- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Strand-specific Single-stranded DNA Sequencing (4S-seq) of E. coli genomes
(*contributed equally to this work) Published: Vol 9, Iss 15, Aug 5, 2019 DOI: 10.21769/BioProtoc.3329 Views: 7531
Reviewed by: Elizabeth LibbyMichael TschernerTimo A Lehti
Original research article
The authors used this protocol in:

Nov 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Most bacterial genomes have biased nucleotide composition, and the asymmetry is considered to be caused by a single-stranded DNA (ssDNA) deamination arising from the bacterial replication machinery. In order to evaluate the relationship experimentally, the position and frequency of ssDNA formed during replication must be verified clearly. Although many ssDNA detection technologies exist, almost all methods have been developed for eukaryotic genomes. To apply these to bacterial genomes, which harbor a smaller amount of DNA than those of eukaryotes, more efficient, new methods are required. Therefore, we developed a novel strand-specific ssDNA sequencing method, called 4S-seq, for the bacterial genome. The 4S-seq method enriches ssDNA in the extracted genomic DNA by a dsDNA-specific nuclease and implements a strand-specific library using a biotin label with a customized tag. As a result, the 4S-seq is able to calculate the ssDNA content in each strand (Watson/Crick) at each position of the genome efficiently.
Keywords: Bacterial genomeBackground
Nascent and template strand tracking is an important approach to investigate bacterial genome evolution. Since biased mutations in bacterial genome arise from asymmetric replication machinery, DNA strands that have been exposed to a single-stranded state for a long time will accumulate more mutations. Several strand-specific sequencing technologies are available for eukaryotic genomes, with various selection strategies for nascent DNA (Hyrien, 2015). In general, the nascent DNA is caught by immunoprecipitation with BrdU (Karnani et al., 2010) or a combination of antibodies against strand-specific binding proteins (Yu et al., 2014). Other protocols utilize a customized label (Smith and Whitehouse, 2012) or an agarose gel trap (Mesner et al., 2011) to select nascent DNA of a certain size. However, these strand-specific sequencing technologies for eukaryotic genomes are not readily applicable to bacterial genomes because the BrdU incorporation efficiency or the amount of DNA is overwhelmingly low in bacteria. Therefore, we developed a novel 4S-seq method for the bacterial genome. Overall, the 4S-seq method is broadly divided into ssDNA enrichment and strand-specific sequencing steps (Figure 1). The ssDNA is enriched in the fragmented genomic DNA using a duplex-specific nuclease. The strand-specific sequencing is implemented using a biotinylated adapter with an exclusively unique designed tag. At this time, the library is prepared using a low input library preparation kit because a low amount of enriched ssDNA is expected. Sequenced reads are analyzed with a custom Perl script, and the ssDNA location and strand are calculated. The new 4S-seq method for the bacterial genome described here does not require specialized equipment and can be applied from plasmid to mega-scale genomes. This 4S-seq was applied to research investigating the relationship between a base compositional bias in a bacterial genome and a spontaneous mutation in ssDNA, and revealed that there is a strong correlation between ssDNA frequency and the mutation rate (Kono et al., 2018). We show the protocol optimized for Escherichia coli.

Figure 1. Overview of the 4S-seq protocol. Steps A-C: purification of the sheared genomic DNA from E. coli cultures; Steps D-F: preparation of dephosphorylated ssDNA and the phosphorylated adapter; Steps G-J: purification of tagged ssDNA fragments using streptavidin beads; Step K: preparation of the library for the Illumina sequencer.
Materials and Reagents
- 1.5 ml tubes (Eppendorf, catalog number: 30108051)
- 0.2 ml tubes (NIPPON GENE CO., LTD., catalog number: BPB79211)
- 1000 ml pipette tips (Sorenson, catalog number: 34000)
- 200 ml pipette tips (NIPPON GENE CO., LTD., catalog number: FG-302RS)
- 10 ml pipette tips (Sorenson, catalog number: 37660)
- TapeStation D1000 Screen Tape (Agilent Technologies, catalog number: 5067-5582)
- 14 ml tube (CORNING, catalog number: 352057)
- 50 ml tube (Greiner Bio-One, catalog number: 227281)
- milliTUBE 1 ml AFA Fiber (Covaris, catalog number: 520131)
- PCR Clean-up Column
- E. coli strains to be analyzed
- BactoTM Tryptone (Thermo Fisher Scientific, catalog number: 211705)
- BactoTM Yeast Extract (Thermo Fisher Scientific, catalog number: 212750)
- NaCl (FUJIFILM Wako Pure Chemical Corporation, catalog number: 192-13925)
- Appropriate antibiotics
- Milli-Q water
- 10x PBS (NIPPON GENE CO., LTD., catalog number: 314-90185)
- NucleoSpin® Gel and PCR Clean-up (MACHEREY-NAGEL, catalog number: 74069.250)
- Binding Buffer NTI
- Wash Buffer NT3
- Elution Buffer NE
- NucleoSpin® Gel and PCR Clean-up Columns
- 2 ml Collection Tube
- Buffer NTC (MACHEREY-NAGEL, catalog number: 740654.100)
