- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Novel Dual Lentiviral CRISPR-based Transcriptional Activation System for Gene Expression Regulation in Neurons

Published: Vol 9, Iss 17, Sep 5, 2019 DOI: 10.21769/BioProtoc.3348 Views: 7328
Reviewed by: Alessandro DidonnaPamela MaherMarzia Di Donato
Original research article
The authors used this protocol in:

Mar 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
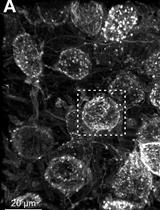

Abstract
Robust and efficient gene expression control enables the study of a gene’s function in the central nervous system. Advances in CRISPR-based technology provide new avenues not only for gene editing, but for complex transcriptional control. Here, we describe a protocol to generate high-titer lentiviruses with neuron-optimized CRISPR-activation constructs (dual lentiviruses consisting of a gene-specific single guide RNA and the CRISPR-activator) for use in primary neurons in vitro or in the adult brain in vivo. This protocol enables modular, scalable, and multiplexable gene regulation in the nervous system and does not require a transgenic model organism.
Keywords: CRISPR-Cas9Background
The study of gene expression in the central nervous system is challenging due to difficulties in transgene delivery in post-mitotic neurons. Approaches to induce gene expression have typically relied on the use of transgenic animal models (Ericsson et al., 2013) and viral overexpression vectors (Prelich, 2012). These techniques are resource and time-consuming to make, neglect the endogenous gene locus, are limited by viral cargo capacities, and generally only target one gene at a time. CRISPR-Cas9 gene editing technology has been adapted to recruit effector proteins, such as transcriptional activators, to enable precise control over transcription (Chavez et al., 2015; Savell and Day, 2017). We recently optimized a CRISPR-activation (CRISPRa) system for robust, specific, titratable, and multiplexable gene induction in neurons both in vitro and in vivo by adapting available CRISPRa tools to be expressed with a dual lentivirus approach (Savell et al., 2019). This dual lentivirus system is composed of lentiviruses containing a gene-specific single guide RNA (sgRNA) and the dCas9-VPR (a strong transcriptional activator). Multiplexing at a single gene involves recruiting multiple dCas9-VPR effectors to a single gene’s promoter to boost transcription of that gene by pooling sgRNAs targeting the same gene. Multiplexing multiple genes is possible through combining sgRNAs targeting each of the desired genes.
This protocol is divided into subsections to outline 1) identification of sgRNA targeting sequences for CRISPRa and cloning the targeting sequence into the sgRNA scaffold, 2) maintaining and using HEK293T cells for lentivirus production, 3-4) lentivirus preparations on different scales for use in vitro and in vivo, 5) transduction guidelines for in vitro applications, 6) Transduction of adult neurons in vivo.
Part I: Identification of sgRNA targeting sequence for CRISPRa at gene promoters and cloning the target sequence into the sgRNA scaffold
Materials and Reagents
- Pipettes (Rainin, catalog number: 17014415 & 17014393)
- 1-2 L vented baffled flask (Fisher Scientific, catalog numbers: 50-121-4761 & 50-153-2267)
- Pipette tips (Denville, catalog numbers: 1159M43, 1159M40, 1159M42; Rainin, catalog number: 30389274)
