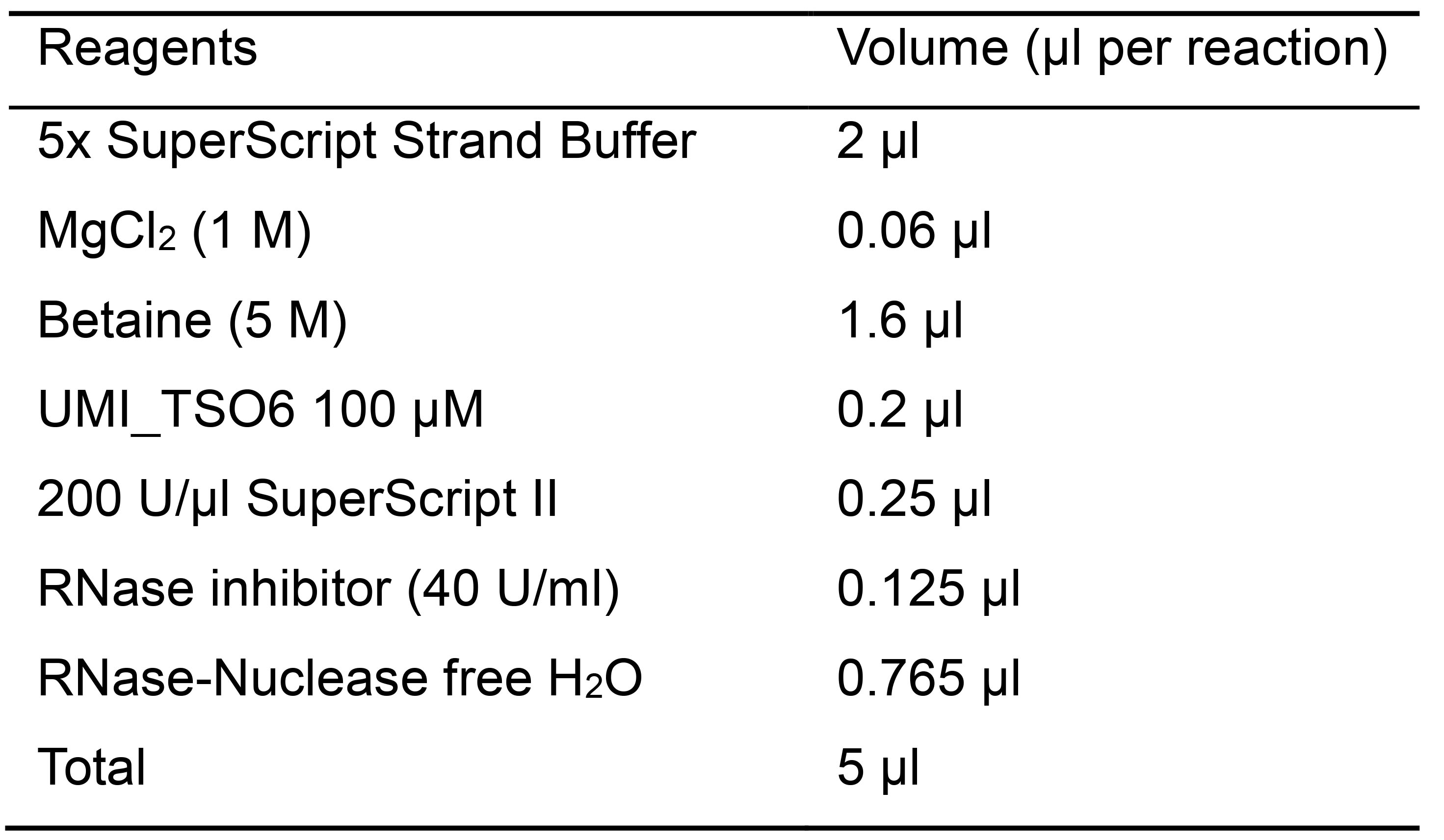
- Spin down the plate and perform the following cycles (Table 3):
Table 3. Incubation temperatures for cell lysis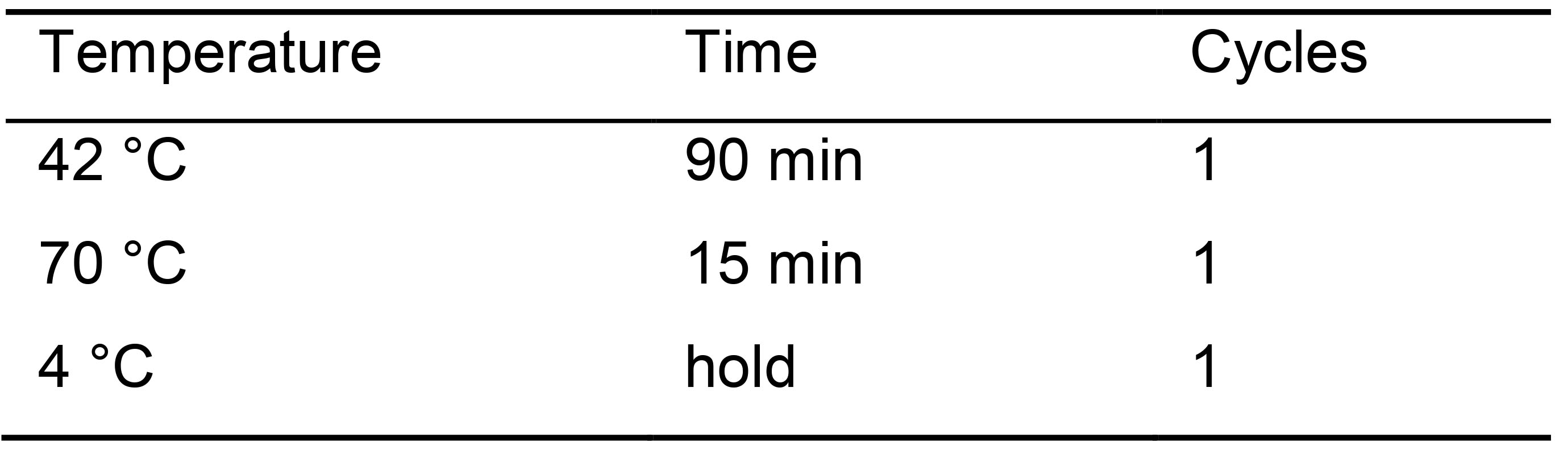
- Add 15 µl PCR mix for library amplification to each well (Table 4).