- QubitTM dsDNA BR Assay Kit (Thermo Fisher Scientific, catalog number: Q32850)
- Duplex-specific nuclease (Evrogen, catalog number: EA001)
- DSN enzyme
- DSN storage buffer
- 10x DSN master buffer
- TapeStation D1000 Reagents (Agilent Technologies, catalog number: 5067-5583)
- 0.5 M EDTA (FUJIFILM Wako Pure Chemical Corporation, catalog number: 311-90075)
- Agencourt AMPure XP (BECKMAN COULTER Life Sciences, catalog number: A63881)
- EtOH (FUJIFILM Wako Pure Chemical Corporation, catalog number: 054-7225)
- Sucrose (FUJIFILM Wako Pure Chemical Corporation, catalog number: 196-00015)
- Lysozyme (FUJIFILM Wako Pure Chemical Corporation, catalog number: 123-06721)
- 100 mg/ml RNase A (QIAGEN, catalog number: 19101)
- 1 M Tris-HCl (pH 8.0) (NIPPON GENE CO., LTD., catalog number: 312-90061)
- 1 M Tris-HCl (pH 7.5) (NIPPON GENE CO., LTD., catalog number: 312-90221)
- FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientific, catalog number: EF0651)
- FastAP Thermosensitive Alkaline Phosphatase
- 10x FastAP Buffer
- Oligo nucleotides
- Adapter_N8B 5′-GGGAANNNNNNNNTAGGGATAACAGGGTAATAGGAGGA-3′ (Eurofins Genomics, custom made)
- 1stAdapter_primer 5′-TCCTCCTATTACCCTGTTATCCCTA-3′ (Eurofins Genomics, custom made)
- T4 Polynucleotide Kinase (T4 PNK) and 10x T4 PNK buffer (NEW ENGLAND BioLab, catalog number: M0201S)
- 100 mM ATP (TAKARA BIO INC., catalog number: 4041)
- T4 DNA ligase and 10x T4 DNA ligase buffer (Enzymatics, catalog number: L6030-HC-L)
- Dynabeads MyOne Streptavidin C1 (Invitrogen, catalog number: DB65001)
- Tks Gflex DNA Polymerase (Gflex) and 2x Gflex PCR Buffer (TAKARA BIO INC., catalog number: R060A)
- I-Sce I and 10x CutSmart buffer (NEW ENGLAND BioLab, catalog number: R0694S)
- SMARTer ThruPLEX DNA-Seq Kit (TAKARA BIO INC., catalog number: RB4674)
- (Optional) Low input Library Prep Kit (Clontech Laboratories, Inc., catalog number: 634947)
- Qubit ssDNA Assay Kit (Thermo Fisher Scientific, Invitrogen, catalog number: Q10212)
- NextSeq 500 High Output Kit (75 cycles) (Illumina, Inc., catalog number: TG-160-2005)
- Luria-Bertani broth (LB) (see Recipes)
- TE buffer, pH 8.0 (see Recipes)
- Lysis buffer (see Recipes)
- 80% EtOH (see Recipes)
- 2x Binding and Washing (B&W) buffer (see Recipes)
- 1x B&W buffer (see Recipes)
Equipment
- 200 ml flask (Sansyo Co., ltd, catalog number: 82-0086)
- Pipettes (GILSON, catalog number: F167380)
- Digital photometer (TAITEC, catalog number: 0040889-000)
- Milli-Q water purification system (Merck, model: Direct-Q UV 5)
- Autoclave (TOMY, model: LSX-300)
- Centrifuge (TOMY, model: MX-307)
- Incubator (TAIYO COMPANY CO., LTD, catalog number: 2-6795-02)
- CovarisTM M220 Focused-ultrasonicatorTM Instrument (Thermo Fisher Scientific, catalog number: 4482277)
- Qubit 3.0 Fluorometer (Thermo Fisher Scientific, catalog number: Q33216)
- Heat block (WAKENBTECH CO., LTD, catalog number: WKN-9603)
- Thermal Cycler (LifeECO ver2.0) (NIPPON Genetics Co., Ltd, catalog number: TC-96GHbC)
- Agilent 2200 TapeStation (Agilent Technologies, catalog number: G2965AA)
- Magnetic stand for 0.2 ml tube (NIPPON Genetics Co., Ltd, catalog number: FG-SSMAG2)
- Magnetic stand for 1.5 ml tube (Thermo Fisher Scientific, catalog number: 12321D)
- NextSeq 500 instrument (Illumina, Inc., catalog number: SY-415-1001)
Software
- bcl2fastq (v. 2.18.0.12) (https://support.illumina.com/sequencing/sequencing_software/bcl2-fastq-conversion-software.html)
- BWA (v. 0.7.1-r1034) (http://bio-bwa.sourceforge.net)
- Samtools (v. 1.2) (http://samtools.sourceforge.net)
- FastQC (v. 0.10.1) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
Procedure
- Cell collection
- E. coli strains are inoculated by picking a single colony and cultured in 2 ml of LB with appropriate antibiotics using 14 ml tubes until they reach an optical density (OD) at 600 nm of 0.4 with a digital photometer.
- Fifty microliters of culture is inoculated to 50 ml of LB with appropriate antibiotics in a 200 ml flask and cultured until they reach an OD at 600 nm of 0.4 with a digital photometer.
- Transfer 50 ml of culture to a 50 ml tube and collect cells by centrifugation at 4,000 x g for 5 min at 4 °C, then remove the supernatant.
- After washing the cell pellet twice each with chilled 1 ml of PBS (pH 8.0), resuspend the cell pellet in 2 ml of Lysis buffer and incubate at 37 °C for 30 min.
- Collect cells by centrifugation at 8,000 x g for 5 min and gently remove the supernatant.
- DNA shearing
- Resuspend the cell pellet in 1 ml of TE buffer (pH 8.0) and transfer the cell lysate to a milliTUBE 1 ml AFA Fiber.
- Transfer the milliTUBE into a CovarisTM M220 Focused-ultrasonicatorTM Instrument and conduct the following shearing protocol [peak power (W): 75.0; duty factor (%): 5.0; cycles/burst: 200; treatment (sec): 1200; temperature (°C): 9.0 (maximum), 7.0 (setpoint), 5.0 (minimum)] to dissociate the genomic DNA into 300-400 bp fragments.
- Purification of sheared genomic DNA
- Dispense a 125 μl aliquot of the total 1 ml sheared DNA solution into eight 1.5 ml
Note: The capacity of the NucleoSpin Gel is limited (≤ 25 μg, 50 bp-20 kb). Purification of sheared genomic DNA is performed at room temperature. - Mix sheared DNA solution with 250 μl Binding Buffer NTC.