- Corning Falcon round-bottom polystyrene tubes (Fisher, catalog number: 14-959-7)
- Nalgene General Long Term Storage Cryogenic Tube, sterile, 1.5 ml (Fisher, catalog number: 03-337-7Y)
- Custom oligos with correct overhangs (outlined in Part II), standard desalted purification
U6 validation primer: TTTCTTGGGTAGTTTGCAGTTTT - T4 polynucleotide kinase (PNK) (NEB, catalog number: M0201S)
- One Shot Stbl3 Chemically Competent E. coli (Invitrogen, catalog number: C737303)
- DNA ligase (NEB, catalog number: M0202S)
- Lenti U6-sgRNA/EF1a-mCherry (Addgene, catalog number:114199)
- NEBuffer 2 (NEB, catalog number: B7002S)
- FastDigest BpiI (Bbsi) (Thermo Fisher, catalog number: FD1014)
- Alkaline Phosphatase, Calf Intestinal (CIP) (NEB, catalog number: M0290S)
- Autoclaved MilliQ H2O
- PCR Purification Kit (Qiagen, catalog number: 28106)
- T4 ligation reaction buffer (NEB, catalog number: B0202S)
- imMedia Growth Medium, agar, ampicillin (Invitrogen, catalog number: 450034)
Note: Follow manufacturer’s instructions for preparation of the plates. - S.O.C. Medium (Invitrogen, catalog number: 15544034)
- Ampicillin sodium salt (Gibco, catalog number: 11593-027)
Note: Resuspend in sterile H2O, store aliquots at -20 °C. - Qiaprep Spin Miniprep Kit (Qiagen, catalog number: 27106)
- OneTaq 2x master mix with standard buffer (NEB, catalog number: M0482S)
- PureLink HiPure Plasmid Filter Midiprep Kit (Fisher, catalog number: K210015)
- PureLink HiPure Plasmid Filter Maxiprep Kit (Fisher, catalog number: K210017)
- TE buffer
- 2XYT broth with ampicillin (see Recipes)
- Tryptone (Fisher, catalog number: BP1421-500)
- Yeast extract (Fisher, catalog number: BP1422-500)
- Sodium chloride (Fisher, catalog number: S271-1)
Equipment
- -80 °C freezer (Thermo Fisher Scientific, model: UXF60086A63)
- Thermal cycler (Bio-Rad, C1000 Touch Thermal Cycler)
- Heat block (Fisher Scientific, Mini Heat Block 14955218)
- Large incubating orbital shaker set to 37 °C and 225 rpm (New Brunswick, G-25 Controlled Environment Incubator Shaker)
- Small shaker/incubator set to 37 °C (Fisher Scientific, Mini-Shaker 02217753)
- Tabletop centrifuge (Eppendorf, model: 5340)
- Refrigerating centrifuge that can accommodate up to 50ml conical tubes and a speed up to 12,000 x g (Eppendorf, model: 5810R)
- Gel electrophoresis equipment (Bio-Rad, Min-Sub Cell GT Cell)
Procedure
A general overview of this process is outlined in Figure 1.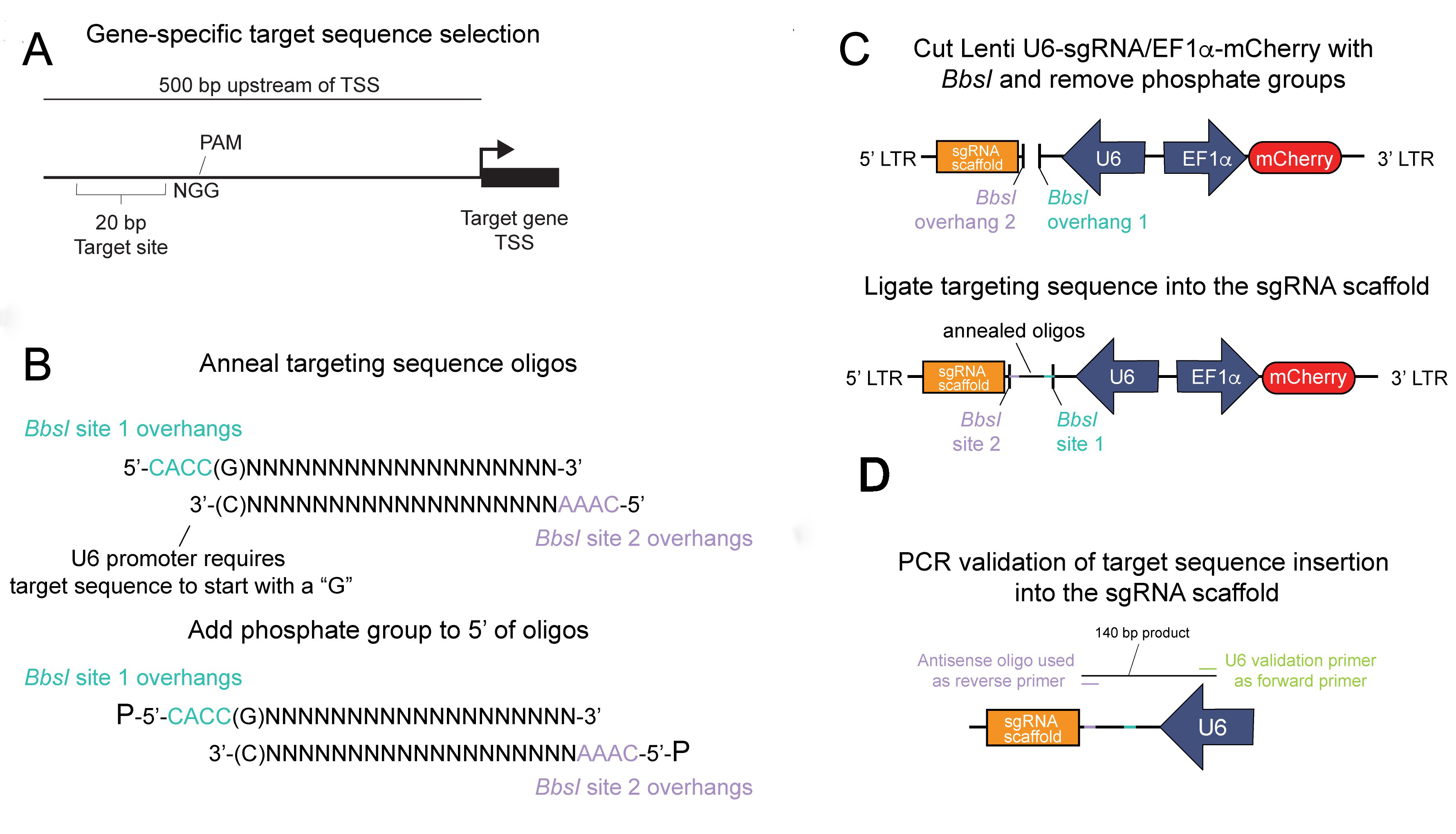
Figure 1. Insertion of the gene-specific targeting sequence into the sgRNA scaffold. A. A targeting region is selected within 500 bp of the transcriptional start site (TSS) of a gene of interest by a publicly available CRISPR/dCas9 target site selection tool. The targeting sequence is immediately adjacent to a protospacer adjacent motif (PAM) and is required for spCas9 targeting. B. Oligos containing the target site and the unique BbsI overhangs are (top) annealed and (bottom) phosphorylated to create a dsDNA fragment for insertion into the sgRNA scaffold plasmid. C. The Lenti U6-sgRNA/Ef1a-mCherry plasmid is digested by BbsI and dephosphorylated (top), then the annealed targeting sequence is ligated into the cut backbone (bottom). D. After the plasmid is transformed and amplified, a PCR reaction is performed to validate correct insertion of the targeting sequence.