Table 4. Master mix reagents for library amplification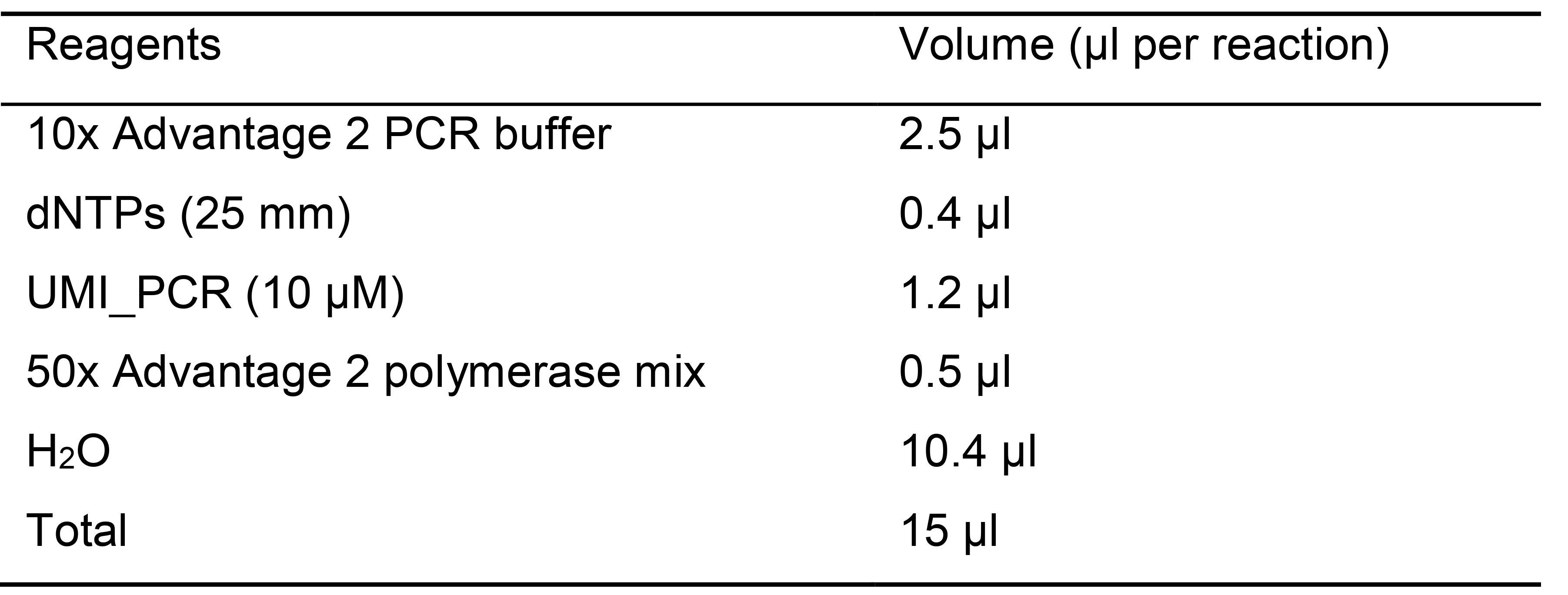
- Spin down the plate and do the following cycles (Table 5):
Table 5. PCR cycling conditions for library amplificationNote: This is a safe stopping point (at 4 °C O/N, or frozen -20 °C for 1-2 months).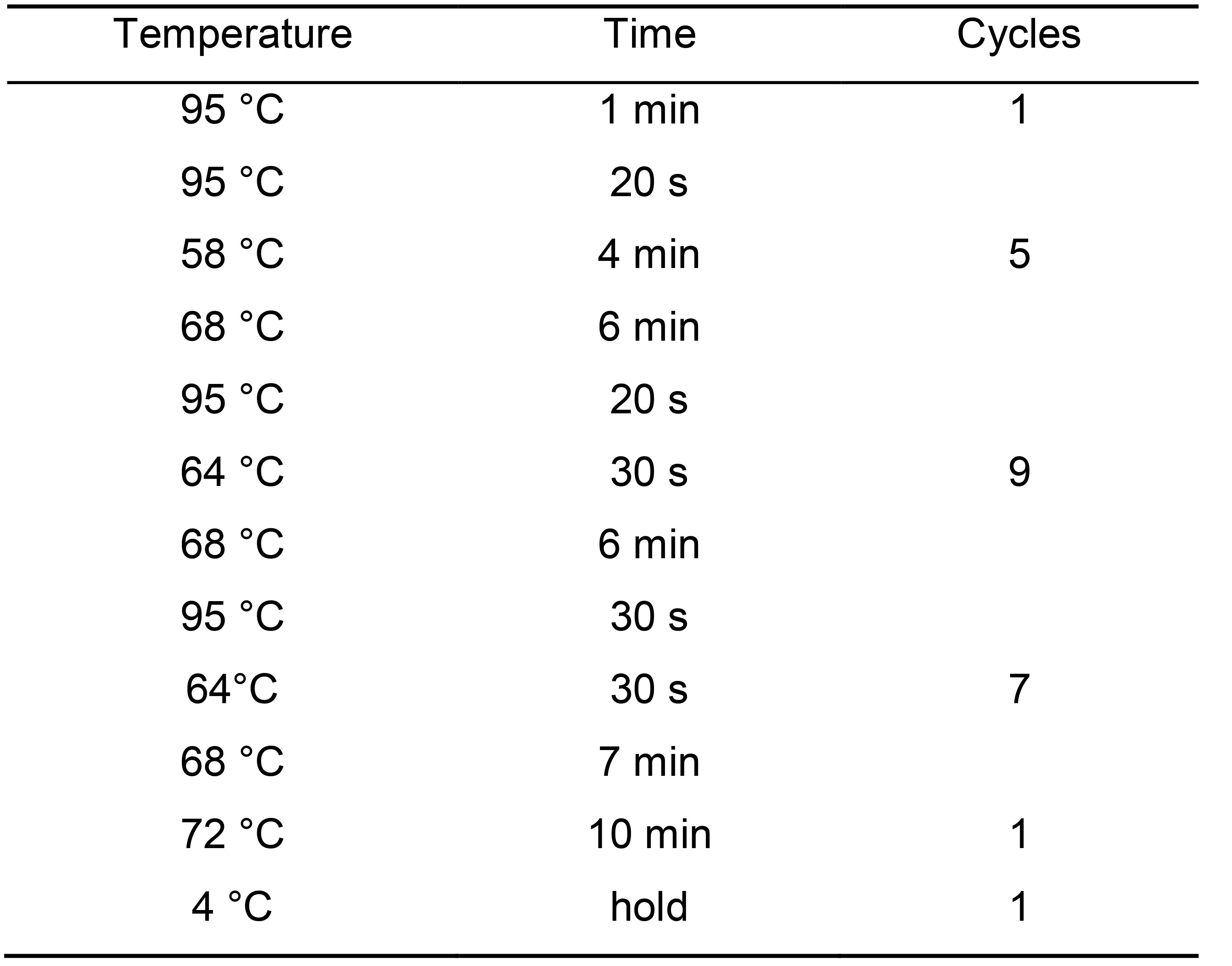
- (Optional) Dilute the amplified library 1:20 and check the percentage of positive cells from Step B15 (Figure 1; Tables 6 and 7).
Note: Use primers against a housekeeping gene (we use TDH3 but we have obtained same results with ADH1) to measure the number of positive cells per well. Use primers against ERCC as a positive control for amplification that should be even across all reactions. This step is especially useful during the protocol set up as it allows inspecting the samples/libraries before moving forward (check for the number of positive libraries). See Table S1 for primer sequences.Table 6. Master mix for qPCR assessment
Note: Primer mix refers to the mixture of Fw and Rv primers (10 µM each) diluted in TE 1x.