- Place a NucleoSpin® Gel and PCR Clean-up Column into a 2 ml Collection Tube and load sample.
- Centrifuge for 30 s at 11,000 x g.
- Discard flow-through and place the column back into the collection tube.
- Add 700 μl Wash Buffer NT3 buffer to the NucleoSpin® Gel and PCR Clean-up Column.
- Centrifuge for 30 s at 11,000 x g.
- Discard flow-through and place the column back into the collection tube.
- Centrifuge for 1 min at 11,000 x g to remove Wash Buffer NT3.
- Place a NucleoSpin® Gel and PCR Clean-up Column into a new 1.5 ml tube.
- Add 20 μl Elution Buffer NE and incubate at room temperature (18-25 °C) for 1 min.
- Centrifuge for 1 min at 11,000 x g.
- Pool all eight eluted DNA solutions into a new 1.5 ml tube.
- Dispense a 125 μl aliquot of the total 1 ml sheared DNA solution into eight 1.5 ml
- Duplex-specific nuclease reaction
- Determine the DNA concentration of purified genomic DNA with QubitTM dsDNA BR Assay Kit according to the manufacturer's instructions.
- Add 10x DSN master buffer and DSN solution (final conc. 0.05 U for 1 μg of purified DNA fragments) to purified DNA fragments, and adjust the total reaction volume to 200 μl.
Note: The efficiency of the dsDNA cleaving is very high. We have demonstrated that 0.05 U is enough to cleave the 2 μg of dsDNA completely (Figure 2). In addition, since quantification of ssDNA with the fluorescent dye of the Qubit ssDNA Assay Kit (Thermo Fisher Scientific, Invitrogen) might not be absolutely accurate, in this protocol, treated DNA samples are qualitatively assessed.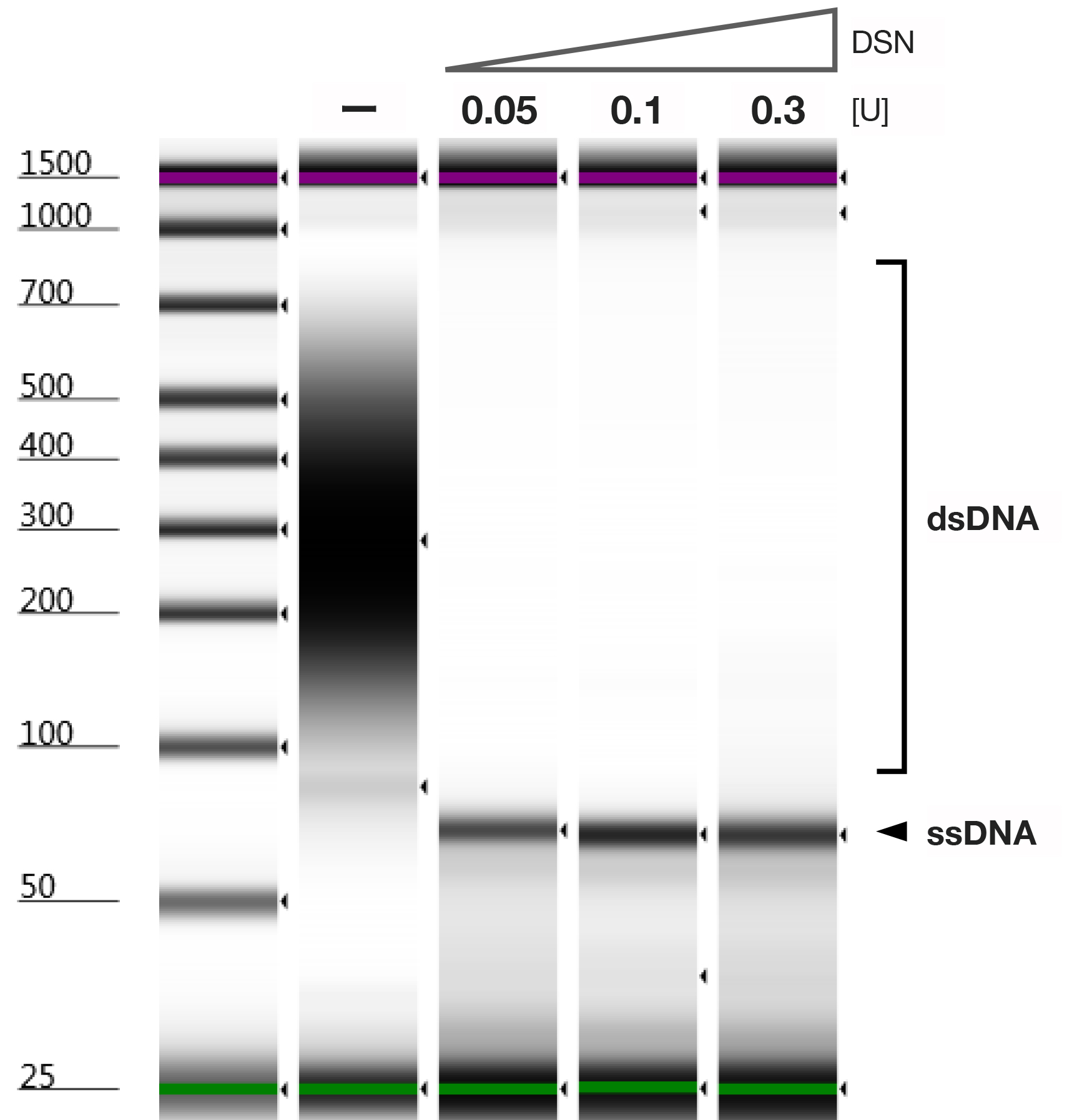
Figure 2. DSN reaction efficiency. dsDNA cleaving reaction from 2 μg of DNA pool including 70-mer ssDNA was either mock-treated or treated with 0.05, 0.1, or 0.3 U of DSN and analyzed by TapeStation with the D1000 ScreenTape. - Incubate the reactions at 65 °C for 20 min to specifically cleave the dsDNA on the heat block.