- Utilize a publicly available CRISPR/Cas9 sgRNA prediction tool (We recommend CHOPCHOP or e-CRISP) to target within 2 kb upstream of the transcriptional start site (TSS) of the gene, typically referred to as the promoter region. While we have observed successful CRISPRa with sgRNAs up to 2 kb from a TSS (or even distal gene enhancers), we have observed the most robust results with sgRNAs targeting within 500 bp upstream of the TSS.
- sgRNA prediction tools will typically output a 20 bp targeting sequence immediately before a 3 bp PAM sequence. The PAM sequence is specific to the species of Cas9/dCas9 used. The CRISPR-activation construct in this protocol utilizes a PAM sequence of NGG. Do not include the PAM site as part of the inserted target sequence.
- Alternatively, the tool can search specific genomic coordinates, which can be obtained using publicly available databases such as the UCSC genome browser or NCBI Gene.
- Choose targeting sequences with no predicted mismatches at other sites in the genome.
- Repeat process to obtain 3-5 additional sgRNA targets covering both DNA strands.
- Order oligos with the following overhangs so that the target sequences are compatible with the Lenti U6-sgRNA/EF1a-mCherry construct.
- The U6 promoter requires a “G” nucleotide to initiate transcription. Therefore, if your target sequence does not begin with a G, you must replace the first base pair.
- Oligos should be ordered with the following added overhangs:
Sense: 5′-CACC(G)NNNNNNNNNNNNNNNNNNN-3′
Antisense: 5′-AAACNNNNNNNNNNNNNNNNNNN(C)-3′- The antisense is the reverse-complement of the sense targeting sequence.
- N: targeting sequence before the NGG PAM sequence.
- CACC/AAAC: overhangs that must be included to ligate into the sgRNA scaffold.
- (G)/(C): replace the first nucleotide of the targeting sequence if it does not start with a G.
- Resuspend oligos (if necessary) and mix the sense and antisense oligos in a 20 µl reaction in a PCR tube, diluted to 10 M each in 1x PBS. Anneal the oligos with the following thermal cycler program: 95 °C for 180 s, 85 °C for 20 s, 75 °C for 20 s, 65 °C for 20 s, 55 °C for 20 s, 45 °C for 20 s, 35 °C for 20 s and 25 °C for 20 s with a 0.1 °C/s decrease rate in between steps. End with 4 °C hold.
- Perform a standard 20 µl PNK reaction in a PCR tube, using 5 µl of the annealed oligo reaction as DNA input:
2 µl NEB T4 ligation buffer
5 µl annealed oligo reaction from Step 3
12 µl MilliQ H2O
1 µl T4 PNK
Note: Substitute NEB T4 PNK buffer for NEB T4 ligation buffer due to the requirement of ATP.- Incubate reaction with the following thermal cycler program: 37 °C for 30 min, 65 °C for 20 min, 4 °C hold.
- After the reaction is complete, dilute the reaction 1:50 (reaction:MilliQ H2O).
- Digest 1 µg of the Lenti U6-sgRNA/EF1a-mCherry with BpiI/Bbsi in a standard 30 µl reaction supplemented with CIP:
1 µg Lenti U6-sgRNA/EF1a-mCherry
3 µl NEBuffer 2
0.1 µl CIP
1 µl BpiI/Bbsi
MilliQ H2O to 30 µl
Incubate reaction for 1 h at 37 °C.
Notes:- The plasmid must be dissolved in water or a buffer without EDTA, which will inhibit the reaction.
- Substitute the Thermo digest buffer for NEBuffer 2.
- Perform PCR cleanup on the digested plasmid from Step 5 using the Qiagen PCR Purification Kit. Elute the digested plasmid in 20-30 µl Buffer EB. Determine the concentration of the digested plasmid.
Note: There is no need to run the product out on a gel and extract. The excised product will be ~20 bp and will not be retained by the cleanup column. - Ligate the annealed target sequence to the digested sgRNA scaffold with a standard T4 ligase reaction:
2 µl 10x T4 DNA ligase buffer
1.5 µl diluted PNK reaction
50 ng of the eluted, digested sgRNA scaffold plasmid
1 µl T4 DNA ligase (add last)
MilliQ water to 20 µl
Run the reaction in a thermal cycler: 23 °C for 15 min, 65 °C for 20 min, 4 °C hold. - Chill ligation on ice when finished and remove a Stbl3 tube from the -80 °C and place on ice.
Note: Always use Stbl3 or similar E. coli strain to maintain the lentiviral LTRs. - Transform the ligated sgRNA into Stbl3s:
- Aseptically add 4 µl of ligation reaction to the tube and tap gently. Do not pipette mix or vortex.
- Rest on ice for 30 min.
- Heat shock cells by incubating at 42 °C on heat block for 30 s. Immediately return to ice and rest for at least 2 min.
- Aseptically add 250 µl S.O.C. media and incubate in a large incubator at 37 °C for 1 h with 225 rpm orbital shaking. Place a prepared ampicillin agar plate to warm in a small 37 °C incubator.
- With a sterile L-shaped spreader (glass or disposable plastic), evenly distribute 50 µl of the transformant onto the warmed ampicillin agar plate. Flip the plate so the agar is on the top and incubate overnight at 37 °C in a small incubator.
- Aseptically inoculate a 14 ml round bottom tube with 3 ml of 2XYT + ampicillin broth by touching a single colony with a sterile pipette tip and dropping it into the round bottom flask. Incubate overnight (no longer than 16 h total) at 37 °C with 250 rpm orbital shaking in a large incubator.