Table 7. PCR cycling conditions for qPCR
Primer sequences for qPCR:
SOMN17 Fw_TDH3_probe: TCGTCAAGTTGGTCTCCTGG
SOMN18 Rv_TDH3_probe: GGCAACGTGTTCAACCAAGT
SOMN21 Fw_ADH1_probe: TGGTGCCAAGTGTTGTTCTG
SOMN22 Rv_ADH1_probe: GGCGAAGAAGTCCAAAGCTT
SOMN310 Fw_5_ERCC_00130: CGGAAAAGTACTGACCAGCG
SOMN311 Rv_5_ERCC_00130: TGCCAATGACTTCAGCTGAC
Note: A good plate will have around 70% positive wells considering the Ct values of the housekeeping gene. Plates with less than 50% positive cells are rare. To determine if low efficiency is due to sorting or due reaction efficiency, perform a qPCR using a 1:10 dilution of the cDNA library against ERCCs. Failure to amplify ERCCs, or uneven amplification (represented by wildly different Ct values), is indicative of incorrect library preparation. In the case of low number of positive cells per plate, you can generate a new plate by combining positive from different plates into a new plate before proceeding to tagmentation and try to improve sorting efficiency.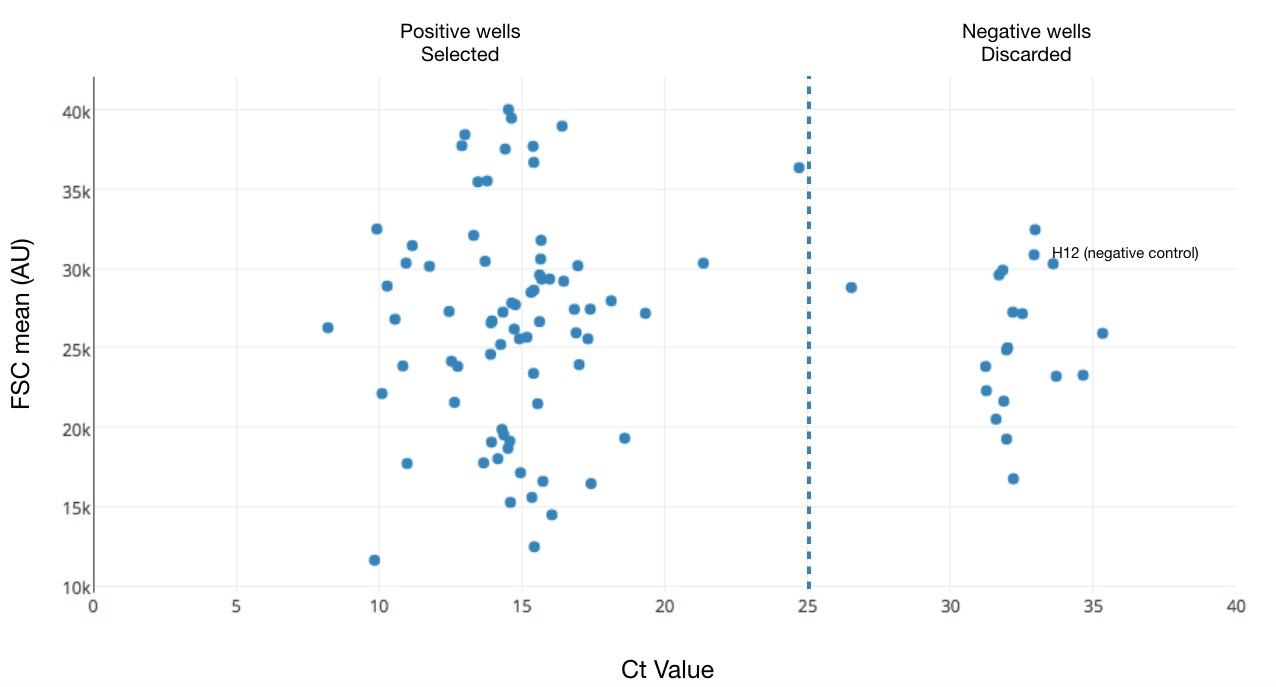
Figure 1. Representative example of Ct Values obtained by qPCR. The scatter plot represents the cycle amplification (Ct value) of a yscRNA-seq 96-well plate. Each dot represents the value obtained using a yeast housekeeping gene (TDH3, x-axis) as a function of the index-sorting value for cell size (Forward Scatter (FSC), y-axis). Dotted line (Ct > 25) displays the threshold used to discriminate positive and negative wells. The label displays the Ct value for the not sorted well H12 (which is used as a negative control). - Add 15 µl of room temperature equilibrated and well homogenized Ampure XP beads (1:0.6 sample/ bead ratio) to each well.
Note: Do not increase the volume of beads in the purification step above the 1:1 ratio. A less-than-standard amount of beads ensures that primer dimer carryover is minimal. - Mix by pipetting up and down ten times, or until the solution appears to be homogeneous. Transfer solutions to a 96-well plate with compatible magnet stand.
- Incubate the mixture for 10 min at room temperature to let the DNA bind to Ampure XP beads.
- Place the 96-well plate on the magnetic stand for 5 min, or until the solution is clear and beads have been collected.
- While samples are on the magnet, carefully remove the liquid without disturbing the beads.
- Wash magnet-bound beads with 200 µl of 80% (vol/vol) ethanol solution. Incubate the samples for 30 s and then remove the ethanol with the tube in the magnet, do not overdry the beads.
Note: It is important that the ethanol solution is freshly prepared every time, as ethanol absorbs moisture from the environment, thus changing the final concentration. Read and follow the manufacturer’s instructions. - Repeat the ethanol wash one more time (repeat Steps B20-B22).
- Remove any trace of ethanol and let beads dry completely by leaving the plate at room temperature for 5 min or until ethanol evaporates.
Note: Cover the plate during this step or protect it from any possible source of contamination or airflows. - Once there is no ethanol left, elute dscDNA libraries from Ampure XP beads with 16.5 µl elution buffer at room temperature (EB buffer Qiagen).
- Remove the plate from the magnet and mix vigorously by pipetting up and down three times or until the solution becomes homogeneous.
- Place the plate on a magnetic stand and leave it for 2 min, or until the solution appears clear.
- Recover 15 μl of supernatant from each well and transfer to a new plate. Label the plate correctly, as it will be stored.
Safe stopping point: cDNA libraries can be stored at -20 °C before proceeding to tagmentation for up to 2 weeks.
- Full-length cDNA library quality check
- Run 1 µl of dscDNA libraries (Step B28) to check the size distribution and estimate of concentrations using a High Sensitivity DNA ChIP (2100 Bioanalyzer). If available, use the qPCR results from Step B16 to guide the selection of wells for Bioanalyzer (Figure 2).