- To inactivate the DSN reaction, add 1.5 μl of 0.5 M EDTA (final concentration: 5 mM) and incubate at room temperature for 5 min.
- Add 604.5 μl AMPure XP (3.0x) and mix by pipetting.
Note: Because the enriched ssDNA size is expected to be about 100 bp, a little bit more AMPure is used to recover the whole amount as much as possible. - After incubation at room temperature for 5 min, place the reaction tube into a magnetic stand for 2 min to separate the beads from the solution.
- Aspirate the supernatant from the reaction tube and discard.
- Dispense 200 μl of 80% EtOH and incubate at room temperature for at least 30 s.
- Aspirate out the ethanol and discard.
- Repeat Steps D7-D8 again.
- Take out the tube from the magnetic stand and incubate at room temperature within 5 min to remove EtOH completely.
- Add 17 μl of 10 mM Tris-HCl (pH 8.0) and mix by pipetting.
- Incubate at room temperature for 2 min.
- Place the reaction tube into a magnetic stand for 1 min to separate beads from solution.
- Transfer purified product to a new 0.2 ml tube.
- Examples of purified ssDNA before and after DSN treatment are shown in Figure 3. One microliter of sample is used in this demonstration.
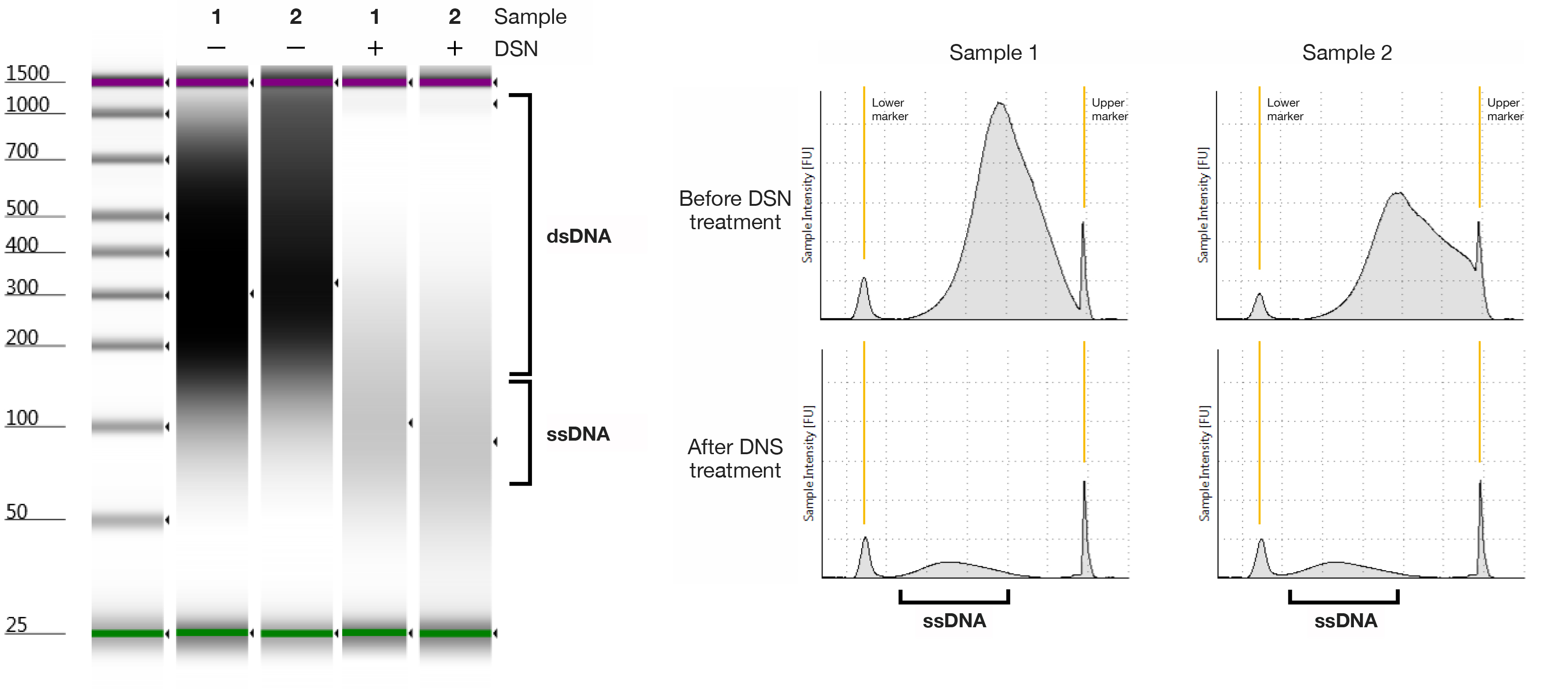
Figure 3. Distribution of ssDNA after the ssDNA enrichment by DSN treatment. Sixty micrograms of genomic DNA was used in a volume of 300 μl for 20 min with 1.5 U of DSN, and the reaction was inactivated by 5 mM EDTA. The enriched ssDNA was observed by the TapeStation with the D1000 ScreenTape.
- Dephosphorylation of ssDNA
- Add 3 μl of 10x FastAP buffer and 2 μl of FastAP to the purified ssDNA and adjust the total reaction volume to 30 μl.
- Transfer the tube to the thermal cycler and incubate at 37 °C for 10 min.
- Inactivate and denature the dephosphorylated ssDNA solution by incubating the reaction at 95 °C for 2 min, quickly transfer the tube onto ice for 2-3 min, and then subsequently transfer it to room temperature (products: 50 μl of [deP-ssDNA]).