- Aseptically prepare a glycerol stock consisting of 500 µl bacterial culture and 500 µl 1:1 (sterile water:glycerol) in a cryovial (store at -80 °C) then prep plasmid with the remaining culture using Qiaprep Spin Miniprep kit and elute in 50 µl buffer EB.
- To validate that the targeting sequence inserted correctly, set up a standard 10 µl OneTaq reaction in a PCR tube:
5 µl OneTaq 2x master mix
1 µl of diluted plasmid (dilute to 1-5 ng/µl)
0.2 µl U6 verification primer (10 µM)
0.2 µl Custom antisense oligo (10 µM)
3.6 µl H2O
Run the reaction in a thermal cycler: 94 °C for 30 s, 30 cycles of (94 °C 15 s, 54 °C 15 s, 68 °C 15 s), 68 °C 5 min, 4 °C hold. - Separate the PCR reaction on a 2% agarose gel.
Notes:- If the insert is correct, there should be a single 140 bp product. Non-target sgRNA antisense oligos can be used as a negative control reaction if desired.
- If Sanger sequencing to validate correct target sequence insertion is desired, the U6 validation primer is validated for that application.
- Ensure all lentivirus helper plasmids (See Part III for all plasmids) are also prepared or if larger preps of the sgRNAs are needed:
- Aseptically streak glycerol stocks onto an ampicillin agar plate, incubate overnight at 37 °C in a small incubator.
- Isolate a single colony from the plate (no more than a day or two old) and inoculate into a 1-2 L vented baffled flask with 100-200 ml 2XYT + ampicillin broth. Incubate overnight at 37 °C with 250 rpm orbital shaking in a large incubator.
- Prep with PureLink HiPure plasmid filter midiprep/maxiprep kit using manufacturer’s instructions. Elute in TE buffer and aliquot plasmid.
Part II: Maintaining HEK293T cells for lentivirus production
Materials and Reagents
- Pipettes (Rainin, catalog number: 17014415 & 17014393)
- Pipette tips (Denville, catalog numbers: 1159M43, 1159M40, 1159M42; Rainin, catalog number: 30389274)
- Stericup Sterile Vacuum Filter Units: 250 ml funnel; PVDF membrane; 250 ml receiver; Pore size: 0.22 µm (Fisher, catalog number: SCGVU02RE)
- Falcon Tissue Culture Treated Flasks: vented cap; canted neck; growth area: 75 sq.cm; 250 ml (Fisher, catalog number: 13-680-65)
- Thermo Scientific Nunc EasYFlasks: filter cap, cell culture treated, 225 sq.cm; 70 ml working volume (Fisher, catalog number: 12-565-221)
- 15 ml conical tube
- Autoclaved disposable borosilicate glass Pasteur pipets: length 5.75 (Fisher, catalog number: 13-678-20B)
- HEK293T Cells (ATCC, catalog number: CRL-3216)
- 1x DPBS (Thermo-Fisher, catalog number: 14190144)
- Trypsin EDTA (0.25%) with Phenol red (Thermo-Fisher, catalog number: 25-200-056)
- 70% ethanol
- HEK293T media (see Recipes)
- 10% volume FBS US Origin (Biofluid Technologies, catalog number: 200-100-Q)
- DMEM, high glucose, pyruvate (Thermo Fisher, Gibco, catalog number: 11-995-081)
- 100x Penicillin-streptomycin (10,000 U/ml) (Thermo Fisher, catalog number: 15-140-122)
Equipment
- Hemocytometer (Sigma-Aldrich, model: Z359629-1EA)
- Class II biosafety cabinet (NuAire, model: NU-425-400)
- Humidity, temperature, and CO2-controlled culture incubator (Thermo Forma Series II Water Jacketed CO2 incubator, set at 37 °C with 5% CO2)
- Water bath set to 37 °C (Fisher Scientific, model: IsoTemp 210)
- Centrifuge with swinging bucket rotor that can accommodate up to 50 ml conical tubes (Eppendorf, model: 5810R)
- Liquid nitrogen Dewar with vial storage boxes within the vapor phase (Thermo, catalog number: 316376)
Procedure
- Revive cells
- Place 11 ml HEK293T media into a T75 flask and place in an incubator to warm and equilibrate pH. Also, warm 10 ml HEK293T media to 37 °C.
- Rapidly revive a vial of HEK293T cells stored in a Dewar by holding the cap with a 70% ethanol pre-soaked tissue in a 37 °C water bath until one or two small ice crystals remain (2 min or less). Disinfect the vial with 70% ethanol and transfer to the biosafety cabinet and work in the hood for remainder of protocol.
- Transfer all of the cell solution (usually 1 ml) to a 15 ml conical tube and dilute 10x with HEK293T media (usually 9 ml).
- Centrifuge cells at 300 x g in a swinging-bucket rotor for 3 min. Remove supernatant and resuspend in 1 ml of warmed HEK293T media. Count the cells with a hemocytometer.
- Seed approximately 2.8 million cells into the media-containing T75 flask. Evenly distribute the cells on over the area of the flask by shifting the flask forward-backward and side-to-side without rocking the flask.
- Check the confluence of the cells daily, but it is likely that seeding that density will result in cells being ready to split around 48 h after seeding.
- Split/passage cells
- Examine cells daily and wait until cells are 60-75% confluence before splitting/passaging. The health of HEK cells is critical to virus titer. Never split cells that are above 80% confluence and generally do not use cells that are above 25 passages. Additionally, do not use cells that are growing unevenly or have any visual morphological changes.
- Place 11.5 ml HEK293T media into a T75 flask and place in an incubator to warm and equilibrate pH. Also, warm 5 ml 1x DPBS, 10 ml HEK293T media, and 2.5 ml of 0.25% trypsin-EDTA to 37 °C.