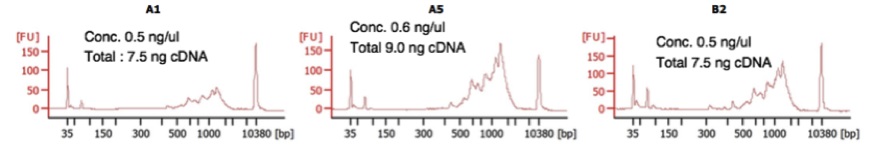
Figure 2. Representative Bioanalyzer traces of full-length cDNA obtained with yscRNA-seq (Step C1). cDNA libraries obtained from step (Step B28) were run on a DNA High Sensitivity CHIP (Agilent 5067-4626) for validation. Library concentration was also measured by Qubit High Sensitivity (Thermo Fisher). Left panel (A1) represents the lower limit of library quality that we sequenced while middle (A5) and right (B2) panel represent average libraries (Figure adapted from Nadal-Ribelles et al., 2019)
Adaptor annealing: In order to generate cell-barcoded libraries, Tn5 needs to be loaded with double stranded DNA (dsDNA) adapters. To do so, 96 different dsDNA adapters need to be annealed.
- Thaw the plate with the 96 STRT barcodes (100 µM) in ice.
- In a new plate, mix 5 μl UMI-TN5-U (100 µM) and 5 μl UMI-TN5_1 to 96 (µM) in TE 1x to final concentration 50 µM (each), a 1:1 dilution.
- Anneal primers by heating the mix at 95 °C for 3 min and gradually cool down to room temperature (0.5 °C/s). Label the plate as “Annealed cell-specific adapters” plate.
Note: Label plates with the amount of annealed primer and the annealing date. Plates can be prepared in advance and stored at -20 °C for up to six months. The amount of annealed adaptors depends on the number of plates that need to be tagmented and the frequency of usage. - To load cell specific adapters to Tn5, prepare a new plate and label it as “loaded Tn5”. In each well, mix the following reagents. We recommend to make a mix with all reagents and add annealed cell-specific adapters individually (Table 8).
Table 8. Master mix to generate 10x transposome
- Load Tn5 by incubating at 37 °C for one hour and immediately cool to 4 °C. Freeze the “loaded Tn5” plate at -20 °C.
Note: Loaded Tn5 plate can be safely stored for 1-2 weeks at -20 °C. Caution, store the plate immediately after use leaving the loaded plate on ice 4 °C will significantly reduce Tn5 activity and will result in inefficient tagmentation.Tagmentation
- Prepare the following mix in a new plate in ice (Table 9).
Table 9. Master mix for library tegmantation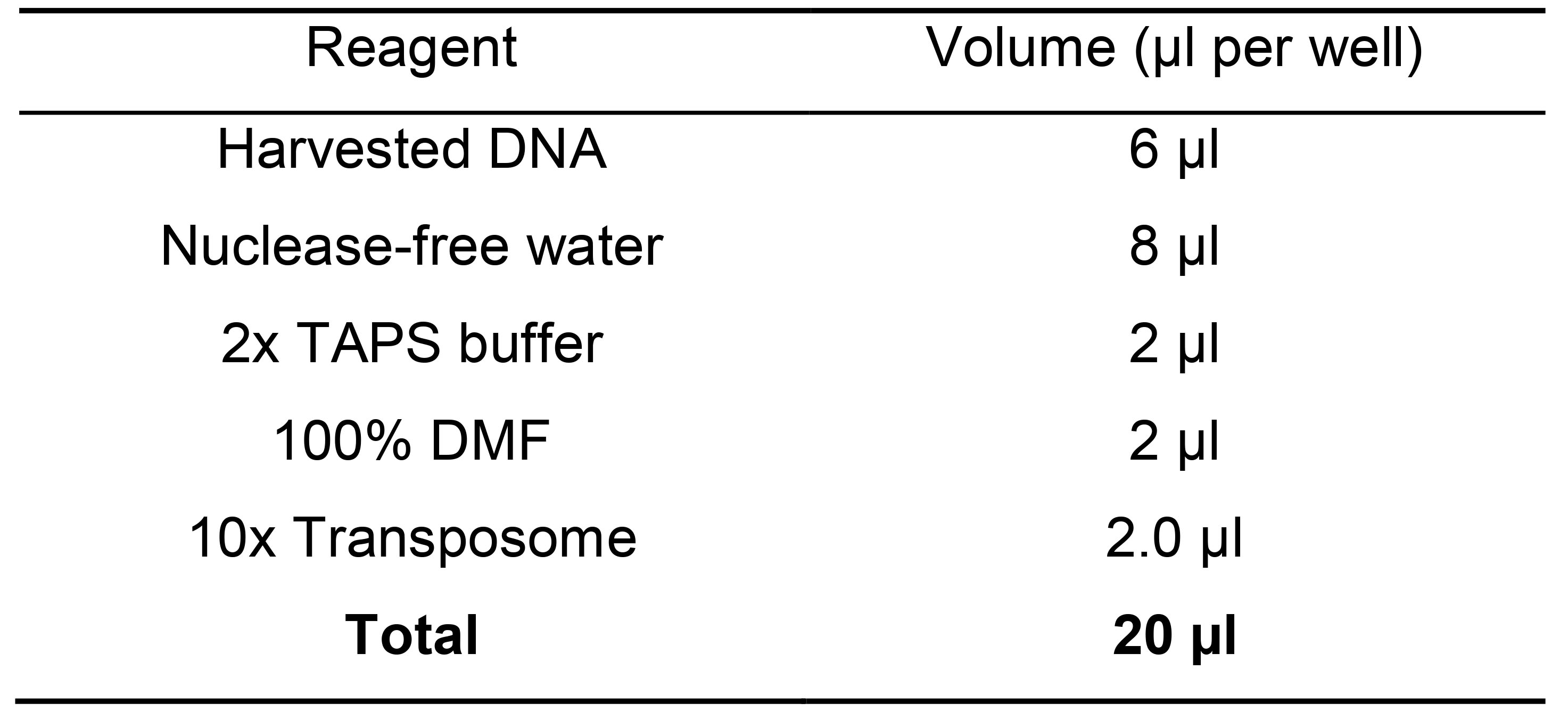
- Incubate at 55 °C for 5 min and 3 min at 85 °C to inactivate Tn5, then cool to 4 °C in a thermocycler. Tagmentation time depends on Tn5 purification efficiency and activity. We have observed that the enzyme loses activity over time. Titration of each batch of Tn5 is strongly suggested by tagmenting the same amount cDNA (Steps B28 and C1) with increasing concentration of loaded Tn5 (Step C6) and check fragmentation profile in a Bioanalyzer.
- Prepare a 1:20 dilution of MyOne C1 Streptavidin per each sample.
- Wash MyOne beads 2 x with 2x BWT buffer and dilute with 20x more volume than the original volume of beads with 2x BWT.
Example: 20 µl beads for 20 samples will be finally diluted with 400 µl 2x BWT. - Add 20 µl of diluted MyOne C1 Streptavidin beads (step C10) to each well and incubate at RT for 5 min at room temperature.
- Pool all samples per plate (up to 96) into a single collecting tube (1.5 or 2 ml).
- Place collecting tube in magnetic rack and allow enough time for the solution to be completely clear.
- While on the magnet, wash beads once with 100 µl of TNT buffer do not mix the beads.
- While on the magnet, wash MyOne C1 Streptavidin once with 100 µl PB Buffer and discard the supernatant.
- While on the magnet, wash beads 3 x with 100 µl TNT buffer again and the discard supernatant.
Remove 3´ ends
- Add 100 µl of the following mix to the washed beads from Step C14 (Table 10).