Note: [deP-ssDNA] can be stored at -20 °C overnight.
- Phosphorylation of 1stAdapter_BIOTEG
- Combine the following reagents in Table 1:
Table 1. Reagents used for phosphorylation of adapter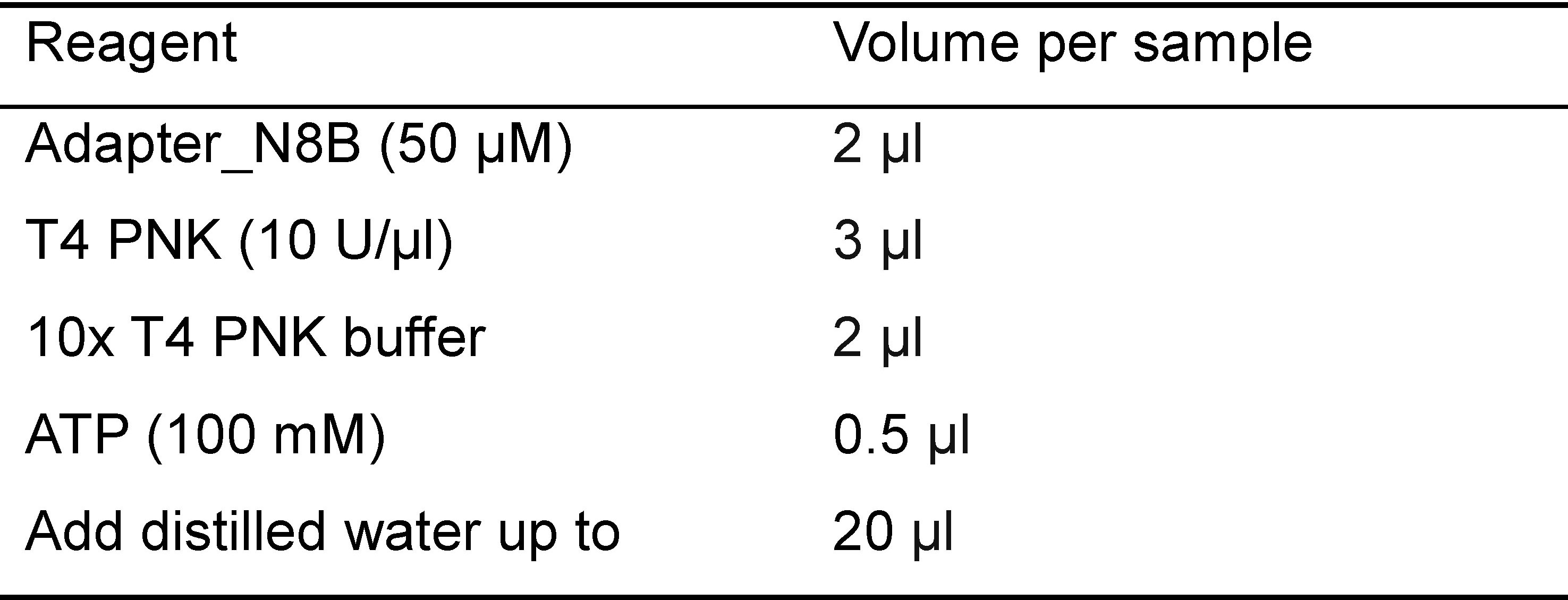
- Incubate the reactions at 37 °C for 1 h, inactivate at 95 °C for 5 min on the thermal cycler, and transfer to room temperature (products: 5 μM of [P-Adapter_N8B]).
Note: [P-adapter_N8B] can be stored at -20 °C overnight.
- Combine the following reagents in Table 1:
- Ligation
- Combine the following reagents in Table 2.
Table 2. Reagents used for ligation of ssDNA and adapter
- Incubate the reactions at 16 °C overnight (products: [ligation product])
- Combine the following reagents in Table 2.
- Immobilization of the biotinylated ssDNA fragment
- Resuspend the Dynabeads MyOne Streptavidin C1 beads in the vial by vortexing.
- Transfer the 25 μl of beads (10 μg/μl) to a 0.2 ml tube.
- Add an equal volume of 1x B&W Buffer and resuspend.
- Place the tube on a magnetic stand for 1 min and discard the supernatant.
- Remove the tube from the magnetic stand and resuspend the washed beads in 25 μl of 1x B&W Buffer.
- Repeat Steps H4-H5 twice, for a total of 3 washes and resuspend the washed beads in 50 μl of 2x B&W Buffer (5 μg/μl).
- Add an equal volume of [ligation product].
- Incubate for 15 min at room temperature using gentle rotation.
- Separate the [ligation product] coated beads on a magnetic stand for 2-3 min.
- Wash the coated beads 2 times with 1x B&W Buffer and discard 1x B&W Buffer (product: [ssDNA-Adapter-C1beads]).