- Aspirate media and gently add the warmed DPBS to the flask to wash cells. Aspirate DPBS and add the trypsin.
- Gently rock the flask until all cells detach, generally in under a minute.
- Stop the trypsin reaction by adding 5 ml warmed HEK293T media. Transfer cells to a 15 ml conical tube and centrifuge cells at 300 x g in a swinging-bucket rotor for 3 min. Remove supernatant and resuspend in 5 ml of warmed HEK293T media. Count the cells with a hemocytometer.
- Seed approximately 2.8 million cells into the media-containing T75 flask. Evenly distribute the cells on over the area of the flask by shifting the flask up/down and side to side without rocking the flask.
- Check the confluence of the cells daily, but it is likely that seeding that density will result in cells being ready to split around 48 h after seeding. For cells to be ready to split approximately 72 h later, seed 1 million cells.
Note: These numbers are a guideline. Carefully watch the growth rate of your HEK293T line and adjust accordingly.
Part III: Small scale lentivirus packaging
This small-scale lentivirus preparation is recommended to validate sgRNA effectiveness at the target gene in vitro. It will yield enough lentivirus to efficiently transduce approximately 375,000 primary rat neurons in culture. A general overview of both small-scale and large-scale virus production is outlined in Figure 2. 
Figure 2. Overview of both small-scale and large-scale lentivirus production preparations
Materials and Reagents
- Pipettes (Rainin, catalog number: 17014415 & 17014393)
- Pipette tips (Denville, catalog numbers: 1159M43, 1159M40, 1159M42; Rainin, catalog number: 30389274)
- Denville cell culture plates: 12-well plate with lid (Thomas Scientific, catalog number: 1156D99)
- 1.5 ml polypropylene microcentrifuge tubes (Fisher, catalog number: 21-402-901)
- Plasmids:
- The custom sgRNA plasmid cloned as described in the Part I (transfer plasmid) (Mini prepped is fine for the small-scale preps)
- pCMV-VSVG (Addgene, catalog number: 8454) (Midi/maxi prepped only)
- psPAX2 (Addgene, catalog number: 12260 ) (Midi/maxi prepped only)
- Dilute plasmids in EB or TE buffer if needed.
- Lenti-X Concentrator (Takara Bio, catalog number: 631231)
- FuGene HD (Promega, catalog number: E2311)
- Opti-MEM I reduced serum medium (Thermo Fisher, catalog number: 31985070)
- Dulbecco’s Phosphate Buffered Saline (DPBS) (Gibco, catalog number: 14190136)
- 10% chlorine bleach (Clorox)
- TE buffer (Thermo-Fisher, catalog number: 12090015)
- EB buffer (Qiagen, catalog number: 19086)
- Lentivirus production media (see Recipes)
- Lonza BioWhittaker UltraCulture Serum-free Medium without L-glutamine (Fisher, catalog number: 12-725F)
- L-glutamine (200 mM) (Thermo Fisher, catalog number: 25030149)
- 7.5% w/v sodium bicarbonate (Fisher, catalog number: S233-500)
- 100 mM sodium pyruvate (Lifetech, catalog number: 11360070)
Equipment
- Tabletop centrifuge with refrigeration (Eppendorf, model: 5424R)
- Fluorescent microscope microscope (Nikon Ti SR)
- Humidity, temperature, and CO2-controlled culture incubator (Thermo Forma Series II Water Jacketed CO2 incubator, set at 37 °C with 5% CO2)
Procedure
- The day before transfection, plate 400k HEK293T cells per well into a 12-well plate with 1 ml HEK293T media.
- When HEK293T cells are approximately 80-95% confluent, prepare for transfection. Warm 1 ml of prepared lentivirus production media per well. The cells are generally ready to transfect within 24 h after plating.
- Allow FuGene to equilibrate to room temperature and vortex briefly to mix. Thaw plasmids. Aseptically transfer 50 µl of OptiMEM to a 1.5 ml polypropylene tube for each well to be transfected.
- Add 350 ng sgRNA plasmid, 250 ng pCMV-VSVG, and 400 ng psPAX2 to the aliquoted OptiMem and vortex to mix.
- Spin down the plasmid solution then add 3 µl FuGene directly into the middle of the liquid, do not run FuGene down the side of the tube as it reacts with plastic. Lightly vortex the reaction.
- Incubate reactions for 10 min. While incubating, gently aspirate media on HEK293T cells and replace with 1 ml warmed lentivirus production media per well.
- Add complete reaction to the well and gently mix by moving the plate forward-backward and side-to-side.
- Incubate transfection reaction on cells for 40-48 h. From this point on, the culture plate contains viable replication-defective lentivirus particles. Adhere to all BSL-2 guidelines or the guidelines outlined by your institution. Disinfect all materials that came into contact with the virus by soaking in 10% chlorine bleach for 30 min before disposing.
- For sgRNAs, validate mCherry expression with a fluorescent microscope if desired.
- Place media into a 1.5 ml microcentrifuge tube and centrifuge at 2,300 x g for 5 min to pellet debris.
- Avoiding the debris pellet at the bottom of the tube, remove 900 µl supernatant and place in a new tube.
- Add 300 µl Lenti-X concentrator, cap, and invert gently to mix.
Note: The Lenti-X concentrator is very viscous, pipette slowly. - Incubate at 4 °C for 6 h to overnight. The sample must be thoroughly chilled.
- Chill a microcentrifuge to 4 °C. Centrifuge each lentivirus at 1,500 x g for 45 min to pellet the lentivirus.