Table 10. Mastermix for 3´ end removal
- Incubate mix for 1 h at 37 °C in a thermomixer. Mix every 2 min for 30 s at 1,000 rpms, to avoid bead sedimentation.
- Wash beads 3 x with 100 µl of TNT buffer.
Elute single stranded cDNA
- Resuspend in 30 µl Nuclease-free water.
- Incubate 10 min at 70 °C, 850 rpm mix in a thermomixer.
- Immediately bind beads to the magnet and transfer the supernatant to a new tube, which contains the single strand cDNA library in the supernatant (the other strand remains bound to the streptavidin beads).
Single-strand cDNA cleanup
- Add 54 µl of room temperature Ampure XP beads to 30 µl sscDNA library.
- Incubate for 10 min at RT.
- Bind beads to the magnet for 1 min or until the solution is completely clear and discard supernatant (keep the beads).
- Wash once with 200 µl fresh 80% ethanol for 20-30 s. Perform this step with the beads bound to the magnet.
- Air dry beads for approximately 2 min.
- Resuspend in 30 µl EB buffer and incubate for 5 min at RT.
- Bind beads for 2 min, or until the solution is clear, and transfer the supernatant to a new tube.
- Run 1 µl of dscDNA libraries (Step B28) to check the size distribution and estimate of concentrations using a High Sensitivity DNA ChIP (2100 Bioanalyzer). If available, use the qPCR results from Step B16 to guide the selection of wells for Bioanalyzer (Figure 2).
- Assess library concentration
- Set up a KAPA quantification reaction with a 1:100, 1:1,000 and 1:10,000 dilutions of the eluted cDNA library (Step C27). All regents except for your DNA library are provided in the kit (Table 11).
Note: This kit can be substituted by your favorite quantification method or by a qPCR using P5-P7 primer pairs with known standards (PhiX is strongly recommended) SYBRGreen 2x mastermix.
Table 11. Master mix for qPCR library quantification
- qPCR cycling conditions for KAPA and homemade Sybergreen (Table 12).
Table 12. PCR cycling conditions for qPCR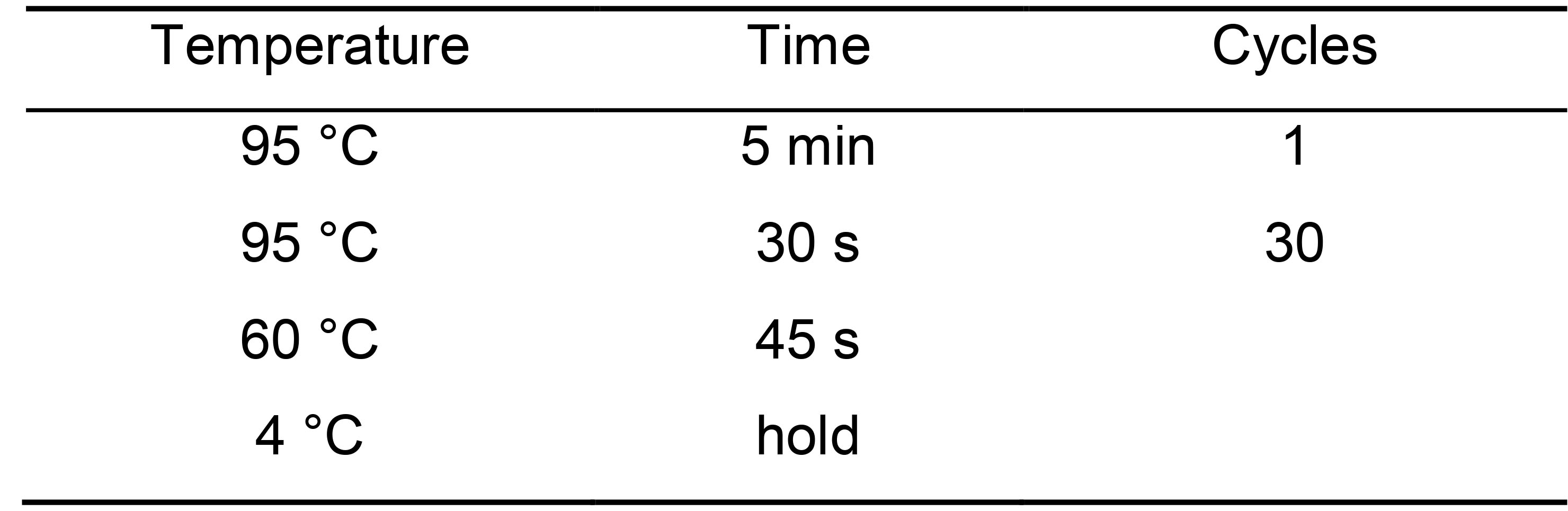
- Use the qPCR to calculate library quantification using the template provided by KAPA biosystems or the instructions provided from your manufacturer.
Note: We have used KAPA, NEB and homemade systems with similar results. - Set up a separate PCR to run a bioanalyzer to determine the final size distribution. Prepare the following mix, one separate reaction per each library to be loaded (Table 13).
Table 13. Master mix for to assess library size after tagmentation
- Run the same PCR as in Step D2 but for 11 cycles.
- Run 1 µl into a High sensitivity DNA CHIP to obtain an average library size based on the Bioanalyzer profile (Figure 3).
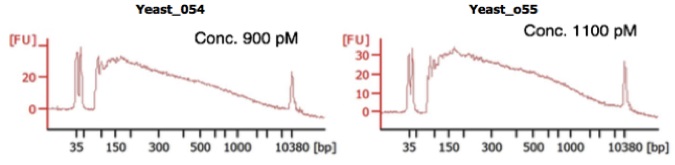
Figure 3. Representative Bioanalyzer traces obtained from yscRNA-seq. Two representative samples obtained from approximately 80 cells. Concentration of each library is shown and was determined by qPCR (Step D3). Figure from Nadal-Ribelles et al., 2019. - Sequence the library on the HiSeq 2000 High output using the custom Read 1 primer and UMI-TN5-U as the Index read primer.
- To run the libraries on the HiSeq rapid run, use lock nucleic acid (LNA) primers. Spike in the primer at 0.5 µM.
Index 1 primer into HP8
Read 1 primer into HP9
Notes:- Double-check this information with your sequencing kit/instrument and/or sequencing core facility.
- For a High Output Run, custom primers are required as well, but without LNA due to differences in sequencing chemistry.
UMI_PCR_read1: +GAATGA+TACGGCG+ACCA +CCGA+T – custom 250 nmole. DNA oligo, HPLC Purification
Index1: CTGT+CT+CTT+ATA+CA +CA+TCTGA+CG+C – custom 250 nmole DNA oligo, HPLC Purification - Load around 8-14 pmol of each library per lane. Libraries are single-stranded DNA, thus, no denaturing is required.
Note: Once the protocol is optimized, there is no need to run a PhiX control, if PhiX is loaded take into consideration a denaturing step for the double-stranded PhIX control, which is not required for yscRNA-seq libraries.