- Primer annealing and extension
- Combine the following reagents in Table 3:
Table 3. Reagents used for primer annealing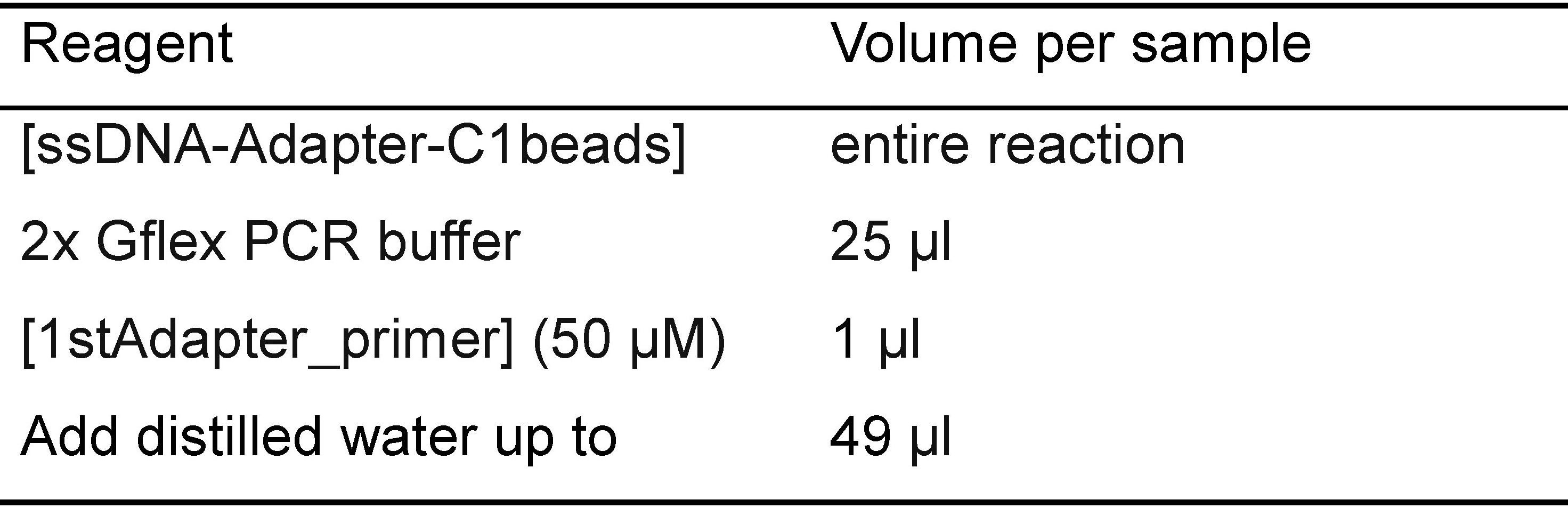
- Incubate the reactions at 65 °C for 2 min and place on ice for 1 min.
- Transfer the tube to a precooled thermal cycler at 15 °C (leave the lid open) and add 1 μl of Gflex.
- Increase the temperature by 0.1 °C/s, from 15 °C to 68 °C, and maintain the reaction at 68 °C for 5 min.
- Wash the beads with 1x B&W buffer (product: [dsDNA-beads]).
- Combine the following reagents in Table 3:
- I-SceI restriction digest to release the product from the beads
- Add 1 μl of I-SceI and 5 μl of 10x CutSmart buffer to [dsDNA-beads], and bring the volume up to 50 μl.
- Incubate at 37 °C for 1 h, and transfer the supernatant to a new tube.
- Add 150 μl AMPure XP (3.0x) and mix by pipetting.
Note: The fragment size is expected to be about 100 bp, a little bit more AMPure is used to recover the whole amount as much as possible. - After incubation at room temperature for 5 min, place the reaction tube into a magnetic stand for 2 min to separate the beads from the solution.
- Aspirate the supernatant from the reaction tube and discard.
- Dispense 200 μl of 80% EtOH and incubate at room temperature for at least 30 s.
- Aspirate out the ethanol and discard.
- Repeat Steps J6-J7 again.
- Take out the tube from the magnetic stand and incubate at room temperature within 5 min to remove EtOH completely.
- Add 10 μl of 10 mM Tris-HCl (pH 8.0) and mix by pipetting.
- Incubate at room temperature for 2 min.
- Place the reaction tube into a magnetic stand for 1 min to separate the beads from solution.
- Transfer purified product to a new 0.2 ml tube.
- Library preparation and sequencing
- The library is prepared using the ThruPLEX DNA-seq Kit according to the manufacturer's instructions.