- Carefully remove all supernatant without disturbing the lentiviral pellet (may be invisible) and resuspend in 10 µl cold 1x sterile DPBS by gentle pipetting. Avoid making bubbles.
- Allow the lentivirus to resuspend for 6 h to overnight before transducing. This is enough virus to efficiently transduce 375,000 primary rat neurons in culture.
Part IV: Large-scale lentivirus packaging
This large-scale lentivirus preparation is recommended to make both the sgRNA and dCas9-VPR lentiviruses for use both in vitro and in vivo. With this protocol, we routinely obtain 150-450 µl of > 5 x 1011 GC/ml (genome copies/ml) for sgRNA viruses and > 1 x 1011 GC/ml for the dCas9-VPR virus for in vitro use. For in vivo, we routinely yield 30-50 µl of > 1 x 1012 GC/ml for sgRNA viruses and > 5 x 1011 GC/ml for the dCas9-VPR virus. This protocol was adapted from a publicly available lentivirus protocol (Synthetic Neurobiology Group, 2009).
Materials and Reagents
- Pipettes (Rainin, catalog number: 17014415 & 17014393)
- Pipette tips (Denville, catalog numbers: 1159M43, 1159M40, 1159M42; Rainin, catalog number: 30389274)
- Ultra-centrifuge tube; thinwall; 38.5 ml; 25 x 89 mm (Beckman Coulter, catalog number: 344058)
- Ultra-centrifuge tube; thinwall; 13.2 ml; 14 x 89 mm (Beckman Coulter, catalog number: 344059)
- 50 ml conical tube
- 0.45 µm filter unit
- Falcon Tissue Culture Treated Flasks: vented cap; canted neck; growth area: 75 sq.cm; 250 ml (Fisher, catalog number: 13-680-65)
- Thermo Scientific Nunc EasYFlasks: filter cap, cell culture treated, 225 sq.cm; 70 ml working volume (Fisher, catalog number: 12-565-221)
- EMD Millipre Millex-HP filter unit: pore size: 0.45 µm; filter diameter: 33 mm (Fisher, catalog number: SLHP033RB)
- BD General-use syringes: Leur lock; volume: 30 ml (Fisher, catalog number: 22-124-969)
- HEK293T cells
- Opti-MEM I reduced serum medium (Thermo Fisher, catalog number: 31985070)
- Plasmids:
- Custom sgRNA plasmid cloned as described in Part I (transfer plasmid)
- Lenti SYN-FLAG-dCas9-VPR (Addgene, catalog number: 114196) (transfer plasmid)
- pCMV-VSVG (Addgene, catalog number: 8454)
- psPAX2 (Addgene, catalog number: 12260 )
- Dilute plasmids in EB or TE buffer if needed
- FuGene HD (Promega, catalog number: E2311)
- Dulbecco’s Phosphate Buffered Saline (DPBS) (Gibco, catalog number: 14190136)
- Lenti-X qRT-PCR Titration Kit (Takara, catalog number: 631235)
- TE buffer (Thermo-Fisher, catalog number: 12090015)
- EB buffer (Qiagen, catalog number: 19086)
- Lentivirus production media (see Recipes)
- Lonza BioWhittaker UltraCulture Serum-free Medium without L-glutamine (Fisher, catalog number: 12-725F)
- L-glutamine (200 mM) (Thermo Fisher, catalog number: 25030149)
- 7.5% w/v sodium bicarbonate (Fisher, catalog number: S233-500)
- 100 mM sodium pyruvate (Lifetech, catalog number: 11360070)
Equipment
- -80 °C freezer (Thermo Fisher Scientific, model: UXF60086A63)
- Ultracentrifuge with appropriate rotors and tube holders (Beckman Coulter, model: Optima L-100K Ultracentrifuge with SW32 and SW41 rotors)
- Tabletop centrifuge with refrigeration (Eppendorf, model: 5424R)
- Fluorescent microscope (Nikon Ti SR)
Procedure
- Forty-eight hours before transfection, plate approximately 3.3 million HEK293T cells per T75 and 10 million cells per T225.
Note: Ensure even distribution of cells across the flask, do not use if growth is uneven. - When HEK293T cells are approximately 80-95% confluent, prepare for transfection. Warm prepared lentivirus production media.
T75: 13 ml
T225: 33 ml - Allow FuGene to equilibrate to room temperature and vortex briefly to mix. Thaw plasmids. Aseptically transfer OptiMEM to a polypropylene conical tube for each flask to be transfected.
T75: 1.5 ml
T225: 4.5 ml - Add plasmids (midi/maxi prepped only) to the OptiMEM, vortex, and spin down:
T75: 9 µg transfer plasmid, 6 µg pCMV-VSVG, and 9 µg psPAX2
T225: 27 µg transfer plasmid, 18 µg pCMV-VSVG, and 27 µg psPAX2
Notes:- A molar ratio can be calculated for each transfer plasmids, but these amounts seem to work well for most constructs.
- The transfer plasmids are either the sgRNA or the dCas9-VPR plasmid.
- Spin down the plasmid solution then add FuGene directly into the middle of the liquid, do not run FuGene down the side of the tube as it reacts with plastic. Lightly vortex the reaction.
T75: 65 µl
T225: 200 µl - Incubate reactions for 10 min. While incubating, gently aspirate media on HEK293T cells and replace with 1 ml warmed lentivirus production media per well.
Note: At this confluence, cells can detach easily. Gently add the media by tilting the flask and slowly running the media down a side that does not contain cells. - Add complete reaction to the flask and gently mix by moving the plate forward-backward and side-to-side.