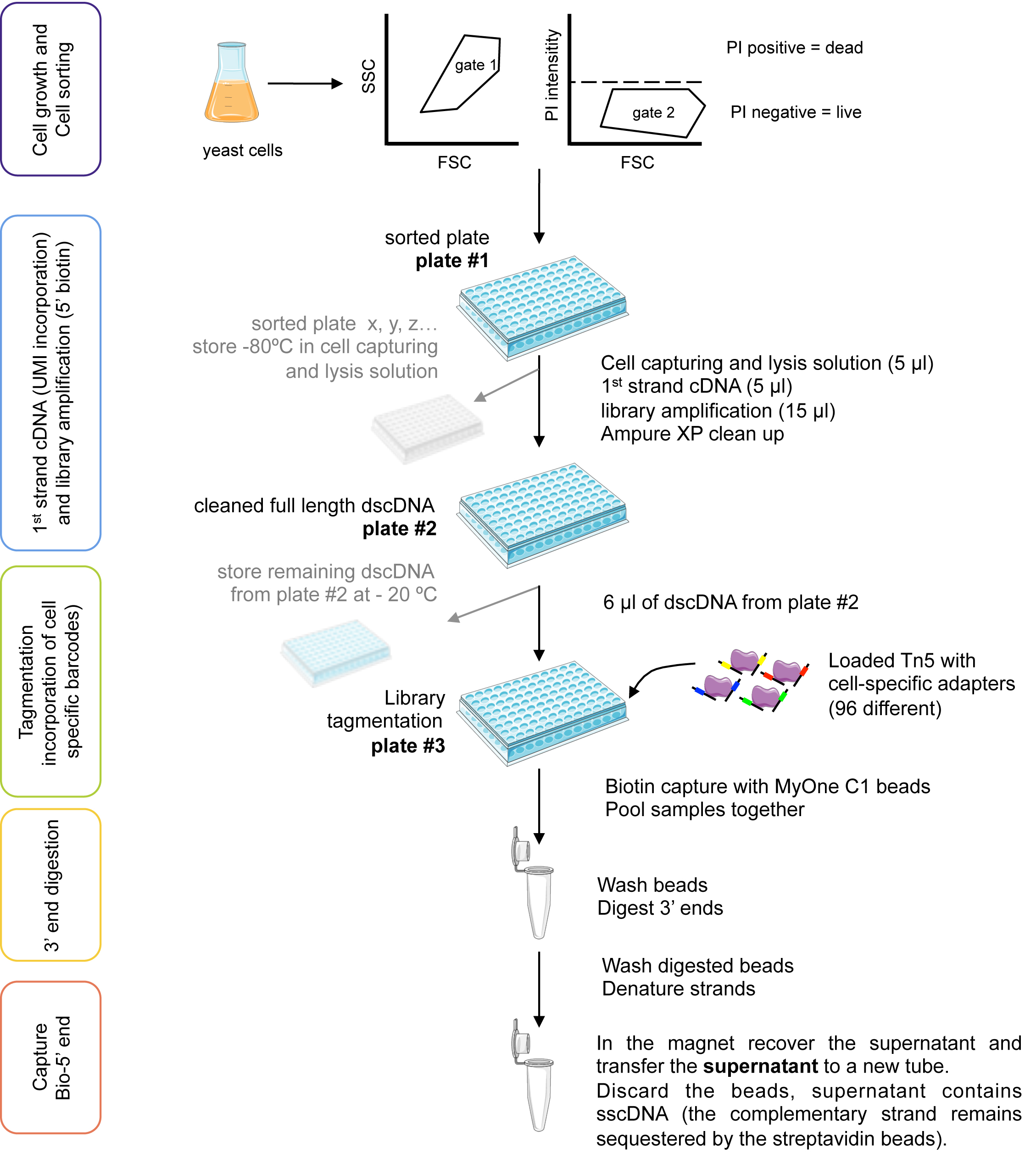
Figure 4. Schematic representation of yscRNA-seq. Images from Smart Medical server (Les Laboratoires Servier, SMART Servier Medical Art.). - Set up a KAPA quantification reaction with a 1:100, 1:1,000 and 1:10,000 dilutions of the eluted cDNA library (Step C27). All regents except for your DNA library are provided in the kit (Table 11).
Data analysis
The initial steps of yscRNA-seq data analysis are very similar to those applied for bulk RNA-seq (Conesa et al., 2016), but with certain particularities. YscRNA-seq generates two reads per sequenced cDNA molecule. The first read (Index 1) contains the cell-specific barcode that associates a given molecule with a specific cell. The second read (Read 1) contains the UMI, and maps to the TSS of a specific transcript, allowing for absolute molecule-counting and transcript identification, respectively. To properly estimate gene expression from yscRNA-seq data, both reads have to be computationally associated to their respective references. The former association is performed by default in Illumina’s BaseSpace demultiplexing tool. This generates individual FASTQ files (one per each yeast cell) that are the starting point of yscRNA-seq analysis. After this, we further pre-process the FASTQ files to identify the UMIs and perform the alignment and quantification to obtain the gene expression table (Figure 5). Here, we detail the steps that we follow for analyzing yscRNA-seq data, starting from FASTQ files:
Note: The analysis pipeline was executed in a Linux 64 bit machine with 32 Gb of RAM and 6 CPUs. We recommend to have at least 12 Gb of RAM. A machine with several computing cores is also preferable for speeding up the processes.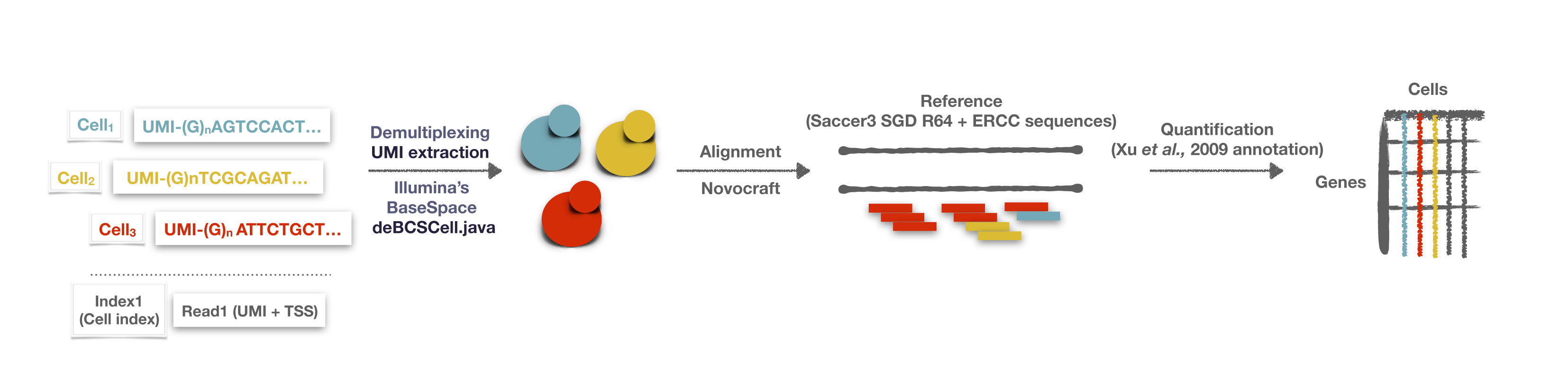
Figure 5. Schematic representation of analysis pipeline of yscRNA-seq
- Read pre-processing
To extract the UMI from the beginning of the reads, we used a custom JavaScript program (deBCSCell).