Note: Because a low amount of adapter-ligated DNA fragment is expected, the low input library preparation kit (ThruPLEX DNA-seq Kit) is used. Of course, other kits may be used (such as Low input Library Prep Kit, Clontech Laboratories, Inc.). Using this protocol we successfully prepared the library with 14 cycle amplification. - Quality check of the library is performed by the TapeStation with D1000 ScreenTape (Figure 4).
- Sequencing is performed on the NextSeq 500 instrument using a NextSeq 500 High Output Kit (75 bp single-end read).
Note: The read length should be 70 bp or more in consideration of the adapter size. The optimal amount of sequence read is 1 million for E. coli genome. The library is pooled according to the amount of read required to analysis. In the case of NextSeq 500 High Output Kit (75 bp single-end read), you can pool approximately 1% of the total.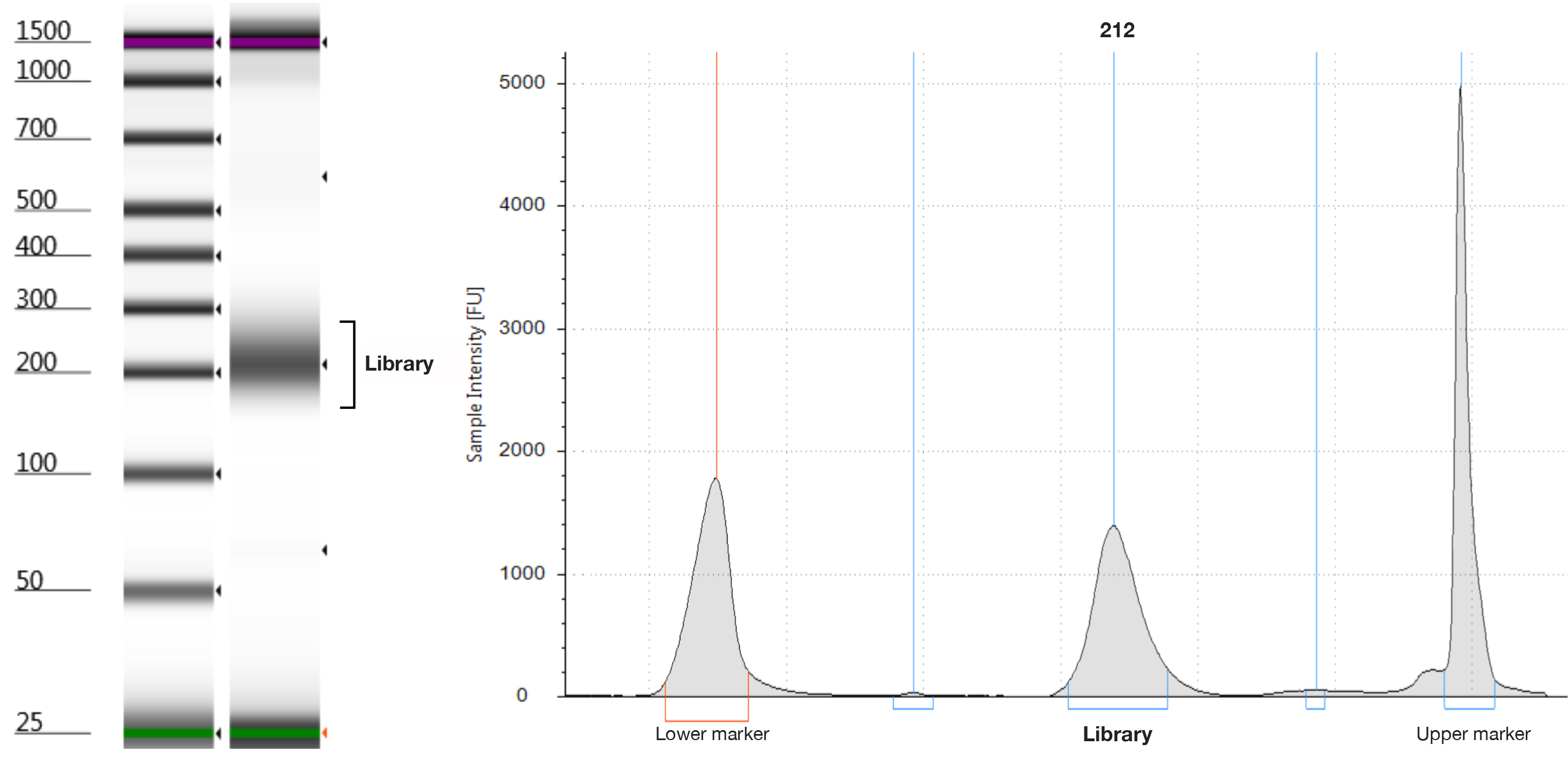
Figure 4. Final library of ThruPLEX with 14 cycle amplification was analyzed by the TapeStation with the D1000 ScreenTape assay. The electropherogram shows a peak with a maximum at 212 bp.
- The library is prepared using the ThruPLEX DNA-seq Kit according to the manufacturer's instructions.
Data analysis
- Perform a demultiplex of sequenced raw data to fastq format using bcl2fastq.
- To assess the sequence read quality, run a FastQC (v. 0.10.1).
- Align sequence reads to a reference genome using BWA-MEM (v. 0.7.1-r1034; Li and Durbin, 2009), and create SAM format file without unmapped read using samtools (v. 1.2).
- The 4S-seq reads contain a molecular tag (5’-GGGAANNNNNNNNTAGGG-3’) in addition to the adapter, and this tag sequence can be used to determine whether the read originated from the Watson or Crick strand.
Note: The ssDNA skew, represented as (Watson - Crick)/(Watson + Crick), can be calculated by a custom Perl script (available at 4Spgm.pl, https://github.com/nkono/4S-seq). Figure 5 represents the example commands of the analysis pipeline and output.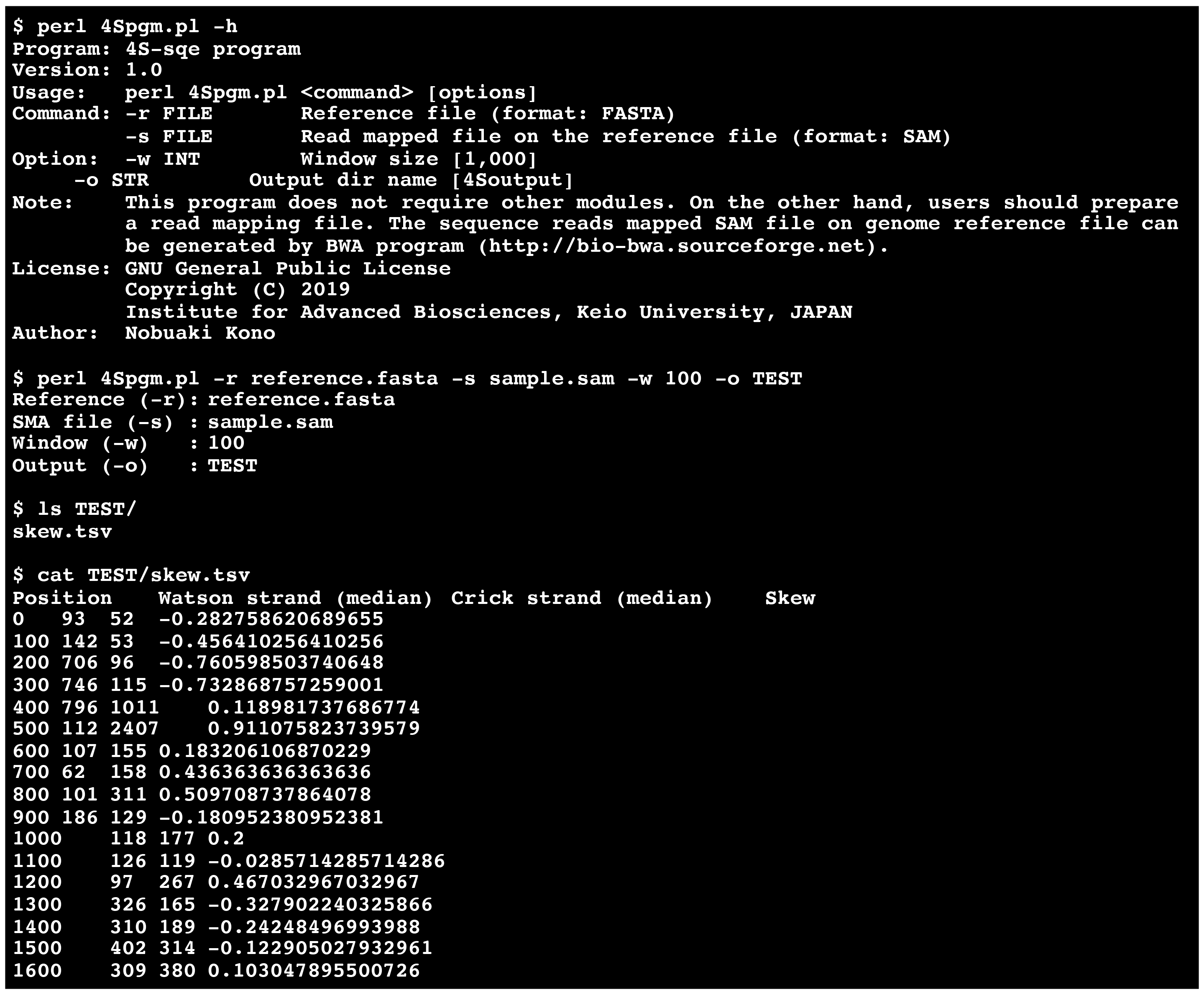
Figure 5. The example of 4Spgm. You can obtain the ssDNA skew data just by using a reference genome file and a SAM file.