- Incubate transfection reaction on cells for 40-48 h. From this point on, the culture plate contains viable replication-defective lentivirus particles.
Note: Adhere to all BSL-2 guidelines or the guidelines outlined by your institution. Disinfect all materials that came into contact with the virus by soaking in 10% chlorine bleach for 30 min before disposing. - Pre-chill the ultracentrifuge, rotor, and tube holders to 4 °C. Clean tube holders and allow to thoroughly dry.
- For sgRNAs, validate mCherry expression with a fluorescent microscope if desired.
- Place media into a 50 ml conical tube and centrifuge at 2,300 x g for 5 min to pellet debris.
- Remove the plunger from the syringe and assemble a 0.45 µm filter unit to a 30 ml syringe for each virus and place ultracentrifuge tubes in the biosafety hood.
- Avoiding the debris pellet at the bottom of the tube, remove supernatant and place in the syringe held over an ultra-centrifuge tube. Replace the plunger and slowly run the supernatant through the filter unit.
T75: 12 ml for the Beckman 344059 tube
T225: 36 ml for the Beckman 344058 tube
Notes:- It is critical to fill the ultracentrifuge tubes to approximately 90% full, otherwise they will collapse during the spin.
- If you need to spin at a later time, tubes can be tightly sealed and stored at 4 °C for a few hours.
- Weigh the tubes and adjust mass with prepared lentivirus production media, ensuring that all tubes are within 0.2 g of each other.
- Place tubes in tube holders, mount tube holders in the rotor, and place into the ultracentrifuge. Spin for 1.75 h at 106,883 x g (25,000 rpm) at 4 °C with a vacuum. Set the rotor to maximum acceleration and slow deceleration.
- Prepare a flask of 10% bleach. Once the spin is complete, take the tube holders into the biosafety hood and decant the supernatant into the bleach. Keeping the tube overturned, use a Kimwipe to remove any residual supernatant. Do not touch the pellet at the bottom of the tube.
- Resuspend pellet in 1x DPBS and gently pipette mix, avoiding creating any bubbles. Tightly seal the tube and allow the pellet to resuspend overnight at 4 °C.
T75: 150 µl for in vitro
T225: 450 µl for in vitro; 30-50 µl for in vivo
Note: Adjust volumes based on your desired GC/ml and expected yield if desired, but these volumes typically work well for both the sgRNA and dCas9-VPR lentiviruses. If even higher titers are desired, multiple culture plates can be combined during resuspension to increase GC/ml. - The next day, pre-chill a microcentrifuge to 4 °C. Transfer all supernatant to a chilled 1.5 ml microcentrifuge tube and centrifuge at 2,300 x g for 5 min at 4 °C to pellet debris.
- Avoiding the pellet, transfer the supernatant to a new 200 µl microcentrifuge tube. Aliquot the virus into single use vials with at least 10 µl/aliquot
- Spin down single use aliquots and flash freeze on dry ice. Store at -80 °C.
Note: We have not found a significant reduction in titer for at least a year if viruses are stored in an interior box within the freezer and rapidly placed on dry ice when accessed. - Obtain the GC/ml titer with Lenti-X qRT-PCR titration kit, using 1 µl virus serially diluted in 1x DPBS as the 150 µl input for the kit.
Part V: Transduction of primary cell cultures in vitro
Materials and Reagents
- Pipette tips (Denville, catalog numbers: 1159M43, 1159M40, 1159M42; Rainin, catalog number: 30389274)
- 50 ml conical tube (Fisher, catalog number: 14-959-49A)
- Primary neurons and their neuronal media
Note: Primary neuronal cultures can be prepared using a variety of established protocols. We use culture preparations described in Savell et al., 2019.
Equipment
- Pipettes (Rainin, catalog number: 17014415 & 17014393)
- Humidity, temperature, and CO2-controlled culture incubator (Thermo Forma Series II Water Jacketed CO2 incubator, set at 37 °C with 5% CO2)
Procedure
- Transduction with a multiplicity of infection (MOI) of 1,000 works well for both the sgRNA and dCas9-VPR constructs, but we have had success at lower MOIs as well. Calculate the virus volume needed for transduction. Transduction should occur 3-5 days after seeding primary neurons. Our standard procedure involves the transduction of an MOI of 1,000 for both the sgRNA and the dCas9-VPR lentiviruses of 125,000 primary neurons seeded into each well of a 24-well plate (Savell et al., 2019).
- We have not exhaustively tested different ratios of sgRNA:dCas9-VPR and find that a 1:1 seems to work well. For some gene targets, an increased MOI for both viruses results in boosted transcription, so you may want to try a range of MOIs to get the desired gene induction levels.
- If multiplexing, add equal MOIs for each sgRNA virus. You may also add a higher MOI of the dCas9-VPR lentiviruses to account for each gene, but we have not tested this thoroughly. Include an increased MOI non-targeting control (We recommend the bacterial LacZ gene).
Example: If targeting 2 genes at once, transduce with an MOI of 1000 for each gene-specific sgRNA and 1000-2000 for the dCas9-VPR construct. Transduce an MOI of 2000 for the non-targeting control to account for pooling sgRNA viruses.
- Remove approximately 55% of the primary neuron culture media from each well and place in a tube. Keep in cell culture incubator until later.
- Add viruses with predetermined volumes to the culture wells by pipetting directing into the remaining media. Gently mix and place back in the incubator.
- Eight to twelve hours later, prepare and warm neuronal media.