Command: $java -cp directoryOfdeBCSCell rootNameOfdeBCSCell F1=fastqFile BL="”
This script performs the following actions:- Extracts the first 6nt (UMI) and incorporates them into the read name for later use.
- Extracts and counts N’s and G’s following the UMI, up to 14. The read is discarded if it contains 14 or more N’s or G’s.
- Alignment
Note: In our case, the reference sequences (referenceSequencesName.fa) are a combination of the S. cerevisiae genome (Saccer3, SGD R64 version; www.yeastgenome.org) and the ERCC control sequences. The ERCC control sequences provided by the manufacturer do not include the restriction sites used during the cloning of the sequences downstream of the T7 promoter. Since yscRNA-seq is TSS-specific, this fact considerably decreases the mapping rate due to misalignments at the beginning of the read. To overcome that, we generated a new ERCC reference that includes these sequences. These modified references could be downloaded from the Gene Expression Omnibus (under GSE122392 accession number).
- In our case, we have sequenced each library to a median depth of ~720,000 reads. We assessed, with down-sampling, that this depth is substantially above the needed to obtain a high resolution of the yeast transcriptome. In fact, at ~500,000 reads per cell in the number of transcripts detected saturation is reached (Supplementary Figure 2C [Nadal-Ribelles et al., 2019]). In addition, we observe a median of ~74% uniquely mapped reads for all sequenced libraries. These values are useful reference values using this protocol.
- We used Novocraft for the alignment of the reads, but open-source aligners such as HISAT2 (Kim et al., 2015) or STAR (Dobin et al., 2013) can be used without obtaining major differences in the results.
- Create the index for Novocraft using novoindex command:
$novoindex referenceSequencesName.nix referenceSequencesName.fa
- Align reads using novoalign command:
$novoalign -f fastqFileUMIproccesed.fastq -d referenceSequencesName -o SAM |
- Convert to BAM format and sort it using Samtools view and Samtools sort: (piped from the previous command)
| samtools view -bS - | samtools sort - fileName.sorted.bam.
- Quantification
Although we are aware of the existence of public software for quantifying UMI-based single-cell data (such as UMI-tools [Smith et al., 2017]), we performed quantification using R custom scripts to have full flexibility and control over data inspection and data filtering. The starting point of the quantification is the sorted BAM files obtained in the previous step, one for each sequenced cell.- Process the BAM files.
Script: readAlignUMI.R. $Rscript readAlignUMI.R
Each BAM file is loaded into R using the Genomic Alignments package. Then, low quality mapping reads (MAPQ < 30) and reads that map with soft-clipping in the 5′ end are filtered out. Finally, reads that map to the same position and that have the same UMI are grouped for collapsing them in the next steps. - Process UMI grouped data.
Script: bard2rds.R. $Rscript bard2rds.R
At this step, UMIs supported with less than 3 reads are filtered out since they could represent sequencing errors. Then, different UMIs that map to the same genomic position are grouped. Different UMIs mapping to the same position represent different molecules of the same transcript. This information is stored into a RangedData object. - Overlap with annotation and count table generation.
Script: txAnno.R (#link). $Rscript txAnno.R.
If there is overlap, each molecule is assigned to a genomic feature from an annotation. In this study, we used the annotation from (Xu et al., 2009) since it includes a comprehensive categorization of genes into different classes (coding, CUTs, SUTs and others). The output obtained is an absolute gene expression table with genes as rows and cell as columns.
- Process the BAM files.
- (Optional) Filtering
To avoid the presence of low-quality cells (dead, stressed or apoptotic), we decided to keep only those with more than 500,000 sequenced reads, and those in which we detect more than 1,000 different transcripts. In our case, this filter eliminates cells with a high ratio of mitochondrial RNA, which has been associated with low quality cells (Ilicic et al., 2016). However, this filter is not imperative and could be tuned depending on the type of experiment and on the biological question under study.
Recipes
Note: All reagents and water used must be nuclease-free and only used for single cell protocols.
- Cell capturing and lysis solution (Table 14)
Table 14. Composition of “Cell capturing and lysis solution”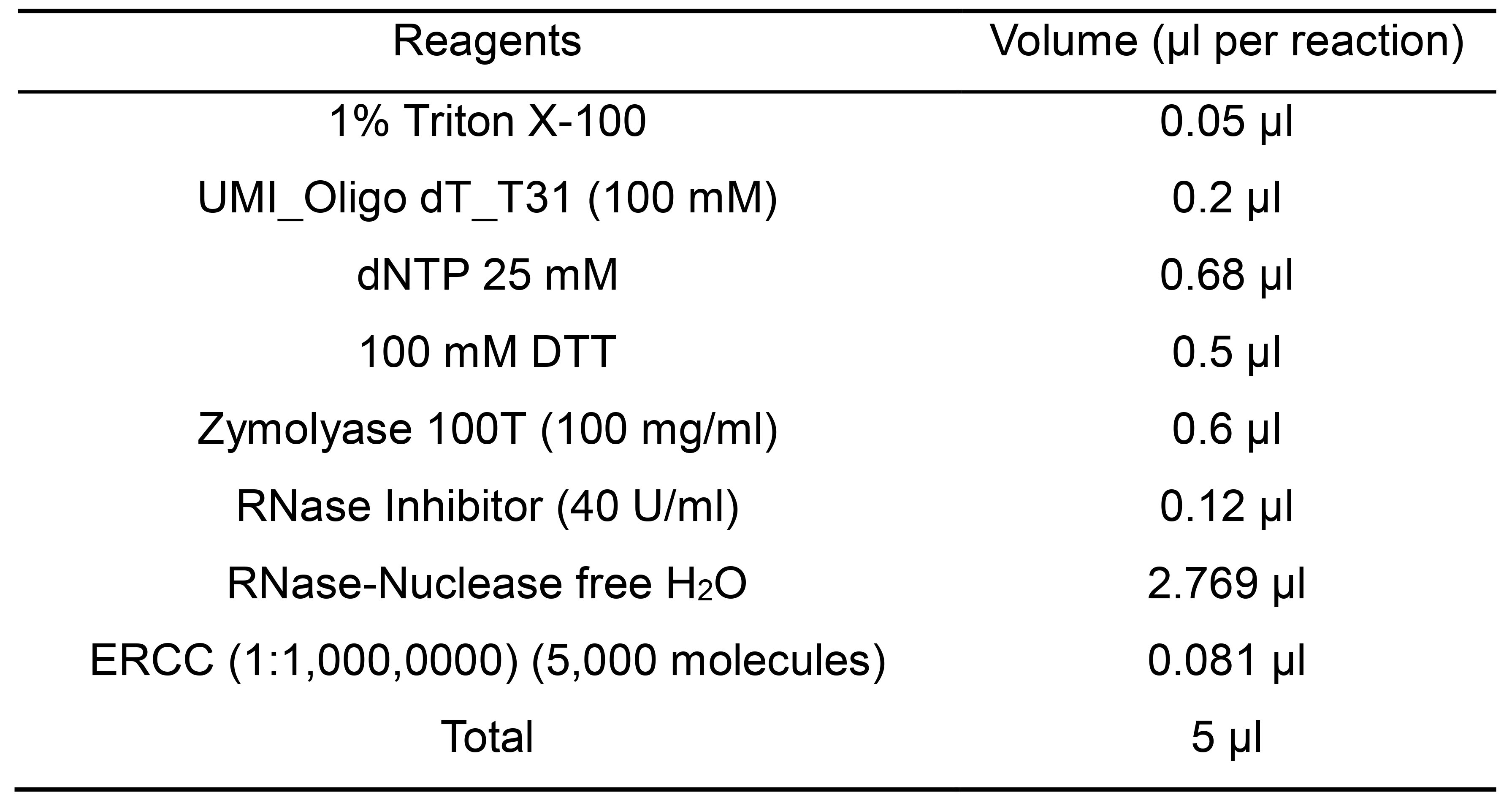
- 2x BWT Buffer
10 mM Tris-HCl pH 7.5
1 mM EDTA
2 M NaCl
0.02% Tween-20 - TNT Buffer
20 mM Tris pH 7.5
50 mM NaCl
0.02% Tween
Acknowledgments