Recipes
- LB
2 g of BactoTM Tryptone (Thermo Fisher SCIENTIFIC)
1 g of BactoTM Yeast Extract (Thermo Fisher SCIENTIFIC)
1 g of NaCl (FUJIFILM Wako Pure Chemical Corporation)
in 200 ml of milli-Q water
After sterilizing with autoclave and adding appropriate antibiotics, store at room temperature - TE buffer
1 ml of 1 M Tris-HCl (pH 8.0, 10 mM final concentration) (NIPPON GENE CO., LTD)
200 μl of 0.5 M EDTA (1 mM final concentration) (FUJIFILM Wako Pure Chemical Corporation)
in 100 ml of Milli-Q water
Store at room temperature - Lysis buffer
1 ml of 1 M Tris-HCl (pH 8.0, 10 mM final concentration) (NIPPON GENE CO., LTD)
2 ml of 0.5 M EDTA (10 mM final concentration) (FUJIFILM Wako Pure Chemical Corporation)
2 g of Sucrose (20% [w/v] final concentration) (FUJIFILM Wako Pure Chemical Corporation)
292.2 mg of NaCl (50 mM final concentration) (FUJIFILM Wako Pure Chemical Corporation)
20 mg of Lysozyme (200 μg/ml final concentration) (FUJIFILM Wako Pure Chemical Corporation)
10 μl of 100 mg/ml RNase A (10 μg/ml final concentration) (QIAGEN)
in 100 ml of Milli-Q water
Freshly prepare before use - 80% (v/v) EtOH
8 ml of EtOH (FUJIFILM Wako Pure Chemical Corporation) in 10 ml of Milli-Q water
Store at room temperature - 2x B&W buffer
1 ml of 1 M Tris-HCl (pH 7.5, 10 mM final concentration) (NIPPON GENE CO., LTD)
200 μl of 0.5 M EDTA (1 mM final concentration) (FUJIFILM Wako Pure Chemical Corporation)
11.688 g of NaCl (2 M final concentration) (FUJIFILM Wako Pure Chemical Corporation)
in 100 ml of Milli-Q water
Store at room temperature - 1x B&W buffer
500 μl of 1 M Tris-HCl (pH 7.5, 5 mM final concentration) (NIPPON GENE CO., LTD)
100 μl of 0.5 M EDTA (500 μM final concentration) (FUJIFILM Wako Pure Chemical Corporation)
5.844 g of NaCl (1 M final concentration) (FUJIFILM Wako Pure Chemical Corporation)
in 100 μl of milli-Q water
Store at room temperature
Acknowledgments
This protocol was adapted from our established publication (Kono, et al., 2018). We thank Nozomi Abe and Yuki Takai for technical assistance, and James Fleming for the English language proof-reading. This work was supported by research funds from the Yamagata Prefectural Government and Tsuruoka City, Japan.
Competing interests
The authors declare no competing interests.
References
- Hyrien, O. (2015). Peaks cloaked in the mist: the landscape of mammalian replication origins. J Cell Biol 208(2): 147-160.
- Karnani, N., Taylor, C. M., Malhotra, A. and Dutta, A. (2010). Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell 21(3): 393-404.
- Kono, N., Tomita, M. and Arakawa, K. (2018). Accelerated Laboratory Evolution Reveals the Influence of Replication on the GC Skew in Escherichia coli. Genome Biol Evol 10(11): 3110-3117.
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14): 1754-1760.
- Mesner, L. D., Valsakumar, V., Karnani, N., Dutta, A., Hamlin, J. L. and Bekiranov, S. (2011). Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res 21(3): 377-389.
- Smith, D. J. and Whitehouse, I. (2012). Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483(7390): 434-438.
- Yu, C., Gan, H., Han, J., Zhou, Z. X., Jia, S., Chabes, A., Farrugia, G., Ordog, T. and Zhang, Z. (2014). Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell 56(4): 551-563.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Masuda, T., Kono, N., Tomita, M. and Arakawa, K. (2019). Strand-specific Single-stranded DNA Sequencing (4S-seq) of E. coli genomes. Bio-protocol 9(15): e3329. DOI: 10.21769/BioProtoc.3329.
Category
Microbiology > Microbial genetics > DNA > DNA sequencing
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.