Notes:- Calculate enough media to wash with 80% of the original volume of neuronal media in the well.
- Calculate enough media to replace with one half fresh neuronal media.
- Aspirate the media on the cells (that contains the virus) and immediately (but gently) add the neuronal media wash.
- Aspirate the wash and add 50% fresh media, then add back the 50% conditioned media that was removed during Step 2.
- Allow constructs to express for at least 5-7 days before extracting RNA and measuring gene expression changes.
Part VI: Transduction of adult neurons in vivo
Materials and Reagents
- Large-scale production (T225) of both the sgRNA and the dCas9-VPR lentiviruses
- Microliter and Gastight Neuros Syringes (Hamilton, catalog number: 7653-01)
- 1x DPBS (Thermo-Fisher, catalog number: 14190144)
Procedure
- First, validate in vitro that a gene-specific sgRNA successfully induces your gene of interest (See Part V).
- Produce large-scale lentivirus preparations in a T225 for each construct and resuspend in only 30-50 µl sterile 1x DPBS.
- Titer viruses as described above and dilute sgRNA viruses with 1x sterile DPBS as necessary so that all sgRNAs used in the study are the same titer.
- Infuse a mixture of the sgRNA and the dCas9-VPR construct into the brain area of interest at a rate of 0.25 µl/min. For our studies, we used a total volume of 1.5 µl, with 0.5 µl of the sgRNA plasmid mixed with 1.0 µl of dCas9-VPR.
- Expression of the transgenes has been validated in rats 2 weeks after infusion.
Recipes
- 2XYT broth with ampicillin (per liter)
16 g tryptone
10 g yeast extract
5 g sodium chloride
Adjust the pH to 7.0- Autoclave and cool to room temperate
- Aseptically add 100 µg/ml ampicillin once solution cools to room temperature
- Store at 4 °C for up to 1 month
- HEK293T media
Gibco DMEM, high glucose, pyruvate
10% heat-inactivated FBS
1x Penicillin-streptomycin (100 U/ml)- 10% volume FBS US Origin, heat inactivated for 30 min at 50 °C immediately before making media
- Prepare media by adding 1% volume of pen-strep [100x Penicillin-streptomycin (10,000 U/ml)] and 10% volume of FBS to DMEM
- Sterile filter and store for up to a month at 4 °C. Protect from light
- Lentivirus production media (make fresh each time)
Lonza BioWhittaker UltraCulture Serum-free Medium without L-glutamine
2 mM L-glutamine
1 mM sodium pyruvate- Prepare media by adding 1/100th volume of L-glutamine, sodium bicarbonate, and sodium pyruvate stock solutions to UltraCulture Serum-free Medium without L-glutamine and sterile filter
- Protect from light and prepare fresh before each transfection
- 200 mM L-glutamine
- 100 mM sodium pyruvate
- 7.5% (w/v) sodium bicarbonate: Prepare a 7.5% solution by dissolving 7.5 g per 100 ml water and sterile filter
Acknowledgments
We would like to thank all current and past members of the Day Lab for their helpful suggestions on how to improve these protocols.
Competing interests
The authors have no competing interests to disclose.
Ethics
All experimental procedures involving animal subjects were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. All experimental procedures involving hazardous materials were approved by the Occupational Health and Safety Biosafety Committee at the University of Alabama at Birmingham.
References
- Chavez, A., Scheiman, J., Vora, S., Pruitt, B. W., Tuttle, M., Iyer E, P. R. , Lin, S., Kiani, S., Guzman, C. D., Wiegand, D. J., Ter-Ovanesyan, D., Braff, J. L., Davidsohn, N., Housden, B. E., Perrimon, N., Weiss, R., Aach, J., Collins, J. J. and Church, G. M. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12(4): 326-328.
- Ericsson, A. C., Crim, M. J. and Franklin, C. L. (2013). A brief history of animal modeling. Mo Med 110(3): 201-205.
- Synthetic Neurobiology Group. (2009). Lentivirus production for high-titer, cell-specific, in vivo neural labeling. http://edboyden.org/09.01.lentivirus.whitepaper.v2.pdf.
- Prelich, G. (2012). Gene overexpression: uses, mechanisms, and interpretation. Genetics 190(3): 841-854.
- Savell, K. E., Bach, S. V., Zipperly, M. E., Revanna, J. S., Goska, N. A., Tuscher, J. J., Duke, C. G., Sultan, F. A., Burke, J. N., Williams, D., Ianov, L. and Day, J. J. (2019). A neuron-optimized CRISPR/dCas9 activation system for robust and specific gene regulation. eNeuro 6(1). doi: 10.1523/ENEURO.0495-18.2019.
- Savell, K. E. and Day, J. J. (2017). Applications of CRISPR/Cas9 in the mammalian central nervous system. Yale J Biol Med 90(4): 567-581.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Savell, K. E., Sultan, F. A. and Day, J. J. (2019). A Novel Dual Lentiviral CRISPR-based Transcriptional Activation System for Gene Expression Regulation in Neurons. Bio-protocol 9(17): e3348. DOI: 10.21769/BioProtoc.3348.
- Savell, K. E., Bach, S. V., Zipperly, M. E., Revanna, J. S., Goska, N. A., Tuscher, J. J., Duke, C. G., Sultan, F. A., Burke, J. N., Williams, D., Ianov, L. and Day, J. J. (2019). A neuron-optimized CRISPR/dCas9 activation system for robust and specific gene regulation. eNeuro 6(1). doi: 10.1523/ENEURO.0495-18.2019.
Category
Neuroscience > Cellular mechanisms > Tissue isolation and culture
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.