The authors would like to thank Sten Linnarsson for kindly providing reagents during the initial tests with yscRNA-seq. We thank the Protein Expression and Purification Core Facility at EMBL, Bianca Hennig and Lars Velten for kindly providing in-house purified Tn5. We thank Derek Caetano-Anollés for editing and refining the manuscript. M.N.R. was a recipient of an EMBO long-term fellowship (Stanford University) and later of a Maria de Maeztu Postdoctoral Fellowship (Doctores Banco de Santander-María de Maeztu at Universitat Pompeu Fabra). P.L. is a recipient of an FI Predoctoral Fellowship (Generalitat de Catalunya). This work was supported by the National Institutes of Health (NIH) and a European Research Council Advanced Investigator Grant (AdG-294542) to L.M.S. and the National Key Research and Development Program of China (2017YFC0908405) to W.W. The study was also supported by grants from the Spanish Ministry of Economy and Competitiveness (PGC2018-094136-B-I00 and FEDER to F.P.; BFU2017-85152-P and FEDER to E.N.), the Catalan Government (2017 SGR 799), and the Unidad de Excelencia Maria de Maeztu, MDM-2014-0370. We gratefully acknowledge institutional funding from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO) through the Centres of Excellence Severo Ochoa award, and from the CERCA Programme of the Catalan Government. F.P. is recipient of an ICREA Acadèmia (Generalitat de Catalunya).
Competing interests
The authors declare no financial or non-financial interests.
References
- Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., Szczesniak, M. W., Gaffney, D. J., Elo, L. L., Zhang, X. and Mortazavi, A. (2016). A survey of best practices for RNA-seq data analysis. Genome Biol 17: 13.
- David, L., Huber, W., Granovskaia, M., Toedling, J., Palm, C. J., Bofkin, L., Jones, T., Davis, R. W. and Steinmetz, L. M. (2006). A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci U S A 103(14): 5320-5325.
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15-21.
- Gasch, A. P., Yu, F. B., Hose, J., Escalante, L. E., Place, M., Bacher, R., Kanbar, J., Ciobanu, D., Sandor, L., Grigoriev, I. V., Kendziorski, C., Quake, S. R. and McClean, M. N. (2017). Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biol 15(12): e2004050.
- Gresham, D., Bonneau, R., Jackson, C. A., Castro, D. M. and Saldi, G. A. (2019). Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. bioRxiv doi: https://doi.org/10.1101/581678.
- Hennig, B. P., Velten, L., Racke, I., Tu, C. S., Thoms, M., Rybin, V., Besir, H., Remans, K. and Steinmetz, L. M. (2018). Large-scale low-cost NGS library preparation using a Robust Tn5 purification and tagmentation protocol. G3 (Bethesda) 8(1): 79-89.
- Ilicic, T., Kim, J. K., Kolodziejczyk, A. A., Bagger, F. O., McCarthy, D. J., Marioni, J. C. and Teichmann, S. A. (2016). Classification of low quality cells from single-cell RNA-seq data. Genome Biol 17: 29.
- Islam, S., Zeisel, A., Joost, S., La Manno, G., Zajac, P., Kasper, M., Lonnerberg, P. and Linnarsson, S. (2014). Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11(2): 163-166.
- Kim, D., Langmead, B. and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4): 357-360.
- Lawrence, Michael, Wolfgang Huber, Hervé Pagès, Patrick Aboyoun, Marc Carlson, Robert Gentleman, Martin T. Morgan, and Vincent J. Carey. (2013). Software for Computing and Annotating Genomic Ranges. PLoS Computational Biology 9(8):e1003118.
- Li, Heng, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, and Richard Durbin. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25(16):2078–79.
- Nadal-Ribelles, M., Islam, S., Wei, W., Latorre, P., Nguyen, M., de Nadal, E., Posas, F. and Steinmetz, L. M. (2019). Sensitive high-throughput single-cell RNA-seq reveals within-clonal transcript correlations in yeast populations. Nat Microbiol 4(4): 683-692.
- Pelechano, V., Wei, W. and Steinmetz, L. M. (2013). Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497(7447): 127-131.
- Pelechano, V., Wei, W. and Steinmetz, L. M. (2015). Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161(6): 1400-1412.
- Pelechano, V., Wei, W., Jakob, P. and Steinmetz, L. M. (2014). Genome-wide identification of transcript start and end sites by transcript isoform sequencing. Nat Protoc 9(7): 1740-1759.
- Saint, M., Bertaux, F., Tang, W., Sun, X. M., Game, L., Koferle, A., Bahler, J., Shahrezaei, V. and Marguerat, S. (2019). Single-cell imaging and RNA sequencing reveal patterns of gene expression heterogeneity during fission yeast growth and adaptation. Nat Microbiol 4(3): 480-491.
- Skinnider, M. A., Squair, J. W. and Foster, L. J. (2019). Evaluating measures of association for single-cell transcriptomics. Nat Methods 16(5): 381-386.
- Smith, T., Heger, A. and Sudbery, I. (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27(3): 491-499.
- Xu, Z., Wei, W., Gagneur, J., Perocchi, F., Clauder-Munster, S., Camblong, J., Guffanti, E., Stutz, F., Huber, W. and Steinmetz, L. M. (2009). Bidirectional promoters generate pervasive transcription in yeast. Nature 457(7232): 1033-1037.
- Zhang, X., Li, T., Liu, F., Chen, Y., Yao, J., Li, Z., Huang, Y. and Wang, J. (2019). Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol Cell 73(1): 130-142 e5.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nadal-Ribelles, M., Islam, S., Wei, W., Latorre, P., Nguyen, M., de Nadal, E., Posas, F. and Steinmetz, L. M. (2019). Yeast Single-cell RNA-seq, Cell by Cell and Step by Step. Bio-protocol 9(17): e3359. DOI: 10.21769/BioProtoc.3359.
Category
Microbiology > Microbial genetics > Gene expression
Microbiology > Microbial cell biology > Cell isolation and culture
Molecular Biology > RNA > Transcription
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.